
Repurposing of Commercially Existing Molecular Target Therapies to Boost the Clinical Efficacy of Immune Checkpoint Blockade
 , ,
, ,  , and
, and
Abstract
Simple Summary
Abstract
1. Introduction
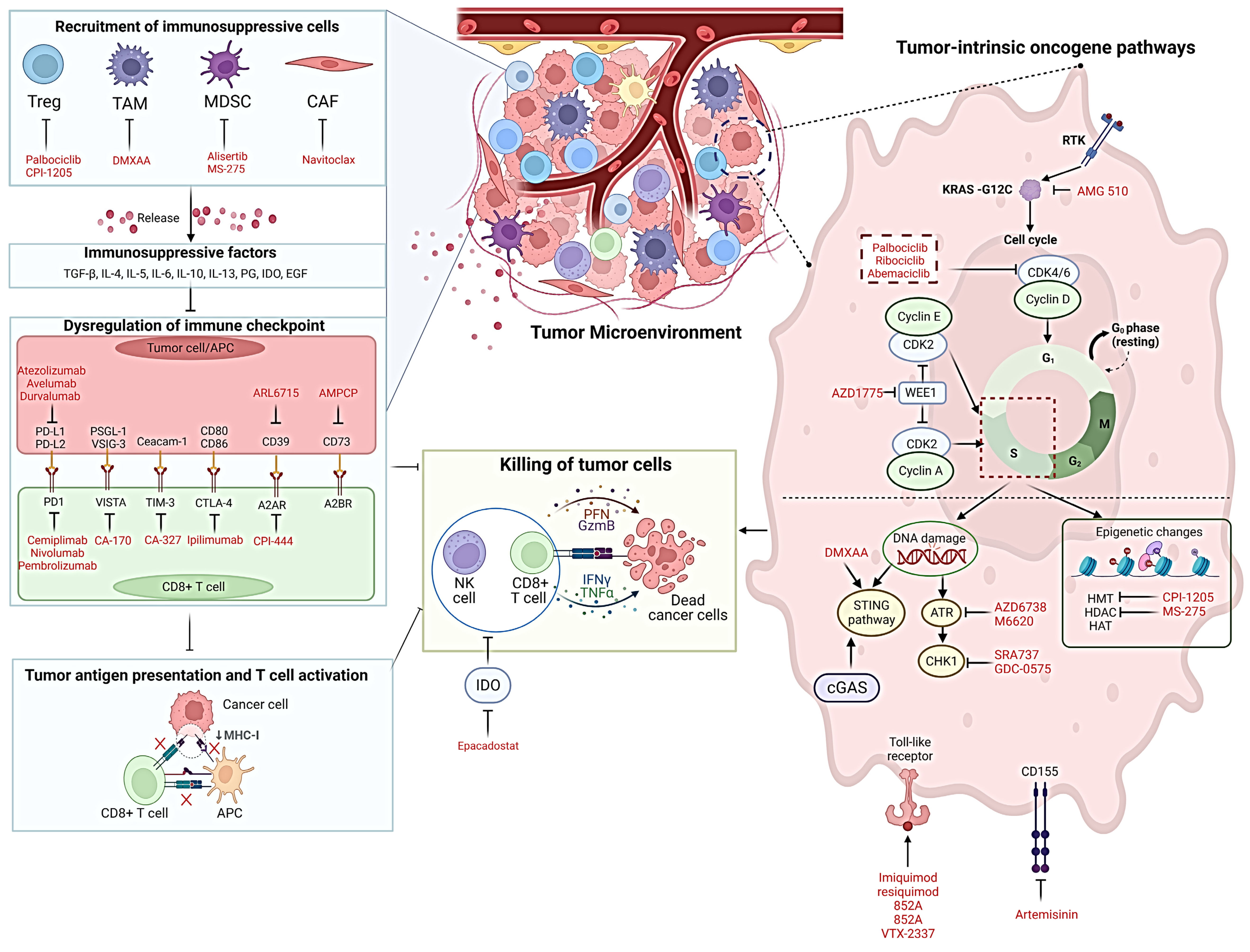
2. Mechanism Driving Resistance to ICB
3. Repurposing SMIs to Improve Efficacy of ICB
4. Taking Advantage of Cell Cycle Inhibitors
5. Potential Application SMIs against DNA Damage Regulatory Proteins
6. Use of SMIs Which Induce Epigenetic Changes
7. SMIs Paving Way for Cytotoxic Lymphocytes to Transform into Super Killers
8. Utilising SMIs to Induce Immunogenic Cell Death
9. Future Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.-L.; Wang, Y.-T.; Fu, W.-J.; Lu, N.; Liu, Z.-S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernández-Rubio, L.; Arasanz, H.; Echaide, M.; Garnica, M.; Ramos, P.; Piñeiro-Hermida, S.; Vera, R.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells 2022, 11, 2351. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Kuang, Z.; Li, L.; Zhang, P.; Chen, B.; Wu, M.; Ni, H.; Yi, S.; Zou, J.; Liu, J. A novel antibody targeting TIM-3 resulting in receptor internalization for cancer immunotherapy. Antib. Ther. 2020, 3, 227–236. [Google Scholar] [CrossRef]
- Kuo, T.C.; Chen, A.; Harrabi, O.; Sockolosky, J.T.; Zhang, A.; Sangalang, E.; Doyle, L.V.; Kauder, S.E.; Fontaine, D.; Bollini, S.; et al. Targeting the myeloid checkpoint receptor SIRPα potentiates innate and adaptive immune responses to promote anti-tumor activity. J. Hematol. Oncol. 2020, 13, 160. [Google Scholar] [CrossRef]
- Mehta, N.; Maddineni, S.; Kelly, R.L.; Lee, R.B.; Hunter, S.A.; Silberstein, J.L.; Sperberg, R.A.P.; Miller, C.L.; Rabe, A.; Labanieh, L.; et al. An engineered antibody binds a distinct epitope and is a potent inhibitor of murine and human VISTA. Sci. Rep. 2020, 10, 15171. [Google Scholar] [CrossRef]
- Li, J.; Yang, Y.; Inoue, H.; Mori, M.; Akiyoshi, T. The expression of costimulatory molecules CD80 and CD86 in human carcinoma cell lines: Its regulation by interferon γ and interleukin-10. Cancer Immunol. Immunother. 1996, 43, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.M.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, Double-Blind, Phase III Trial of Ipilimumab Versus Placebo in Asymptomatic or Minimally Symptomatic Patients With Metastatic Chemotherapy-Naive Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Bristol-Myers Squibb Announces Phase 3 CheckMate-498 Study Did Not Meet Primary Endpoint of Overall Survival with Opdivo (nivolumab) Plus Radiation in Patients with Newly Diagnosed MGMT-Unmethylated Glioblastoma Multiforme. Available online: https://news.bms.com/news/corporate-financial/2019/Bristol-Myers-Squibb-Announces-Phase-3-CheckMate--498-Study-Did-Not-Meet-Primary-Endpoint-of-Overall-Survival-with-Opdivo-nivolumab-Plus-Radiation-in-Patients-with-Newly-Diagnosed-MGMT-Unmethylated-Glioblastoma-Multiforme/default.aspx (accessed on 5 September 2019).
