
Metformin as a Potential Treatment Option for Endometriosis

 ,
,  ,
,
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
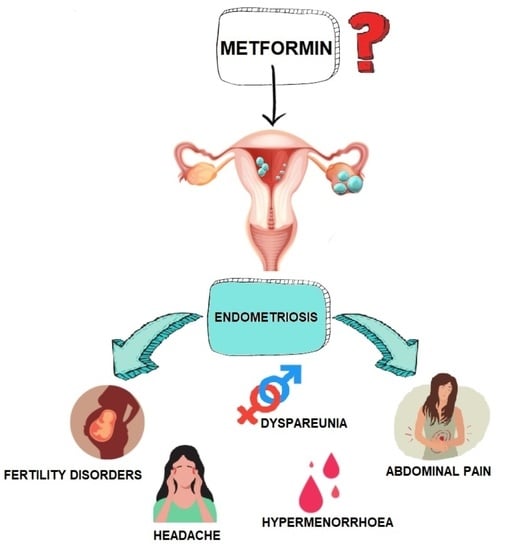
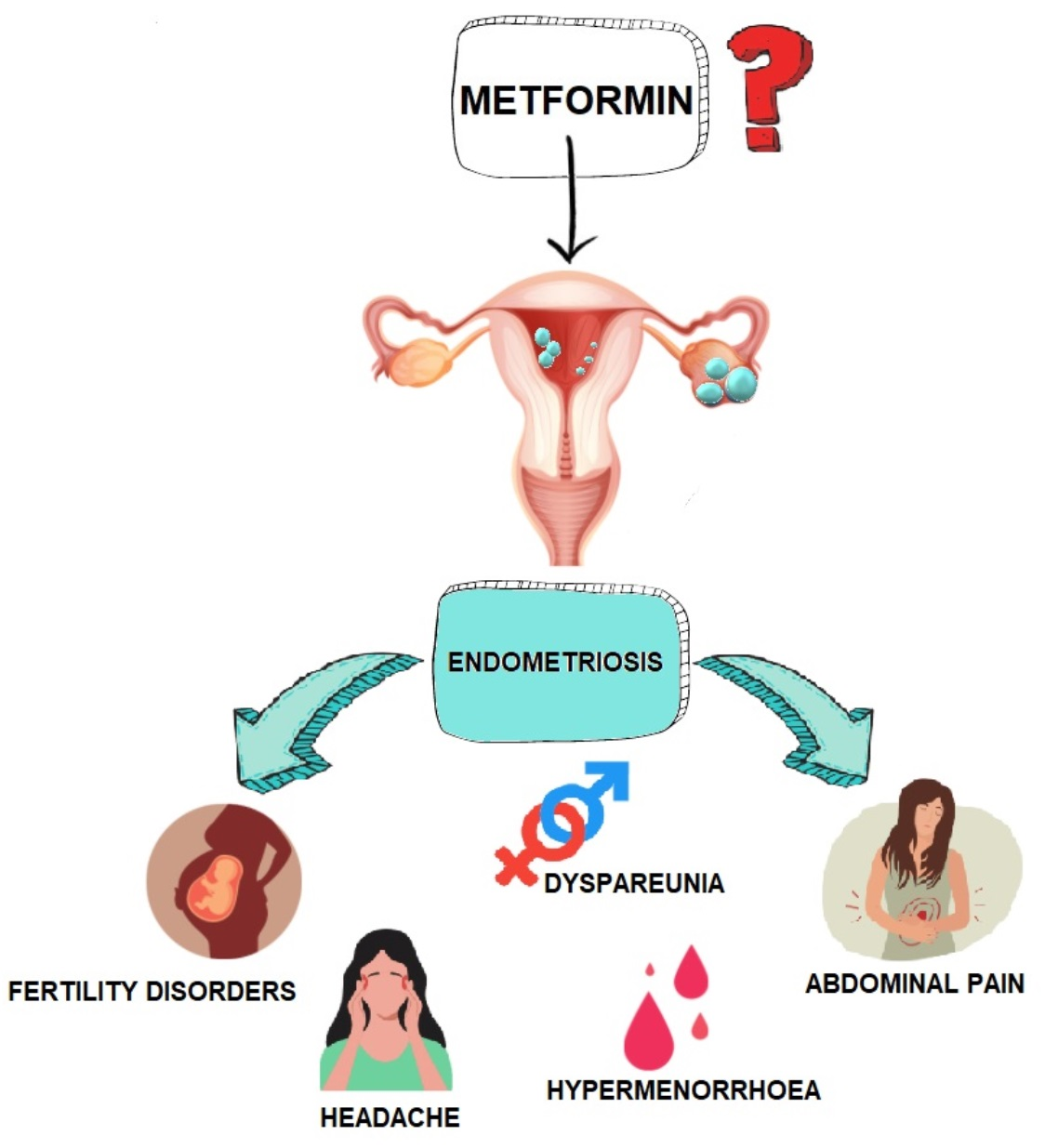
1.1. Pathophysiology of Endometriosis
1.2. Current Treatment Options for Endometriosis
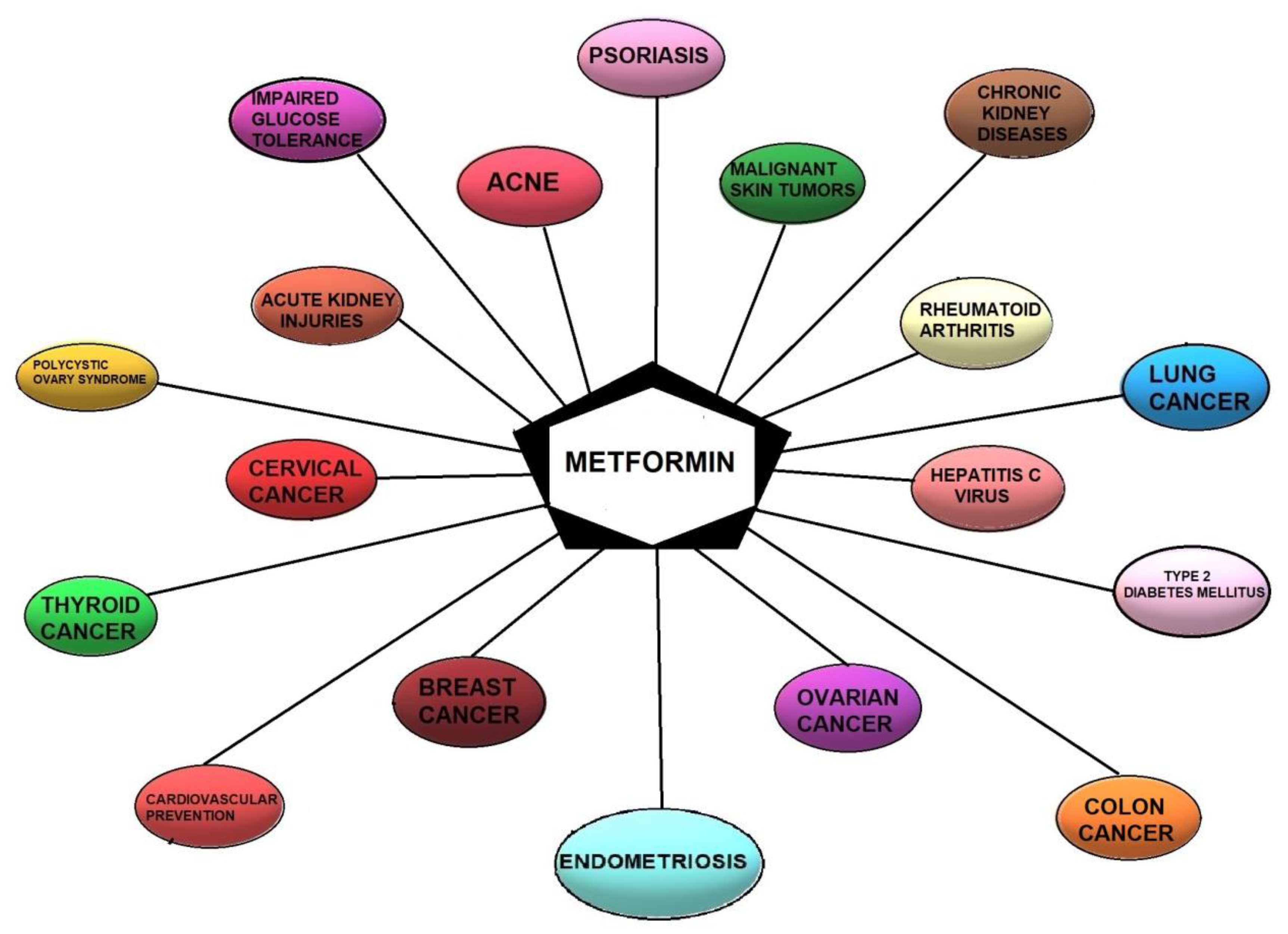
2. Metformin—State of the Art
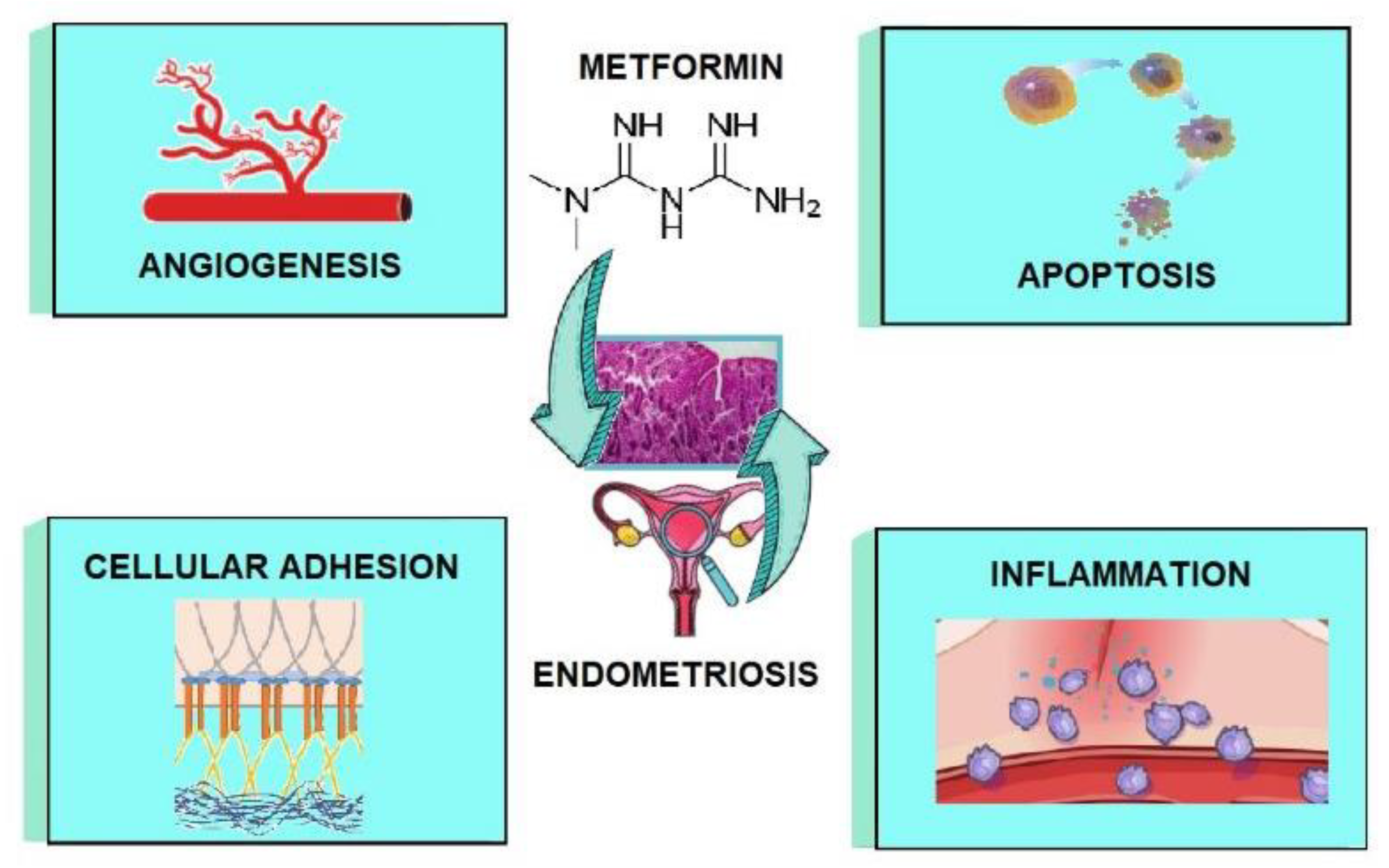
3. Metformin as a Potential Treatment Option for Endometriosis—Mechanisms
3.1. The Impact of Metformin on Inflammation
3.2. The Impact of Metformin on Angiogenesis
3.3. The Impact of Metformin on Adhesion and Invasion
3.4. The Impact of Metformin on Apoptosis
4. Metformin and Estrobolome
5. The Use of Metformin as Pharmacological Therapy in Endometriosis—Clinical Research
6. Possible Beneficial Indications for Using Metformin in Endometriosis
7. Side Effects of Using Metformin
8. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Vallée, A.; Vallée, J.N.; Le Blanche, A.; Lecarpentier, Y. PPARγ Agonists: Emergent Therapy in Endometriosis. Pharmaceuticals 2021, 14, 543. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Yamada, Y.; Morioka, S.; Niiro, E.; Shigemitsu, A.; Ito, F. Mechanism of Pain Generation for Endometriosis-Associated Pelvic Pain. Arch. Gynecol. Obstet. 2014, 289, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Saunders, P.T.K.; Horne, A.W. Endometriosis: Etiology, pathobiology, and therapeutic prospects. Cell 2021, 184, 2807–2824. [Google Scholar] [CrossRef] [PubMed]
- Dull, A.M.; Moga, M.A.; Dimienescu, O.G.; Sechel, G.; Burtea, V.; Anastasiu, C.V. Therapeutic Approaches of Resveratrol on Endometriosis via Anti-Inflammatory and Anti-Angiogenic Pathways. Molecules 2019, 24, 667. [Google Scholar] [CrossRef] [PubMed]
- Barcz, E.; Milewski, Ł.; Dziunycz, P.; Kamiński, P.; Płoski, R.; Malejczyk, J. Peritoneal Cytokines and Adhesion Formation in Endometriosis: An Inverse Association with Vascular Endothelial Growth Factor Concentration. Fertil. Steril. 2012, 97, 1380–1386. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.Y.; Chen, H.Y.; Chen, W.; Liu, Y.N.; Fu, Y.; Wang, L.N. Expression of Inflammatory Cytokines in Serum and Peritoneal Fluid from Patients with Different Stages of Endometriosis. Gynecol. Endocrinol. 2018, 34, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Sikora, J.; Smycz-Kubańska, M.; Mielczarek-Palacz, A.; Kondera-Anasz, Z. Abnormal Peritoneal Regulation of Chemokine Activation-The Role of IL-8 in Pathogenesis of Endometriosis. Am. J. Reprod. Immunol. 2017, 77, e12622. [Google Scholar] [CrossRef]
- Sikora, J.; Mielczarek-Palacz, A.; Kondera-Anasz, Z. Association of the Precursor of Interleukin-1β and Peritoneal Inflammation-Role in Pathogenesis of Endometriosis. J. Clin. Lab. Anal. 2016, 30, 831–837. [Google Scholar] [CrossRef]
- Kimber-Trojnar, Ż.; Pilszyk, A.; Niebrzydowska, M.; Pilszyk, Z.; Ruszała, M.; Leszczyńska-Gorzelak, B. The Potential of Non-Invasive Biomarkers for Early Diagnosis of Asymptomatic Patients with Endometriosis. J. Clin. Med. 2021, 10, 2762. [Google Scholar] [CrossRef]
- Vetvicka, V.; Laganà, A.S.; Salmeri, F.M.; Triolo, O.; Palmara, V.I.; Vitale, S.G.; Sofo, V.; Králíčková, M. Regulation of Apoptotic Pathways during Endometriosis: From the Molecular Basis to the Future Perspectives. Arch. Gynecol. Obstet. 2016, 294, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, K.Y.; Chang, N.; Lin, S.C.; Li, Y.H.; Wu, M.H. Inhibition of Dual Specificity Phosphatase-2 by Hypoxia Promotes Interleukin-8-Mediated Angiogenesis in Endometriosis. Hum. Reprod. 2014, 29, 2747–2755. [Google Scholar] [CrossRef] [PubMed]
- Kalu, E.; Sumar, N.; Giannopoulos, T.; Patel, P.; Croucher, C.; Sherriff, E.; Bansal, A. Cytokine Profiles in Serum and Peritoneal Fluid from Infertile Women with and without Endometriosis. J. Obstet. Gynaecol. Res. 2007, 33, 490–495. [Google Scholar] [CrossRef]
- Kyama, C.M.; Overbergh, L.; Mihalyi, A.; Meuleman, C.; Mwenda, J.M.; Mathieu, C.; D’Hooghe, T.M. Endometrial and Peritoneal Expression of Aromatase, Cytokines, and Adhesion Factors in Women with Endometriosis. Fertil. Steril. 2008, 89, 301–310. [Google Scholar] [CrossRef]
- Borghese, B.; Mondon, F.; Noël, J.C.; Fayt, I.; Mignot, T.M.; Vaiman, D.; Chapron, C. Gene Expression Profile for Ectopic versus Eutopic Endometrium Provides New Insights into Endometriosis Oncogenic Potential. Mol. Endocrinol. 2008, 22, 2557–2562. [Google Scholar] [CrossRef]
- Hadler-Olsen, E.; Winberg, J.O.; Uhlin-Hansen, L. Matrix Metalloproteinases in Cancer: Their Value as Diagnostic and Prognostic Markers and Therapeutic Targets. Tumor Biol. 2013, 34, 2041–2051. [Google Scholar] [CrossRef]
- Jana, S.; Rudra, D.S.; Paul, S.; Snehasikta, S. Curcumin Delays Endometriosis Development by Inhibiting MMP-2 Activity. Indian J. Biochem. Biophys. 2012, 49, 342–348. [Google Scholar]
- Yao, M.; Kargman, S.; Lam, E.C.; Kelly, C.R.; Zheng, Y.; Luk, P.; Kwong, E.; Evans, J.F.; Wolfe, M.M. Inhibition of Cyclooxygenase-2 by Rofecoxib Attenuates the Growth and Metastatic Potential of Colorectal Carcinoma in Mice. Cancer Res. 2003, 63, 586–592. [Google Scholar]