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.; Lim, M.; Wick, A.; Baehring, J.; Ahluwalia, M.S.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Curioni-Fontecedro, A.; Dafni, U.; Shah, R.; O’Brien, M.; Pope, A.; Fisher, P.; Spicer, J.; Roy, A.; Gilligan, D.; et al. A multicentre randomised phase III trial comparing pembrolizumab versus single-agent chemotherapy for advanced pre-treated malignant pleural mesothelioma: The European Thoracic Oncology Platform (ETOP 9-15) PROMISE-meso trial. Ann. Oncol. 2020, 31, 1734–1745. [Google Scholar] [CrossRef]
- Winer, E.P.; Lipatov, O.; Im, S.-A.; Goncalves, A.; Muñoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Shitara, K.; Özgüroğlu, M.; Bang, Y.-J.; Di Bartolomeo, M.; Mandalà, M.; Ryu, M.-H.; Fornaro, L.; Olesiński, T.; Caglevic, C.; Chung, H.C.; et al. Pembrolizumab versus paclitaxel for previously treated, advanced gastric or gastro-oesophageal junction cancer (KEYNOTE-061): A randomised, open-label, controlled, phase 3 trial. Lancet 2018, 392, 123–133. [Google Scholar] [CrossRef]
- Shitara, K.; Van Cutsem, E.; Bang, Y.-J.; Fuchs, C.; Wyrwicz, L.; Lee, K.-W.; Kudaba, I.; Garrido, M.; Chung, H.C.; Lee, J.; et al. Efficacy and Safety of Pembrolizumab or Pembrolizumab Plus Chemotherapy vs Chemotherapy Alone for Patients With First-line, Advanced Gastric Cancer: The KEYNOTE-062 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1571–1580. [Google Scholar] [CrossRef]
- Powles, T.; Csőszi, T.; Özgüroğlu, M.; Matsubara, N.; Géczi, L.; Cheng, S.Y.-S.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Barrera, R.M.; et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 931–945. [Google Scholar] [CrossRef]
- Finn, R.S.; Ryoo, B.-Y.; Merle, P.; Kudo, M.; Bouattour, M.; Lim, H.Y.; Breder, V.; Edeline, J.; Chao, Y.; Ogasawara, S.; et al. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J. Clin. Oncol. 2020, 38, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Update on KESTREL Phase III Trial of Imfinzi with or without Tremelimumab in the 1st-Line Treatment of Recurrent or Metastatic Head and Neck Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2021/update-on-kestrel-phase-iii-trial-for-imfinzi.html (accessed on 5 February 2021).
- Lee, N.Y.; Ferris, R.L.; Psyrri, A.; I Haddad, R.; Tahara, M.; Bourhis, J.; Harrington, K.; Chang, P.M.-H.; Lin, J.-C.; Razaq, M.A.; et al. Avelumab plus standard-of-care chemoradiotherapy versus chemoradiotherapy alone in patients with locally advanced squamous cell carcinoma of the head and neck: A randomised, double-blind, placebo-controlled, multicentre, phase 3 trial. Lancet Oncol. 2021, 22, 450–462. [Google Scholar] [CrossRef] [PubMed]
- On the Phase III NEPTUNE Trial of Imfinzi Plus Tremelimumab in Stage IV Non-Small Cell Lung Cancer. Available online: https://www.astrazeneca.com/media-centre/press-releases/2019/update-on-the-phase-iii-neptune-trial-of-imfinzi-plus-tremelimumab-in-stage-iv-non-small-cell-lung-cancer-21082019.html#! (accessed on 21 August 2019).
- Owonikoko, T.K.; Park, K.; Govindan, R.; Ready, N.; Reck, M.; Peters, S.; Dakhil, S.R.; Navarro, A.; Rodríguez-Cid, J.; Schenker, M.; et al. Nivolumab and Ipilimumab as Maintenance Therapy in Extensive-Disease Small-Cell Lung Cancer: CheckMate 451. J. Clin. Oncol. 2021, 39, 1349–1359. [Google Scholar] [CrossRef]
- Phase 3 Trial in Squamous Non Small Cell Lung Cancer Subjects Comparing Ipilimumab Plus Paclitaxel and Carboplatin Versus Placebo Plus Paclitaxel and Carboplatin. Available online: https://clinicaltrials.gov/ct2/show/results/NCT02279732 (accessed on 28 August 2019).
- Govindan, R.; Szczesna, A.; Ahn, M.-J.; Schneider, C.-P.; Mella, P.F.G.; Barlesi, F.; Han, B.; Ganea, D.E.; Von Pawel, J.; Vladimirov, V.; et al. Phase III Trial of Ipilimumab Combined With Paclitaxel and Carboplatin in Advanced Squamous Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2017, 35, 3449–3457. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Luft, A.; Szczesna, A.; Havel, L.; Kim, S.-W.; Akerley, W.; Pietanza, M.C.; Wu, Y.-L.; Zielinski, C.; Thomas, M.; et al. Phase III Randomized Trial of Ipilimumab Plus Etoposide and Platinum versus Placebo Plus Etoposide and Platinum in Extensive-Stage Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3740–3748. [Google Scholar] [CrossRef]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Pitt, J.; Marabelle, A.; Eggermont, A.; Soria, J.-C.; Kroemer, G.; Zitvogel, L. Targeting the tumor microenvironment: Removing obstruction to anticancer immune responses and immunotherapy. Ann. Oncol. 2016, 27, 1482–1492. [Google Scholar] [CrossRef]
- Hunter, M.C.; Teijeira, A.; Halin, C. T Cell Trafficking through Lymphatic Vessels. Front. Immunol. 2016, 7, 613. [Google Scholar] [CrossRef]
- Chow, K.T.; Gale, M. SnapShot: Interferon Signaling. Cell 2015, 163, 1808–1808.e1. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Three Es of Cancer Immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Huang, R.-Y.; Francois, A.; McGray, A.R.; Miliotto, A.; Odunsi, K. Compensatory upregulation of PD-1, LAG-3, and CTLA-4 limits the efficacy of single-agent checkpoint blockade in metastatic ovarian cancer. Oncoimmunology 2016, 6, e1249561. [Google Scholar] [CrossRef]
- Wang, M.; Liu, Y.; Cheng, Y.; Wei, Y.; Wei, X. Immune checkpoint blockade and its combination therapy with small-molecule inhibitors for cancer treatment. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2018, 1871, 199–224. [Google Scholar] [CrossRef]
- Kwilas, A.R.; Donahue, R.N.; Tsang, K.Y.; Hodge, J.W. Immune consequences of tyrosine kinase inhibitors that synergize with cancer immunotherapy. Cancer Cell Microenviron. 2015, 2, e677. [Google Scholar] [CrossRef]
- Sinha, D.; Smith, C.; Khanna, R. Joining Forces: Improving Clinical Response to Cellular Immunotherapies with Small-Molecule Inhibitors. Trends Mol. Med. 2020, 27, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Pembrolizumab in Combination with Epacadostat or Placebo in Cisplatin-ineligible Urothelial Carcinoma (KEYNOTE-672/ECHO-307). Available online: https://clinicaltrials.gov/ct2/show/results/NCT03361865 (accessed on 16 December 2021).
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.-J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Efficacy and Safety Study of Pembrolizumab (MK-3475) with or without Lenvatinib (MK-7902/E7080) in Adults with Programmed Cell Death-Ligand 1 (PD-L1)-Positive Treatment-naïve Nonsmall Cell Lung Cancer (NSCLC) (MK-7902-007/E7080-G000-314/LEAP-007). Available online: https://clinicaltrials.gov/ct2/show/results/NCT03829332 (accessed on 28 October 2022).
- Makker, V.; Colombo, N.; Herráez, A.C.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Powles, T.; Plimack, E.R.; Soulières, D.; Waddell, T.; Stus, V.; Gafanov, R.; Nosov, D.; Pouliot, F.; Melichar, B.; Vynnychenko, I.; et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (KEYNOTE-426): Extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020, 21, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Kim, T.W.; Bendell, J.; Argilés, G.; Tebbutt, N.C.; Di Bartolomeo, M.; Falcone, A.; Fakih, M.; Kozloff, M.; Segal, N.H.; et al. Atezolizumab with or without cobimetinib versus regorafenib in previously treated metastatic colorectal cancer (IMblaze370): A multicentre, open-label, phase 3, randomised, controlled trial. Lancet Oncol. 2019, 20, 849–861. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2021, 384, 829–841. [Google Scholar] [CrossRef]
- LBA76—Overall Survival (OS) Results from the Phase III TROPiCS-02 Study of Sacituzumab Govitecan (SG) vs. Treatment of Physician’s Choice (TPC) in Patients (pts) with HR+/HER2-Metastatic Breast Cancer (mBC). Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress/overall-survival-os-results-from-the-phase-iii-tropics-02-study-of-sacituzumab-govitecan-sg-vs-treatment-of-physician-s-choice-tpc-in-patient (accessed on 9 September 2022).