- Wu, M.H.; Wang, C.A.; Lin, C.C.; Chen, L.C.; Chang, W.C.; Tsai, S.J. Distinct Regulation of Cyclooxygenase-2 by Interleukin-1β in Normal and Endometriotic Stromal Cells. J. Clin. Endocrinol. Metab. 2005, 90, 286–295. [Google Scholar] [CrossRef]
- Sun, H.S.; Hsiao, K.Y.; Hsu, C.C.; Wu, M.H.; Tsai, S.J. Transactivation of Steroidogenic Acute Regulatory Protein in Human Endometriotic Stromalcells Is Mediated by the Prostaglandin EP2 Receptor. Endocrinology 2003, 144, 3934–3942. [Google Scholar] [CrossRef]
- Buchanan, F.G.; Wang, D.; Bargiacchi, F.; DuBois, R.N. Prostaglandin E2 Regulates Cell Migration via the Intracellular Activation of the Epidermal Growth Factor Receptor. J. Biol. Chem. 2003, 278, 35451–35457. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. The Role of Prostaglandin E(2) in Tumor-Associated Immunosuppression. Trends Mol. Med. 2016, 22, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Gazvani, R.; Templeton, A. Peritoneal Environment, Cytokines and Angiogenesis in the Pathophysiology of Endometriosis. Reproduction 2002, 123, 217–226. [Google Scholar] [CrossRef]
- Lousse, J.C.; Van Langendonckt, A.; González-Ramos, R.; Defrère, S.; Renkin, E.; Donnez, J. Increased Activation of Nuclear Factor-Kappa B (NF-KappaB) in Isolated Peritoneal Macrophages of Patients with Endometriosis. Fertil. Steril. 2008, 90, 217–220. [Google Scholar] [CrossRef]
- Ahn, S.H.; Monsanto, S.P.; Miller, C.; Singh, S.S.; Thomas, R.; Tayade, C. Pathophysiology and Immune Dysfunction in Endometriosis. BioMed Res. Int. 2015, 2015, 795976. [Google Scholar] [CrossRef]
- Wu, M.H.; Shoji, Y.; Wu, M.C.; Chuang, P.C.; Lin, C.C.; Huang, M.F.; Tsai, S.J. Suppression of Matrix Metalloproteinase-9 by Prostaglandin E(2) in Peritoneal Macrophage Is Associated with Severity of Endometriosis. Am. J. Pathol. 2005, 167, 1061–1069. [Google Scholar] [CrossRef]
- Shikone, T.; Yamoto, M.; Kokawa, K.; Yamashita, K.; Nishimori, K.; Nakano, R. Apoptosis of Human Corpora Lutea during Cyclic Luteal Regression and Early Pregnancy. J. Clin. Endocrinol. Metab. 1996, 81, 2376–2380. [Google Scholar] [PubMed]
- Jin, Y.P.; Fishbein, M.C.; Said, J.W.; Jindra, P.T.; Rajalingam, R.; Rozengurt, E.; Reed, E.F. Anti-HLA class I antibody-mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum. Immunol. 2004, 65, 291–302. [Google Scholar] [CrossRef]
- Romashkova, J.A.; Makarov, S.S. NF-KappaB Is a Target of AKT in Anti-Apoptotic PDGF Signalling. Nature 1999, 401, 86–90. [Google Scholar] [CrossRef]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs Block Apoptotic Events Induced by Caspase-8 and Cytochrome c by Direct Inhibition of Distinct Caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef]
- Jones, R.G.; Parsons, M.; Bonnard, M.; Chan, V.S.; Yeh, W.C.; Woodgett, J.R.; Ohashi, P.S. Protein Kinase B Regulates T Lymphocyte Survival, Nuclear Factor KappaB Activation, and Bcl-X(L) Levels in Vivo. J. Exp. Med. 2000, 191, 1721–1734. [Google Scholar] [CrossRef]
- Zong, W.X.; Edelstein, L.C.; Chen, C.; Bash, J.; Gélinas, C. The Prosurvival Bcl-2 Homolog Bfl-1/A1 Is a Direct Transcriptional Target of NF-KappaB That Blocks TNFalpha-Induced Apoptosis. Genes Dev. 1999, 13, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the Tumour Vasculature: Insights from Physiological Angiogenesis. Nat. Rev. Cancer. 2010, 10, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Kerbel, R.S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Curcumin: A Therapeutic Strategy in Cancers by Inhibiting the Canonical WNT/β-Catenin Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 323. [Google Scholar] [CrossRef]
- Vallée, A.; Guillevin, R.; Vallée, J.N. Vasculogenesis and Angiogenesis Initiation under Normoxic Conditions through Wnt/β-Catenin Pathway in Gliomas. Rev. Neurosci. 2018, 29, 71–91. [Google Scholar] [CrossRef]
- Mayerhofer, M.; Valent, P.; Sperr, W.R.; Griffin, J.D.; Sillaber, C. BCR/ABL Induces Expression of Vascular Endothelial Growth Factor and Its Transcriptional Activator, Hypoxia Inducible Factor-1alpha, through a Pathway Involving Phosphoinositide 3-Kinase and the Mammalian Target of Rapamycin. Blood 2002, 100, 3767–3775. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef]
- Reiss, Y.; Machein, M.R.; Plate, K.H. The Role of Angiopoietins during Angiogenesis in Gliomas. Brain Pathol. 2005, 15, 311–317. [Google Scholar] [CrossRef]
- Attar, E.; Bulun, S.E. Aromatase and other steroidogenic genes inendometriosis: Translational aspects. Hum. Reprod. Update 2006, 12, 49–56. [Google Scholar] [CrossRef]
- Sebastian, S.; Bulun, S.E. A highly complex organization of the reg-ulatory region of the human CYP19 (aromatase) gene revealed bythe Human Genome Project. J. Clin. Endocrinol. Metab. 2001, 86, 4600–4602. [Google Scholar] [CrossRef]
- Laschke, M.W.; Menger, M.D. The gut microbiota: A puppet master in the patho-genesis of endometriosis? Am. J. Obstet. Gynecol. 2016, 215, 68. [Google Scholar] [CrossRef] [PubMed]
- Goenka, L.; George, M.; Sen, M. A peek into the drug development scenario of en-dometriosis−Asystematic review. Biomed. Pharmacother. 2017, 90, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Noble, L.S.; Takayama, K.; Zeitoun, K.M.; Putman, J.M.; Johns, D.A.; Hinshelwood, M.M.; Bulun, S.E.; Agarwal, V.R.; Zhao, Y.; Carr, B.R. Prostaglandin E2 sti-mulates aromatase expression in endometriosis-derived stromalcells. J. Clin. Endocrinol. Metab. 1997, 82, 600–606. [Google Scholar] [PubMed]
- Bulun, S.E.; Lin, Z.; Imir, G.; Amin, S.; Demura, M.; Yilmaz, B.; Deb, S.; Martin, R.; Utsunomiya, H.; Thung, S.; et al. Regulation of aromatase expres-sion in estrogen-responsive breast and uterine disease: From benchto treatment. Pharmacol. Rev. 2005, 57, 359–383. [Google Scholar] [CrossRef]
- Hinshelwood, M.M.; Michael, M.D.; Simpson, E.R. The 50-flankingregion of the ovarian promoter of the bovine CYP19 gene con-tains a deletion in a cyclic adenosine 30,50-monophosphate-likeresponsive sequence. Endocrinology 1997, 138, 3704–3710. [Google Scholar] [CrossRef]
- Khan, K.N.; Fujishita, A.; Masumoto, H.; Muto, H.; Kitajima, M.; Masuzaki, H.; Kitawaki, J. Molecular detection of intrauterine microbial colonization in women with endometriosis. Eur. J. Obstet. Gynecol. Reprod. Biol. 2016, 199, 69–75. [Google Scholar] [CrossRef]
- Baker, J.M.; Al-Nakkash, L.; Herbst-Kralovetz, M.M. Estrogen-gut microbiome axis: Physiological and clinical implications. Maturitas 2017, 103, 45–53. [Google Scholar] [CrossRef]
- Laganà, A.S.; Vitale, S.G.; Salmeri, F.M.; Triolo, O.; Ban Frangež, H.; Vrtačnik-Bokal, E.; Stojanovska, L.; Apostolopoulos, V.; Granese, R.; Sofo, V. Unus pro Omnibus, Omnes pro Uno: A Novel, Evidence-Based, Unifying Theory for the Pathogenesis of Endometriosis. Med. Hypotheses 2017, 103, 10–20. [Google Scholar] [CrossRef]
- Laganà, A.S.; Vitale, S.G.; Granese, R.; Palmara, V.; Ban Frangež, H.; Vrtačnik-Bokal, E.; Chiofalo, B.; Triolo, O. Clinical Dynamics of Dienogest for the Treatment of Endometriosis: From Bench to Bedside. Expert Opin. Drug Metab. Toxicol. 2017, 13, 593–596. [Google Scholar] [CrossRef]
- Stochino-Loi, E.; Major, A.L.; Gillon, T.E.R.; Ayoubi, J.M.; Feki, A.; Bouquet de Joliniere, J. Metformin, the Rise of a New Medical Therapy for Endometriosis? A Systematic Review of the Literature. Front. Med. 2021, 8, 581311. [Google Scholar] [CrossRef]
- Patel, B.G.; Rudnicki, M.; Yu, J.; Shu, Y.; Taylor, R.N. Progesterone resistance in endometriosis: Origins, consequences and interventions. Acta Obstet. Gynecol. Scand. 2017, 96, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, A.; Tanabe, A.; Kawabe, S.; Hayashi, M.; Yuguchi, H.; Yamashita, Y.; Okuda, K.; Ohmichi, M. Dienogest increases the progesterone receptor isoform B/A ratio in patients with ovarian endometriosis. J. Ovarian Res. 2012, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Collinet, P.; Fritel, X.; Revel-Delhom, C.; Ballester, M.; Bolze, P.A.; Borghese, B.; Bornsztein, N.; Boujenah, J.; Brillac, T.; Chabbert-Buffet, N.; et al. Management of endometriosis: CNGOF/HAS clinical practice guidelines—Short version. J. Gynecol. Obstet. Hum. Reprod. 2018, 47, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Seracchioli, R.; Raimondo, D.; Arena, A.; Zanello, M.; Mabrouk, M. Clinical use of endovenous indocyanine green during rectosigmoid segmental resection for endometriosis. Fertil. Steril. 2018, 109, 1135. [Google Scholar] [CrossRef]
- Jamali, N.; Zal, F.; Mostafavi-Pour, Z.; Samare-Najaf, M.; Poordast, T.; Dehghanian, A. Ameliorative Effects of Quercetin and Metformin and Their Combination Against Experimental Endometriosis in Rats. Reprod. Sci. 2021, 28, 683–692. [Google Scholar] [CrossRef]
- Park, Y.B.; Heo, G.M.; Moon, H.K.; Cho, S.J.; Shin, Y.C.; Eom, K.S.; Kim, C.H.; Lee, J.Y.; Mo, E.K.; Jung, K.S. Pulmonary endometriosis resected by video-assisted thoracoscopic surgery. Respirology 2006, 11, 221–223. [Google Scholar] [CrossRef]
- Bedaiwy, M.A.; Alfaraj, S.; Yong, P.; Casper, R. New developments in the medical treatment of endometriosis. Fertil. Steril. 2017, 107, 555–565. [Google Scholar] [CrossRef]
- Bina, F.; Soleymani, S.; Toliat, T.; Hajimahmoodi, M.; Tabarrai, M.; Abdollahi, M.; Rahimi, R. Plant-derived medicines for treatment of endometriosis: A comprehensive review of molecular mechanisms. Pharmacol. Res. 2019, 139, 76–90. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Sanz Garcia, N.; Leclerc, J.; Foretz, M.; Andreelli, F. Cellular and molecular mechanisms of metformin: An overview. Clin. Sci. 2012, 122, 253–270. [Google Scholar] [CrossRef]
- Gunton, J.E.; Delhanty, P.J.; Takahashi, S.; Baxter, R.C. Metformin rapidly increases insulin receptor activation in human liver and signals preferentially through insulin-receptor substrate-2. J. Clin. Endocrinol. Metab. 2003, 88, 1323–1332. [Google Scholar] [CrossRef]