- Merck and Eisai Provide Update on Phase 3 LEAP-002 Trial Evaluating KEYTRUDA® (pembrolizumab) Plus LENVIMA® (lenvatinib) versus LENVIMA Monotherapy in Patients with Unresectable Hepatocellular Carcinoma. Available online: https://www.merck.com/news/merck-and-eisai-provide-update-on-phase-3-leap-002-trial-evaluating-keytruda-pembrolizumab-plus-lenvima-lenvatinib-versus-lenvima-monotherapy-in-patients-with-unresectable-hepatocellul/ (accessed on 3 August 2022).
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark. Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Zhao, H.; Song, S.; Ma, J.; Yan, Z.; Xie, H.; Feng, Y.; Che, S. CD47 as a promising therapeutic target in oncology. Front. Immunol. 2022, 13, 757480. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-B.; Ye, Z.-H.; Chen, X.; Shi, J.-J.; Lu, J.-J. The development of small-molecule inhibitors targeting CD47. Drug Discov. Today 2020, 26, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Cabrales, P. RRx-001 Acts as a Dual Small Molecule Checkpoint Inhibitor by Downregulating CD47 on Cancer Cells and SIRP-α on Monocytes/Macrophages. Transl. Oncol. 2019, 12, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Oronsky, B.; Reid, T.R.; Larson, C.; Caroen, S.; Quinn, M.; Burbano, E.; Varner, G.; Thilagar, B.; Brown, B.; Coyle, A.; et al. REPLATINUM Phase III randomized study: RRx-001 + platinum doublet versus platinum doublet in third-line small cell lung cancer. Futur. Oncol. 2019, 15, 3427–3433. [Google Scholar] [CrossRef]
- Tomita, Y.; Oronsky, B.; Abrouk, N.; Cabrales, P.; Reid, T.R.; Lee, M.-J.; Yuno, A.; Baker, J.; Lee, S.; Trepel, J.B. In small cell lung cancer patients treated with RRx-001, a downregulator of CD47, decreased expression of PD-L1 on circulating tumor cells significantly correlates with clinical benefit. Transl. Lung Cancer Res. 2021, 10, 274–278. [Google Scholar] [CrossRef]
- Smith, W.M.; Purvis, I.J.; Bomstad, C.N.; Labak, C.M.; Velpula, K.K.; Tsung, A.J.; Regan, J.N.; Venkataraman, S.; Vibhakar, R.; Asuthkar, S. Therapeutic targeting of immune checkpoints with small molecule inhibitors. Am. J. Transl. Res. 2019, 11, 529–541. [Google Scholar]
- Shaw, G.; Cavalcante, L.; Giles, F.J.; Taylor, A. Elraglusib (9-ING-41), a selective small-molecule inhibitor of glycogen synthase kinase-3 beta, reduces expression of immune checkpoint molecules PD-1, TIGIT and LAG-3 and enhances CD8+ T cell cytolytic killing of melanoma cells. J. Hematol. Oncol. 2022, 15, 1–13. [Google Scholar] [CrossRef]
- Rudd, C.E.; Chanthong, K.; Taylor, A. Small Molecule Inhibition of GSK-3 Specifically Inhibits the Transcription of Inhibitory Co-receptor LAG-3 for Enhanced Anti-tumor Immunity. Cell Rep. 2020, 30, 2075–2082.e4. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8+ T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.-H.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen–specific CD8+ T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Zhou, X.; Jiao, L.; Qian, Y.; Dong, Q.; Sun, Y.; Zheng, W.; Zhao, W.; Zhai, W.; Qiu, L.; Wu, Y.; et al. Repositioning Azelnidipine as a Dual Inhibitor Targeting CD47/SIRPα and TIGIT/PVR Pathways for Cancer Immuno-Therapy. Biomolecules 2021, 11, 706. [Google Scholar] [CrossRef]
- Zhou, X.; Du, J.; Wang, H.; Chen, C.; Jiao, L.; Cheng, X.; Zhou, X.; Chen, S.; Gou, S.; Zhao, W.; et al. Repositioning liothyronine for cancer immunotherapy by blocking the interaction of immune checkpoint TIGIT/PVR. Cell Commun. Signal. 2020, 18, 142. [Google Scholar] [CrossRef]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed]
- Bedognetti, D.; Roelands, J.; Decock, J.; Wang, E.; Hendrickx, W. The MAPK hypothesis: Immune-regulatory effects of MAPK-pathway genetic dysregulations and implications for breast cancer immunotherapy. Emerg. Top. Life Sci. 2017, 1, 429–445. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Tian, H. Current Development Status of MEK Inhibitors. Molecules 2017, 22, 1551. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Dunn, I.S.; Durda, P.J.; Butera, D.; Rose, L.B.; Haggerty, T.J.; Benson, E.M.; Kurnick, J.T. Role of the Mitogen-Activated Protein Kinase Signaling Pathway in the Regulation of Human Melanocytic Antigen Expression. Mol. Cancer Res. 2006, 4, 779–792. [Google Scholar] [CrossRef] [PubMed]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.-N.J.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E Inhibition Enhances T-Cell Recognition of Melanoma without Affecting Lymphocyte Function. Cancer Res 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Peng, W.; Xu, C.; Lou, Y.; Zhang, M.; Wargo, J.A.; Chen, J.Q.; Li, H.S.; Watowich, S.S.; Yang, Y.; et al. BRAF Inhibition Increases Tumor Infiltration by T cells and Enhances the Antitumor Activity of Adoptive Immunotherapy in Mice. Clin. Cancer Res. 2013, 19, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-H.; Keam, B.; Ahn, Y.-O.; Park, H.-R.; Kim, M.; Kim, T.M.; Kim, D.-W.; Heo, D.S. Inhibition of MEK with trametinib enhances the efficacy of anti-PD-L1 inhibitor by regulating anti-tumor immunity in head and neck squamous cell carcinoma. Oncoimmunology 2018, 8, e1515057. [Google Scholar] [CrossRef]