- Maida, A.; Lamont, B.J.; Cao, X.; Drucker, D.J. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia 2011, 54, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Consoli, A.; DeFronzo, R.A. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J. Clin. Endocrinol. Metab. 1996, 81, 4059–4067. [Google Scholar] [PubMed]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- Natali, A.; Ferrannini, E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: A systematic review. Diabetologia 2006, 49, 434–441. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, J.N.; Zeng, C.; Li, X.; Zhou, Y.F.; Qi, Y.; Xue, Q. Metformin Suppresses Prostaglandin E2-Induced Cytochrome P450 Aromatase Gene Expression and Activity via Stimulation of AMP-Activated Protein Kinase in Human Endometriotic Stromal Cells. Reprod. Sci. 2015, 22, 1162–1170. [Google Scholar] [CrossRef]
- Isoda, K.; Young, J.L.; Zirlik, A.; MacFarlane, L.A.; Tsuboi, N.; Gerdes, N.; Schönbeck, U.; Libby, P. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 611–617. [Google Scholar] [CrossRef]
- Attia, G.R.; Rainey, W.E.; Carr, B.R. Metformin directly inhibits androgen production in human thecal cells. Fertil. Steril. 2001, 76, 517–524. [Google Scholar] [CrossRef]
- Mansfield, R.; Galea, R.; Brincat, M.; Hole, D.; Mason, H. Metformin has direct effects on human ovarian steroidogenesis. Fertil. Steril. 2003, 79, 956–962. [Google Scholar] [CrossRef]
- Oner, G.; Ozcelik, B.; Ozgun, M.T.; Serin, I.S.; Ozturk, F.; Basbug, M. The effects of metformin and letrozole on endometriosis and comparison of the two treatment agents in a rat model. Hum. Reprod. 2010, 25, 932–937. [Google Scholar] [CrossRef]
- Bergheim, I.; Luyendyk, J.P.; Steele, C.; Russell, G.K.; Guo, L.; Roth, R.A.; Arteel, G.E. Metformin prevents endotoxin-induced liver injury after partial hepatectomy. J. Pharmacol. Exp. Ther. 2006, 316, 1053–1061. [Google Scholar] [CrossRef]
- Lin, H.Z.; Yang, S.Q.; Chuckaree, C.; Kuhajda, F.; Ronnet, G.; Diehl, A.M. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat. Med. 2000, 6, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Lord, J.; Wilkin, T. Metformin in polycystic ovary syndrome. Curr. Opin. Obstet. Gynecol. 2004, 16, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Rotermund, C.; Machetanz, G.; Fitzgerald, J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. 2018, 9, 400. [Google Scholar] [CrossRef] [PubMed]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.; Gans, R.O.; de Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMP-activated protein kinase: A target for drugs both ancient and modern. Chem. Biol. 2012, 19, 1222–1236. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Xu, J.N.; Zeng, C.; Zhou, Y.; Peng, C.; Zhou, Y.F.; Xue, Q. Metformin inhibits StAR expression in human endometriotic stromal cells via AMPK-mediated disruption of CREB-CRTC2 complex formation. J. Clin. Endocrinol. Metab. 2014, 99, 2795–2803. [Google Scholar] [CrossRef]
- Rice, S.; Pellatt, L.; Ramanathan, K.; Whitehead, S.A.; Mason, H.D. Metformin inhibits aromatase via an extracellular signal regulated kinase-mediated pathway. Endocrinology 2009, 150, 4794–4801. [Google Scholar] [CrossRef]
- Rice, S.; Elia, A.; Jawad, Z.; Pellatt, L.; Mason, H.D. Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: Relevance to polycystic ovary syndrome. J. Clin. Endocrinol. Metab. 2013, 98, E1491–E1500. [Google Scholar] [CrossRef]
- Brown, K.A.; McInnes, K.J.; Hunger, N.I.; Oakhill, J.S.; Steinberg, G.R.; Simpson, E.R. Subcellular localization of cyclic AMP-responsive element binding protein-regulated transcription coactivator 2 provides a link between obesity and breast cancer in postmenopausal women. Cancer Res. 2009, 69, 5392–5399. [Google Scholar] [CrossRef]
- Brown, K.A.; Hunger, N.I.; Docanto, M.; Simpson, E.R. Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res. Treat. 2010, 123, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Kola, B.; Boscaro, M.; Rutter, G.A.; Grossman, A.B.; Korbonits, M. Expanding role of AMPK in endocrinology. Trends Endocrinol. Metab. 2006, 17, 205–215. [Google Scholar] [CrossRef]
- Tosca, L.; Solnais, P.; Ferre, P.; Foufelle, F.; Dupont, J. Metformininduced stimulation of adenosine 50-monophosphate-activated protein kinase (PRKA) impairs progesterone secretion in rat granulosa cells. Biol. Reprod. 2006, 75, 342–351. [Google Scholar] [CrossRef]
- Tosca, L.; Chabrolle, C.; Uzbekova, S.; Dupont, J. Effects of metformin on bovine granulosa cells steroidogenesis: Possible involvement of adenosine 50-monophosphate-activated protein kinase (AMPK). Biol. Reprod. 2007, 76, 368–378. [Google Scholar] [CrossRef]
- Tosca, L.; Uzbekova, S.; Chabrolle, C.; Dupont, J. Possible role of 50AMP-activated protein kinase in the metformin-mediated arrest of bovine oocytes at the germinal vesicle stage during in vitro maturation. Biol. Reprod. 2007, 77, 452–465. [Google Scholar] [CrossRef]
- Takemura, Y.; Osuga, Y.; Harada, M.; Hirata, T.; Koga, K.; Morimoto, C.; Hirota, Y.; Yoshino, O.; Yano, T.; Taketani, Y. Serum adiponectin concentrations are decreased in women with endometriosis. Hum. Reprod. 2005, 20, 3510–3513. [Google Scholar] [CrossRef]