- Hu-Lieskovan, S.; Mok, S.; Moreno, B.H.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef]
- Prasad, M.; Zorea, J.; Jagadeeshan, S.; Shnerb, A.B.; Mathukkada, S.; Bouaoud, J.; Michon, L.; Novoplansky, O.; Badarni, M.; Cohen, L.; et al. MEK1/2 inhibition transiently alters the tumor immune microenvironment to enhance immunotherapy efficacy against head and neck cancer. J. Immunother. Cancer 2022, 10, e003917. [Google Scholar] [CrossRef] [PubMed]
- George, M.A.; Qureshi, S.; Omene, C.; Toppmeyer, D.L.; Ganesan, S. Clinical and Pharmacologic Differences of CDK4/6 Inhibitors in Breast Cancer. Front. Oncol. 2021, 11, 693104. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Deng, J.; Wang, E.S.; Jenkins, R.W.; Li, S.; Dries, R.; Yates, K.; Chhabra, S.; Huang, W.; Liu, H.; Aref, A.R.; et al. CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov. 2018, 8, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef]
- Uzhachenko, R.V.; Bharti, V.; Ouyang, Z.; Blevins, A.; Mont, S.; Saleh, N.; Lawrence, H.A.; Shen, C.; Chen, S.-C.; Ayers, G.D.; et al. Metabolic modulation by CDK4/6 inhibitor promotes chemokine-mediated recruitment of T cells into mammary tumors. Cell Rep. 2021, 35, 108944. [Google Scholar] [CrossRef]
- Heckler, M.; Ali, L.R.; Clancy-Thompson, E.; Qiang, L.; Ventre, K.S.; Lenehan, P.; Roehle, K.; Luoma, A.; Boelaars, K.; Peters, V.; et al. Inhibition of CDK4/6 Promotes CD8 T-cell Memory Formation. Cancer Discov. 2021, 11, 2564–2581. [Google Scholar] [CrossRef]
- Lelliott, E.J.; Sheppard, K.E.; McArthur, G.A. Harnessing the immunotherapeutic potential of CDK4/6 inhibitors in melanoma: Is timing everything? npj Precis. Oncol. 2022, 6, 26. [Google Scholar] [CrossRef]
- Lelliott, E.J.; Kong, I.Y.; Zethoven, M.; Ramsbottom, K.M.; Martelotto, L.G.; Meyran, D.; Jiang Zhu, J.; Costacurta, M.; Kirby, L.; Sandow, J.J.; et al. CDK4/6 inhibition promotes anti-tumor immunity through the induction of T cell memory. Cancer Discov. 2021, 11, 2582–2601. [Google Scholar] [CrossRef]
- Taniguchi, H.; Caeser, R.; Chavan, S.S.; Zhan, Y.A.; Chow, A.; Manoj, P.; Uddin, F.; Kitai, H.; Qu, R.; Hayatt, O.; et al. WEE1 inhibition enhances the antitumor immune response to PD-L1 blockade by the concomitant activation of STING and STAT1 pathways in SCLC. Cell Rep. 2022, 39, 110814. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Z.; Wang, X. Exploration of the Combination of PLK1 Inhibition with Immunotherapy in Cancer Treatment. J. Oncol. 2018, 2018, 3979527. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yang, Q.; Lu, L.; Tuo, Z.; Shou, Z.; Cheng, J. PLK1 Inhibition Induces Immunogenic Cell Death and Enhances Immunity against NSCLC. Int. J. Med Sci. 2021, 18, 3516–3525. [Google Scholar] [CrossRef]
- Yin, T.; Zhao, Z.-B.; Guo, J.; Wang, T.; Yang, J.-B.; Wang, C.; Long, J.; Ma, S.; Huang, Q.; Zhang, K.; et al. Aurora A Inhibition Eliminates Myeloid Cell–Mediated Immunosuppression and Enhances the Efficacy of Anti–PD-L1 Therapy in Breast Cancer. Cancer Res. 2019, 79, 3431–3444. [Google Scholar] [CrossRef] [PubMed]
- Tani, T.; Kitajima, S.; Conway, E.B.; Knelson, E.H.; Barbie, D.A. KRAS G12C inhibition and innate immune targeting. Expert Opin. Ther. Targets 2021, 25, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non–Small Cell Lung Cancer: A Retrospective Analysis. Clin. Cancer Res. 2016, 22, 4585–4593. [Google Scholar] [CrossRef]
- Shi, C.; Qin, K.; Lin, A.; Jiang, A.; Cheng, Q.; Liu, Z.; Zhang, J.; Luo, P. The role of DNA damage repair (DDR) system in response to immune checkpoint inhibitor (ICI) therapy. J. Exp. Clin. Cancer Res. 2022, 41, 268. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2013, 21, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.-J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, T.; Flies, D.B.; Marjon, N.A.; Mantia-Smaldone, G.; Ronner, L.; Gimotty, P.A.; Adams, S.F. CTLA-4 Blockade Synergizes Therapeutically with PARP Inhibition in BRCA1-Deficient Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 1257–1268. [Google Scholar] [CrossRef]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; O’Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit beyond BRCA-Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759–3771. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef]