- Takemura, Y.; Osuga, Y.; Harada, M.; Hirata, T.; Koga, K.; Yoshino, O.; Hirota, Y.; Morimoto, C.; Yano, T.; Taketani, Y. Concentration of adiponectin in peritoneal fluid is decreased in women with endometriosis. Am. J. Reprod. Immunol. 2005, 54, 217–221. [Google Scholar] [CrossRef]
- Takemura, Y.; Osuga, Y.; Yamauchi, T.; Kobayashi, M.; Harada, M.; Hirata, T.; Morimoto, C.; Hirota, Y.; Yoshino, O.; Koga, K.; et al. Expression of adiponectin receptors and its possible implication in the human endometrium. Endocrinology 2006, 147, 3203–3210. [Google Scholar] [CrossRef]
- Manna, P.R.; Chandrala, S.P.; King, S.R.; Jo, Y.; Counis, R.; Huhtaniemi, I.T.; Stocco, D.M. Molecular mechanisms of insulin-like growth factor-I mediated regulation of the steroidogenic acute regulatory protein in mouse leydig cells. Mol. Endocrinol. 2006, 20, 362–378. [Google Scholar] [CrossRef]
- Martinelle, N.; Holst, M.; Soder, O.; Svechnikov, K. Extracellular signal-regulated kinases are involved in the acute activation of steroidogenesis in immature rat Leydig cells by human chorionic gonadotropin. Endocrinology 2004, 145, 4629–4634. [Google Scholar] [CrossRef]
- Eaton, J.L.; Unno, K.; Caraveo, M.; Lu, Z.; Kim, J.J. Increased AKT or MEK1/2 activity influences progesterone receptor levels and localization in endometriosis. J. Clin. Endocrinol. Metab. 2013, 98, E1871–E1879. [Google Scholar] [CrossRef] [PubMed]
- Sirianni, R.; Chimento, A.; Malivindi, R.; Mazzitelli, I.; Ando, S.; Pezzi, V. Insulin-like growth factor-I, regulating aromatase expression through steroidogenic factor 1, supports estrogen-dependent tumor Leydig cell proliferation. Cancer Res. 2007, 67, 8368–8377. [Google Scholar] [CrossRef] [PubMed]
- Hunzicker-Dunn, M.; Maizels, E.T. FSH signaling pathways in immature granulosa cells that regulate target gene expression: Branching out from protein kinase A. Cell. Signal. 2006, 18, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Soares, S.R.; Martínez-Varea, A.; Hidalgo-Mora, J.J.; Pellicer, A. Pharmacologic therapies in endometriosis: A systematic review. Fertil. Steril. 2012, 98, 529–555. [Google Scholar] [CrossRef]
- Paul, S.; Sharma, A.V.; Mahapatra, P.D.; Bhattacharya, P.; Reiter, R.J.; Swarnakar, S. Role of melatonin in regulating matrix metalloproteinase-9 via tissue inhibitors of metalloproteinase-1 during protection against endometriosis. J. Pineal. Res. 2008, 44, 439–449. [Google Scholar] [CrossRef]
- Mori, T.; Yamasaki, S.; Masui, F.; Matsuda, M.; Sasabe, H.; Zhou, Y.F. Suppression of the development of experimentally induced uterine adenomyosis by a novel matrix metalloproteinase inhibitor, ONO-4817, in mice. Exp. Biol. Med. 2001, 226, 429–433. [Google Scholar] [CrossRef]
- Mori, T.; Nakahashi, K.; Kyokuwa, M.; Yamasaki, S.; Nagasawa, H. A matrix metalloproteinase inhibitor, ONO-4817, retards the development of mammary tumor and the progression of uterine adenomyosis in mice. Anticancer. Res. 2002, 22, 3985–3988. [Google Scholar]
- Vitagliano, A.; Noventa, M.; Gizzo, S. Is it time to consider patients suffering from endometriosis-related infertility as “novel candidates” for targeted peri-conceptional D-chiro inositol supplementation? Hypothesis, rationale and some considerations. J. Assist. Reprod. Genet. 2015, 32, 407–408. [Google Scholar] [CrossRef]
- McKinnon, B.; Bertschi, D.; Wotzkow, C.; Bersinger, N.A.; Evers, J.; Mueller, M.D. Glucose transporter expression in eutopic endometrial tissue and ectopic endometriotic lesions. J. Mol. Endocrinol. 2014, 52, 169–179. [Google Scholar] [CrossRef]
- Melo, A.S.; Rosa-e-Silva, J.C.; Rosa-e-Silva, A.C.; Poli-Neto, O.B.; Ferriani, R.A.; Vieira, C.S. Unfavorable lipid profile in women with endometriosis. Fertil. Steril. 2010, 93, 2433–2436. [Google Scholar] [CrossRef]
- Santulli, P.; Marcellin, L.; Noël, J.-C.; Borghese, B.; Fayt, I.; Vaiman, D.; Chapron, C.; Méhats, C. Sphingosine Pathway Deregulation in Endometriotic Tissues. Fertil. Steril. 2012, 97, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Takemura, Y.; Osuga, Y.; Yoshino, O.; Hasegawa, A.; Hirata, T.; Hirota, Y.; Nose, E.; Morimoto, C.; Harada, M.; Koga, K.; et al. Metformin suppresses interleukin (IL)-1beta-induced IL-8 production, aromatase activation, and proliferation of endometriotic stromal cells. J. Clin. Endocrinol. Metab. 2007, 92, 3213–3218. [Google Scholar] [CrossRef] [PubMed]
- Omer, N.A.; Taher, M.A.; Aljebory, H.D.S. Effect ofmetformin treatment on some blood biomarkers in women with endometriosis. Iraq. J. Pharm. Sci. 2016, 25, 28–36. [Google Scholar]
- Klappan, A.K.; Hones, S.; Mylonas, I.; Brüning, A. Proteasome inhibition by quercetin triggers macroautophagy and blocks mTOR activity. Histochem. Cell. Biol. 2012, 137, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Huang, S.; Yin, X.; Zan, Y.; Guo, Y.; Han, L. Quercetin suppresses the mobility of breast cancer by suppressing glycolysis through Akt-mTOR pathway mediated autophagy induction. Life Sci. 2018, 208, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Luo, H. Metformin: The next angiogenesis panacea? SAGE Open Med. 2021, 9, 20503121211001641. [Google Scholar] [CrossRef]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.M.S.A.; Oon, C.E. Angiogenesis: Managing the Culprits behind Tumorigenesis and Metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef]