- Dunphy, G.; Flannery, S.M.; Almine, J.F.; Connolly, D.J.; Paulus, C.; Jønsson, K.L.; Jakobsen, M.R.; Nevels, M.M.; Bowie, A.G.; Unterholzner, L. Non-canonical Activation of the DNA Sensing Adaptor STING by ATM and IFI16 Mediates NF-κB Signaling after Nuclear DNA Damage. Mol. Cell 2018, 71, 745–760. [Google Scholar] [CrossRef]
- Li, T.; Chen, Z.J. The cGAS–cGAMP–STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 2018, 215, 1287–1299. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Demaria, S.; Formenti, S.C.; Galluzzi, L. Cytosolic DNA Sensing in Organismal Tumor Control. Cancer Cell 2018, 34, 361–378. [Google Scholar] [CrossRef]
- Hsieh, R.C.-E.; Krishnan, S.; Wu, R.-C.; Boda, A.R.; Liu, A.; Winkler, M.; Hsu, W.-H.; Lin, S.H.; Hung, M.-C.; Chan, L.-C.; et al. ATR-mediated CD47 and PD-L1 up-regulation restricts radiotherapy-induced immune priming and abscopal responses in colorectal cancer. Sci. Immunol. 2022, 7, eabl9330. [Google Scholar] [CrossRef]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- da Costa, A.A.B.A.; Chowdhury, D.; Shapiro, G.I.; D’Andrea, A.D.; Konstantinopoulos, P.A. Targeting replication stress in cancer therapy. Nat. Rev. Drug Discov. 2022, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Garcia-Muse, T.; Aguilera, A. Replication stress and cancer. Nat. Rev. Cancer 2015, 15, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Plummer, E.R.; Kristeleit, R.S.; Cojocaru, E.; Haris, N.; Carter, L.; Jones, R.H.; Blagden, S.P.; Evans, T.J.; Arkenau, H.-T.; Sarker, D.; et al. A first-in-human phase I/II trial of SRA737 (a Chk1 Inhibitor) in subjects with advanced cancer. J. Clin. Oncol. 2019, 37, 3094. [Google Scholar] [CrossRef]
- Yap, T.A.; O’Carrigan, B.; Penney, M.S.; Lim, J.S.; Brown, J.S.; Luken, M.J.D.M.; Tunariu, N.; Perez-Lopez, R.; Rodrigues, D.N.; Riisnaes, R.; et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 3195–3204. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Della Corte, C.M.; Milutinovic, S.; Cardnell, R.J.; Diao, L.; Ramkumar, K.; Gay, C.M.; Stewart, C.A.; Fan, Y.; Shen, L.; et al. Combination Treatment of the Oral CHK1 Inhibitor, SRA737, and Low-Dose Gemcitabine Enhances the Effect of Programmed Death Ligand 1 Blockade by Modulating the Immune Microenvironment in SCLC. J. Thorac. Oncol. 2019, 14, 2152–2163. [Google Scholar] [CrossRef]
- Kim, R.; Kwon, M.; An, M.; Kim, S.; Smith, S.; Loembé, A.; Mortimer, P.; Armenia, J.; Lukashchuk, N.; Shah, N.; et al. Phase II study of ceralasertib (AZD6738) in combination with durvalumab in patients with advanced/metastatic melanoma who have failed prior anti-PD-1 therapy. Ann. Oncol. 2021, 33, 193–203. [Google Scholar] [CrossRef]
- Tang, Z.; Pilié, P.G.; Geng, C.; Manyam, G.C.; Yang, G.; Park, S.; Wang, D.; Peng, S.; Wu, C.; Peng, G.; et al. ATR Inhibition Induces CDK1–SPOP Signaling and Enhances Anti–PD-L1 Cytotoxicity in Prostate Cancer. Clin. Cancer Res. 2021, 27, 4898–4909. [Google Scholar] [CrossRef]
- Italiano, A.; Infante, J.; Shapiro, G.; Moore, K.; LoRusso, P.; Hamilton, E.; Cousin, S.; Toulmonde, M.; Postel-Vinay, S.; Tolaney, S.; et al. Phase I study of the checkpoint kinase 1 inhibitor GDC-0575 in combination with gemcitabine in patients with refractory solid tumors. Ann. Oncol. 2018, 29, 1304–1311. [Google Scholar] [CrossRef]
- Wallez, Y.; Dunlop, C.R.; Johnson, T.I.; Koh, S.-B.; Fornari, C.; Yates, J.W.; Fernández, S.B.D.Q.; Lau, A.; Richards, F.M.; Jodrell, D.I. The ATR Inhibitor AZD6738 Synergizes with Gemcitabine In Vitro and In Vivo to Induce Pancreatic Ductal Adenocarcinoma Regression. Mol. Cancer Ther. 2018, 17, 1670–1682. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.; Cruz, J.G.; Daignault-Mill, S.; Veitch, M.; Zeng, B.; Ehmann, A.; Sabdia, M.; Snell, C.; Keane, C.; Dolcetti, R.; et al. Targeting Replication Stress Using CHK1 Inhibitor Promotes Innate and NKT Cell Immune Responses and Tumour Regression. Cancers 2021, 13, 3733. [Google Scholar] [CrossRef] [PubMed]
- Oo, Z.Y.; Proctor, M.; Stevenson, A.J.; Nazareth, D.; Fernando, M.; Daignault, S.M.; Lanagan, C.; Walpole, S.; Bonazzi, V.; Škalamera, D.; et al. Combined use of subclinical hydroxyurea and CHK1 inhibitor effectively controls melanoma and lung cancer progression, with reduced normal tissue toxicity compared to gemcitabine. Mol. Oncol. 2019, 13, 1503–1518. [Google Scholar] [CrossRef]