- Qu, H.; Yang, X. Metformin inhibits angiogenesis induced by interaction of hepatocellular carcinoma with hepatic stellate cells. Cell Biochem. Biophys. 2015, 71, 931–936. [Google Scholar] [CrossRef]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef]
- Wang, J.C.; Li, G.Y.; Wang, B.; Han, S.X.; Sun, X.; Jiang, Y.N.; Shen, Y.W.; Zhou, C.; Feng, J.; Lu, S.Y.; et al. Metformin inhibits metastatic breast cancer progression and improves chemosensitivity by inducing vessel normalization via PDGF-B downregulation. J. Exp. Clin. Cancer Res. 2019, 38, 235. [Google Scholar] [CrossRef]
- Moschetta, M.G.; Leonel, C.; Maschio-Signorini, L.B.; Borin, T.F.; Gelaleti, G.B.; Jardim-Perassi, B.V.; Ferreira, L.C.; Sonehara, N.M.; Carvalho, L.G.S.; Hellmén, E.; et al. Evaluation of Angiogenesis Process after Metformin and LY294002 Treatment in Mammary Tumor. Anticancer Agents Med. Chem. 2019, 19, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, S.; Reggiani, F.; Talarico, G.; Mancuso, P.; Calleri, A.; Gregato, G.; Labanca, V.; Noonan, D.M.; Dallaglio, K.; Albini, A.; et al. The biguanides metformin and phenformin inhibit angiogenesis, local and metastatic growth of breast cancer by targeting both neoplastic and microenvironment cells. Int. J. Cancer 2015, 136, E534–E544. [Google Scholar] [CrossRef]
- Qian, W.; Li, J.; Chen, K.; Jiang, Z.; Cheng, L.; Zhou, C.; Yan, B.; Cao, J.; Ma, Q.; Duan, W. Metformin suppresses tumor angiogenesis and enhances the chemosensitivity of gemcitabine in a genetically engineered mouse model of pancreatic cancer. Life Sci. 2018, 208, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Samimi, M.; Pourhanifeh, M.H.; Mehdizadehkashi, A.; Eftekhar, T.; Asemi, Z. The role of inflammation, oxidative stress, angiogenesis, and apoptosis in the pathophysiology of endometriosis: Basic science and new insights based on gene expression. J. Cell Physiol. 2019, 234, 19384–19392. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, S.; Darcha, C. Co-operation between the AKT and ERK signaling pathways may support growth of deep endometriosis in a fibrotic microenvironment in vitro. Hum. Reprod. 2015, 30, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Rooprai, H.K.; McCormick, D. Proteases and Their Inhibitors in Human Brain Tumours: A Review. Anticancer. Res. 1997, 17, 4151–4162. [Google Scholar] [PubMed]
- Wang, N.; Liu, B.; Liang, L.; Wu, Y.; Xie, H.; Huang, J.; Guo, X.; Tan, J.; Zhan, X.; Liu, Y.; et al. Antiangiogenesis therapy of endometriosis using PAMAM as a gene vector in a noninvasive animal model. Biomed. Res. Int. 2014, 2014, 546479. [Google Scholar] [CrossRef]
- Yilmaz, B.; Sucak, A.; Kilic, S.; Aksakal, O.; Aksoy, Y.; Lortlar, N.; Sut, N.; Gungor, T. Metformin regresses endometriotic implants in rats by improving implant levels of superoxide dismutase, vascular endothelial growth factor, tissue inhibitor of metalloproteinase-2, and matrix metalloproteinase-9. Am. J. Obstet. Gynecol. 2010, 202, 368. [Google Scholar] [CrossRef]
- Guo, M.; Zhou, J.J.; Huang, W. Metformin alleviates endometrial hyperplasia through the UCA1/miR-144/TGF-β1/AKT signaling pathway. Int. J. Mol. Med. 2020, 45, 623–633. [Google Scholar] [CrossRef]
- Masuda, S.; Oda, Y.; Sasaki, H.; Ikenouchi, J.; Higashi, T.; Akashi, M.; Nishi, E.; Furuse, M. LSR defines cell corners for tricellular tight junction formation in epithelial cells. J. Cell. Sci. 2011, 124, 548–555. [Google Scholar] [CrossRef]
- Herbsleb, M.; Birkenkamp-Demtroder, K.; Thykjaer, T.; Wiuf, C.; Hein, A.M.; Orntoft, T.F.; Dyrskjøt, L. Increased cell motility and invasion upon knockdown of lipolysis stimulated lipoprotein receptor (LSR) in SW780 bladder cancer cells. BMC Med. Genomics 2008, 1, 31. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Satohisa, S.; Kohno, T.; Takahashi, S.; Hatakeyama, T.; Konno, T.; Tsujiwaki, M.; Saito, T.; Kojima, T. The roles of tricellular tight junction protein lipolysis-stimulated lipoprotein receptor in malignancy of human endometrial cancer cells. Oncotarget 2016, 7, 27735–27752. [Google Scholar] [CrossRef] [PubMed]
- Gebel, H.M.; Braun, D.P.; Tambur, A.; Frame, D.; Rana, N.; Dmowski, W.P. Spontaneous Apoptosis of Endometrial Tissue Is Impaired in Women with Endometriosis. Fertil. Steril. 1998, 69, 1042–1047. [Google Scholar] [CrossRef]
- Harada, A.; Kimura, Y.; Kojima, C.; Kono, K. Effective Tolerance to Serum Proteins of Head-Tail Type Polycation Vectors by PEGylation at the Periphery of the Head Block. Biomacromolecules 2010, 11, 1036–1042. [Google Scholar] [CrossRef] [PubMed]
- Vaskivuo, T.E.; Stenbäck, F.; Karhumaa, P.; Risteli, J.; Dunkel, L.; Tapanainen, J.S. Apoptosis and Apoptosis-Related Proteins in Human Endometrium. Mol. Cell. Endocrinol. 2000, 165, 75–83. [Google Scholar] [CrossRef]
- Iba, Y.; Harada, T.; Horie, S.; Deura, I.; Iwabe, T.; Terakawa, N. Lipopolysaccharide-Promoted Proliferation of Endometriotic Stromal Cells via Induction of Tumor Necrosis Factor Alpha and Interleukin-8 Expression. Fertil. Steril. 2004, 82 (Suppl. S3), 1036–1042. [Google Scholar] [CrossRef]
- Khan, K.N.; Masuzaki, H.; Fujishita, A.; Kitajima, M.; Hiraki, K.; Sekine, I.; Matsuyama, T.; Ishimaru, T. Interleukin-6- and Tumour Necrosis Factor Alpha-Mediated Expression of Hepatocyte Growth Factor by Stromal Cells and Its Involvement in the Growth of Endometriosis. Hum. Reprod. 2005, 20, 2715–2723. [Google Scholar] [CrossRef][Green Version]
- Hsieh Li, S.M.; Liu, S.T.; Chang, Y.L.; Ho, C.L.; Huang, S.M. Metformin causes cancer cell death through downregulation of p53-dependent differentiated embryo chondrocyte 1. J. Biomed. Sci. 2018, 25, 81. [Google Scholar] [CrossRef]
- Xiao, X.; He, Q.; Lu, C.; Werle, K.D.; Zhao, R.X.; Chen, J.; Davis, B.C.; Cui, R.; Liang, J.; Xu, Z.X. Metformin impairs the growth of liver kinase B1-intact cervical cancer cells. Gynecol. Oncol. 2012, 127, 249–255. [Google Scholar] [CrossRef]
- Irie, H.; Banno, K.; Yanokura, M.; Iida, M.; Adachi, M.; Nakamura, K.; Umene, K.; Nogami, Y.; Masuda, K.; Kobayashi, Y.; et al. Metformin: A candidate for the treatment of gynecological tumors based on drug repositioning. Oncol. Lett. 2016, 11, 1287–1293. [Google Scholar] [CrossRef]
- Chen, Y.H.; Yang, S.F.; Yang, C.K.; Tsai, H.D.; Chen, T.H.; Chou, M.C.; Hsiao, Y.H. Metformin induces apoptosis and inhibits migration by activating the AMPK/p53 axis and suppressing PI3K/AKT signaling in human cervical cancer cells. Mol. Med. Rep. 2021, 23, 88. [Google Scholar] [CrossRef]
- Fu, Y.L.; Zhang, Q.H.; Wang, X.W.; He, H. Antidiabetic drug metformin mitigates ovarian cancer SKOV3 cell growth by triggering G2/M cell cycle arrest and inhibition of m-TOR/PI3K/Akt signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1169–1175. [Google Scholar] [PubMed]
- Zhao, Y.; Sun, H.; Feng, M.; Zhao, J.; Zhao, X.; Wan, Q.; Cai, D. Metformin is associated with reduced cell proliferation in human endometrial cancer by inbibiting PI3K/AKT/mTOR signaling. Gynecol. Endocrinol. 2018, 34, 428–432. [Google Scholar] [CrossRef]
- Zhang, H.H.; Zhang, Y.; Cheng, Y.N.; Gong, F.L.; Cao, Z.Q.; Yu, L.G.; Guo, X.L. Metformin in combination with curcumin inhibits the growth, metastasis, and angiogenesis of hepatocellular carcinoma in vitro and in vivo. Mol. Carcinog. 2018, 57, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Storozhuk, Y.; Hopmans, S.N.; Sanli, T.; Barron, C.; Tsiani, E.; Cutz, J.C.; Pond, G.; Wright, J.; Singh, G.; Tsakiridis, T. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br. J. Cancer 2013, 108, 2021–2032. [Google Scholar] [CrossRef]
- Dhillon, S.S.; Groman, A.; Meagher, A.; Demmy, T.; Warren, G.W.; Yendamuri, S. Metformin and not diabetes influences the survival of resected early stage NSCLC patients. J. Cancer Sci. Ther. 2014, 6, 217–222. [Google Scholar] [PubMed]
- Sayed, R.; Saad, A.S.; ElWakeel, L.; Elkholy, E.; Badary, O. Metformin addition to chemotherapy in stage IV non-small cell lung cancer: An open label randomized controlled study. Asian Pac. J. Cancer Prev. 2015, 16, 6621–6626. [Google Scholar] [CrossRef]
- Falcone, T.; Lebovic, D.I. Clinical management of endometriosis. Obstet. Gynecol. 2011, 118, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef]
- Deitch, E.A. The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch. Surg. 1990, 125, 403–404. [Google Scholar] [CrossRef]
- Plottel, C.S.; Blaser, M.J. Microbiome and malignancy. Cell. Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Xu, J.; Lefever, D.E.; Glenn, T.C.; Nagy, T.; Guo, T.L. Genistein prevention of hyperglycemia and improvement of glucose tolerance in adult non-obese diabetic mice are associated with alterations of gut microbiome and immune homeostasis. Toxicol. Appl. Pharmacol. 2017, 332, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Flores, R.; Shi, J.; Fuhrman, B.; Xu, X.; Veenstra, T.D.; Gail, M.H.; Gajer, P.; Ravel, J.; Goedert, J.J. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: A cross-sectional study. J. Transl. Med. 2012, 10, 253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shen, Q.; Celestino, J.; Milam, M.R.; Westin, S.N.; Lacour, R.A.; Meyer, L.A.; Shipley, G.L.; Davies, P.J.; Deng, L.; et al. Enhanced estrogen-induced proliferation in obese rat endometrium. Am. J. Obstet. Gynecol. 2009, 200, e1–e186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bailey, M.T.; Coe, C.L. Endometriosis is associated with an altered profile of intestinal microflora in female rhesus monkeys. Hum. Reprod. 2002, 17, 1704–1708. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef]
- Damman, C.J.; Miller, S.I.; Surawicz, C.M.; Zisman, T.L. The microbiome and inflammatory bowel disease: Is there a therapeutic role for fecal microbiota transplantation? Am. J. Gastroenterol. 2012, 107, 1452–1459. [Google Scholar] [CrossRef]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef]