- Nazareth, D.; Jones, M.J.K.; Gabrielli, B. Everything in Moderation: Lessons Learned by Exploiting Moderate Replication Stress in Cancer. Cancers 2019, 11, 1320. [Google Scholar] [CrossRef] [PubMed]
- Dai, E.; Zhu, Z.; Wahed, S.; Qu, Z.; Storkus, W.J.; Guo, Z.S. Epigenetic modulation of antitumor immunity for improved cancer immunotherapy. Mol. Cancer 2021, 20, 171. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Wilson, C.B.; Rowell, E.; Sekimata, M. Epigenetic control of T-helper-cell differentiation. Nat. Rev. Immunol. 2009, 9, 91–105. [Google Scholar] [CrossRef]
- Kakaradov, B.; Arsenio, J.; Widjaja, C.E.; He, Z.; Aigner, S.; Metz, P.J.; Yu, B.; Wehrens, E.J.; Lopez, J.; Kim, S.H.; et al. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017, 18, 422–432. [Google Scholar] [CrossRef]
- Veazey, K.J.; Muller, D.; Golding, M.C. Prenatal alcohol exposure and cellular differentiation: A role for Polycomb and Trithorax group proteins in FAS phenotypes? Alcohol Res. 2013, 35, 77–85. [Google Scholar]
- Henning, A.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356. [Google Scholar] [CrossRef]
- Nguyen, A.; Ho, L.; Workenhe, S.T.; Chen, L.; Samson, J.; Walsh, S.R.; Pol, J.; Bramson, J.; Wan, Y. HDACi Delivery Reprograms Tumor-Infiltrating Myeloid Cells to Eliminate Antigen-Loss Variants. Cell Rep. 2018, 24, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.-C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell Rep. 2018, 23, 3262–3274. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Taylor, A.; Chin, M.; Huang, H.-R.; Conery, A.R.; Mertz, J.A.; Salmeron, A.; Dakle, P.J.; Mele, D.; Cote, A.; et al. Regulatory T Cell Modulation by CBP/EP300 Bromodomain Inhibition. J. Biol. Chem. 2016, 291, 13014–13027. [Google Scholar] [CrossRef] [PubMed]
- Boyson, S.; Gao, C.; Quinn, K.; Boyd, J.; Paculova, H.; Frietze, S.; Glass, K. Functional Roles of Bromodomain Proteins in Cancer. Cancers 2021, 13, 3606. [Google Scholar] [CrossRef]
- Wang, N.; Wu, R.; Tang, D.; Kang, R. The BET family in immunity and disease. Signal Transduct. Target. Ther. 2021, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET Bromodomain Inhibition as a Therapeutic Strategy to Target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Deng, W.; Liu, Y.; Wang, C. General mechanism of JQ1 in inhibiting various types of cancer. Mol. Med. Rep. 2020, 21, 1021–1034. [Google Scholar] [CrossRef]
- Zhu, H.; Bengsch, F.; Svoronos, N.; Rutkowski, M.R.; Bitler, B.G.; Allegrezza, M.J.; Yokoyama, Y.; Kossenkov, A.V.; Bradner, J.E.; Conejo-Garcia, J.R.; et al. BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep. 2016, 16, 2829–2837. [Google Scholar] [CrossRef]
- Tan, Y.-F.; Wang, M.; Chen, Z.-Y.; Wang, L.; Liu, X.-H. Inhibition of BRD4 prevents proliferation and epithelial–mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Sasikumar, P.G.; Sudarshan, N.S.; Adurthi, S.; Ramachandra, R.K.; Samiulla, D.S.; Lakshminarasimhan, A.; Ramanathan, A.; Chandrasekhar, T.; Dhudashiya, A.A.; Talapati, S.R.; et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Commun. Biol. 2021, 4, 699. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Iams, W.; Cruz, M.R.; Chandra, S.; Choi, J.; Giles, F. Current landscape and future of dual anti-CTLA4 and PD-1/PD-L1 blockade immunotherapy in cancer; lessons learned from clinical trials with melanoma and non-small cell lung cancer (NSCLC). J. Immunother. Cancer 2018, 6, 39. [Google Scholar] [CrossRef]
- Dietsch, G.N.; Randall, T.D.; Gottardo, R.; Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Manjarrez, K.L.; Newkirk, M.; Bryan, J.K.; Hershberg, R.M. Late-Stage Cancer Patients Remain Highly Responsive to Immune Activation by the Selective TLR8 Agonist Motolimod (VTX-2337). Clin. Cancer Res. 2015, 21, 5445–5452. [Google Scholar] [CrossRef] [PubMed]
- Northfelt, D.W.; Ramanathan, R.K.; Cohen, P.A.; Von Hoff, D.D.; Weiss, G.J.; Dietsch, G.N.; Manjarrez, K.L.; Randall, T.D.; Hershberg, R.M. A Phase I Dose-Finding Study of the Novel Toll-like Receptor 8 Agonist VTX-2337 in Adult Subjects with Advanced Solid Tumors or Lymphoma. Clin. Cancer Res. 2014, 20, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.Z.; Yunis, C.; Harrison, L.I.; Kumar, S.; Hawkinson, R.; Cooley, S.; Vasilakos, J.P.; Gorski, K.S.; Miller, J.S. First in Human Phase I Trial of 852A, a Novel Systemic Toll-like Receptor 7 Agonist, to Activate Innate Immune Responses in Patients with Advanced Cancer. Clin. Cancer Res. 2007, 13, 7119–7125. [Google Scholar] [CrossRef] [PubMed]