- Yari, S.; Khoei, H.H.; Saber, M.; Esfandiari, F.; Moini, A.; Shahhoseini, M. Metformin attenuates expression of angiogenic and inflammatory genes in human endometriotic stromal cells. Exp. Cell Res. 2021, 404, 112659. [Google Scholar] [CrossRef]
- Zhang, H.; Xue, J.; Li, M.; Zhao, X.; Wei, D.; Li, C. Metformin regulates stromal-epithelial cells communication via Wnt2/β-catenin signaling in endometriosis. Mol. Cell Endocrinol. 2015, 413, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar]
- Streuli, I.; Gaitzsch, H.; Wenger, J.M.; Petignat, P. Endometriosis after menopause: Physiopathology and management of an uncommon condition. Climacteric 2017, 20, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Holdsworth-Carson, S.J.; Dior, U.P.; Colgrave, E.M.; Healey, M.; Montgomery, G.W.; Rogers, P.A.W.; Girling, J.E. The association of body mass index with endometriosis and disease severity in women with pain. J. Endometr. Pelvic Pain Disord. 2018, 10, 79–87. [Google Scholar] [CrossRef]
- Morin-Papunen, L.; Rantala, A.S.; Unkila-Kallio, L.; Tiitinen, A.; Hippeläinen, M.; Perheentupa, A.; Tinkanen, H.; Bloigu, R.; Puukka, K.; Ruokonen, A.; et al. Metformin improves pregnancy and live-birth rates in women with polycystic ovary syndrome (PCOS): A multicenter, double-blind, placebo-controlled randomized trial. J. Clin. Endocrinol. Metab. 2012, 97, 1492–1500. [Google Scholar] [CrossRef]
- Congiu, T.; Alghrably, M.; Emwas, A.-H.; Jaremko, L.; Lachowicz, J.I.; Piludu, M.; Monica, P.; Faa, G.; Pichirim, G.; Jaremkom, M.; et al. Undercover toxic ménage à trois of Amylin, Copper(II) and Metformin in human embryonic kidney cells. Pharmaceutics 2021, 13, 830. [Google Scholar] [CrossRef] [PubMed]
- Slahor, L. CME: Metformin—Dos und Don’ts CME-Fragen [ME: Metformin—Dos and Don’ts]. Praxis (Bern 1994) 2021, 110, 939–945. [Google Scholar] [CrossRef]
- Tarry-Adkins, J.L.; Grant, I.D.; Ozanne, S.E.; Reynolds, R.M.; Aiken, C.E. Efficacy and Side Effect Profile of Different Formulations of Metformin: A Systematic Review and Meta-Analysis. Diabetes. Ther. 2021, 12, 1901–1914. [Google Scholar] [CrossRef]
- Ma, R.L.; Deng, Y.; Wang, Y.F.; Zhu, S.Y.; Ding, X.S.; Sun, A.J. Short-term combined treatment with exenatide and metformin for overweight/obese women with polycystic ovary syndrome. Chin. Med. J. 2021, 134, 2882–2889. [Google Scholar] [CrossRef]
- Alhowail, A. Potential mechanisms of metformin-induced memory impairment. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 4757–4761. [Google Scholar]
- Fadden, E.J.; Longley, C.; Mahambrey, T. Metformin-associated lactic acidosis. BMJ. Case. Rep. 2021, 14, e239154. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Anand, U.; Nahon-Crystal, E.; Di Carlo, M.; Shteinfer-Kuzmine, A. Adverse Effects of Metformin From Diabetes to COVID-19, Cancer, Neurodegenerative Diseases, and Aging: Is VDAC1 a Common Target? Front. Physiol. 2021, 12, 730048. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.D.; Baker, B.C.; Scott, E.M.; Forbes, K. Interaction between Metformin, Folate and Vitamin B12 and the Potential Impact on Fetal Growth and Long-Term Metabolic Health in Diabetic Pregnancies. Int. J. Mol. Sci. 2021, 22, 5759. [Google Scholar] [CrossRef] [PubMed]
- Lášticová, M.; Víšek, J.; Zima, O.; Šmahelová, A.; Bláha, V. Metformin-associated lactic acidosis. Vnitr. Lek. 2020, 66, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Walsh, D. An old friend turned foe: Metformin-induced diarrhea with resultant symptomatic hypokalemia, hypomagnesemia, hypocalcemia, and hypophosphatemia. Clin. Case. Rep. 2021, 9, e04621. [Google Scholar] [CrossRef]
- Vercellini, P.; Bracco, B.; Mosconi, P.; Roberto, A.; Alberico, D.; Dhouha, D.; Somigliana, E. Norethindrone acetate or dienogest for the treatment of symptomatic endometriosis: A before and after study. Fertil. Steril. 2016, 105, e3–e743. [Google Scholar] [CrossRef]
- Foda, A.A.; Abdel Aal, I.A. Metformin as a new therapy for endometriosis, its effects on both clinical picture and cytokines profile. Middle East. Fertil. Soc. J. 2012, 17, 262–267. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kimber-Trojnar, Ż.; Dłuski, D.F.; Wierzchowska-Opoka, M.; Ruszała, M.; Leszczyńska-Gorzelak, B. Metformin as a Potential Treatment Option for Endometriosis. Cancers 2022, 14, 577. https://doi.org/10.3390/cancers14030577
Kimber-Trojnar Ż, Dłuski DF, Wierzchowska-Opoka M, Ruszała M, Leszczyńska-Gorzelak B. Metformin as a Potential Treatment Option for Endometriosis. Cancers. 2022; 14(3):577. https://doi.org/10.3390/cancers14030577
Chicago/Turabian StyleKimber-Trojnar, Żaneta, Dominik Franciszek Dłuski, Magdalena Wierzchowska-Opoka, Monika Ruszała, and Bożena Leszczyńska-Gorzelak. 2022. "Metformin as a Potential Treatment Option for Endometriosis" Cancers 14, no. 3: 577. https://doi.org/10.3390/cancers14030577
APA StyleKimber-Trojnar, Ż., Dłuski, D. F., Wierzchowska-Opoka, M., Ruszała, M., & Leszczyńska-Gorzelak, B. (2022). Metformin as a Potential Treatment Option for Endometriosis. Cancers, 14(3), 577. https://doi.org/10.3390/cancers14030577