- Donin, N.M.; Chamie, K.; Lenis, A.T.; Pantuck, A.J.; Reddy, M.; Kivlin, D.; Holldack, J.; Pozzi, R.; Hakim, G.; Karsh, L.I.; et al. A phase 2 study of TMX-101, intravesical imiquimod, for the treatment of carcinoma in situ bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2016, 35, 39.e1–39.e7. [Google Scholar] [CrossRef]
- Van Dalen, F.J.; van Stevendaal, M.H.M.E.; Fennemann, F.L.; Verdoes, M.; Ilina, O. Molecular repolarisation of tu-mour-associated macrophages. Molecules 2019, 24, 9. [Google Scholar] [CrossRef] [PubMed]
- Müller, E.; Christopoulos, P.F.; Halder, S.; Lunde, A.; Beraki, K.; Speth, M.; Øynebråten, I.; Corthay, A. Toll-Like Receptor Ligands and Interferon-γ Synergize for Induction of Antitumor M1 Macrophages. Front. Immunol. 2017, 8, 1383. [Google Scholar] [CrossRef]
- Müller, E.; Speth, M.; Christopoulos, P.F.; Lunde, A.; Avdagic, A.; Øynebråten, I.; Corthay, A. Both Type I and Type II Interferons Can Activate Antitumor M1 Macrophages When Combined With TLR Stimulation. Front. Immunol. 2018, 9, 2520. [Google Scholar] [CrossRef]
- Brackett, C.M.; Kojouharov, B.; Veith, J.; Greene, K.F.; Burdelya, L.G.; Gollnick, S.O.; Abrams, S.I.; Gudkov, A.V. Toll-like receptor-5 agonist, entolimod, suppresses metastasis and induces immunity by stimulating an NK-dendritic-CD8+ T-cell axis. Proc. Natl. Acad. Sci. USA 2016, 113, E874–E883. [Google Scholar] [CrossRef]
- Jochems, C.; Fantini, M.; Fernando, R.I.; Kwilas, A.R.; Donahue, R.N.; Lepone, L.M.; Grenga, I.; Kim, Y.-S.; Brechbiel, M.W.; Gulley, J.L.; et al. The IDO1 selective inhibitor epacadostat enhances dendritic cell immunogenicity and lytic ability of tumor antigen-specific T cells. Oncotarget 2016, 7, 37762–37772. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.-F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Adamiak, M.; Bujko, K.; Brzezniakiewicz-Janus, K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Inhibition of CD39 and CD73 Cell Surface Ectonucleotidases by Small Molecular Inhibitors Enhances the Mobilization of Bone Marrow Residing Stem Cells by Decreasing the Extracellular Level of Adenosine. Stem Cell Rev. Rep. 2019, 15, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Leone, R.D.; Sun, I.-M.; Oh, M.-H.; Wen, J.; Englert, J.; Powell, J.D. Inhibition of the adenosine A2a receptor modulates expression of T cell coinhibitory receptors and improves effector function for enhanced checkpoint blockade and ACT in murine cancer models. Cancer Immunol. Immunother. 2018, 67, 1271–1284. [Google Scholar] [CrossRef]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A Adenosine Receptor Gene Deletion or Synthetic A2A Antagonist Liberate Tumor-Reactive CD8+ T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Galluzzi, L.; Apetoh, L.; Baert, T.; Birge, R.B.; Bravo-San Pedro, J.M.; Breckpot, K.; Brough, D.; Chaurio, R.; Cirone, M.; et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front. Immunol. 2015, 6, 588. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef]
- Janowski, A.M.; Ekolb, R.; Ezhang, W.; Sutterwala, F.S. Beneficial and Detrimental Roles of NLRs in Carcinogenesis. Front. Immunol. 2013, 4, 370. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.H.; Ellis, L.Z.; Fujita, M. Inflammasomes as molecular mediators of inflammation and cancer: Potential role in melanoma. Cancer Lett. 2012, 314, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Yatim, N.; Jusforgues-Saklani, H.; Orozco, S.; Schulz, O.; da Silva, R.B.; e Sousa, C.R.; Green, D.R.; Oberst, A.; Albert, M.L. RIPK1 and NF-κB signaling in dying cells determines crosspriming of CD8+ T cells. Science 2016, 350, 328–334. [Google Scholar] [CrossRef]
- Aaes, T.L.; Kaczmarek, A.; Delvaeye, T.; De Craene, B.; De Koker, S.; Heyndrickx, L.; Delrue, I.; Taminau, J.; Wiernicki, B.; De Groote, P.; et al. Vaccination with Necroptotic Cancer Cells Induces Efficient Anti-tumor Immunity. Cell Rep. 2016, 15, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.-D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic GMP-AMP Synthase Is an Innate Immune DNA Sensor for Mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; McAllister, D.; Vonderhaar, E.P.; Palen, K.; Riese, M.J.; Gershan, J.; Johnson, B.D.; Dwinell, M.B. STING agonist inflames the pancreatic cancer immune microenvironment and reduces tumor burden in mouse models. J. Immunother. Cancer 2019, 7, 115. [Google Scholar] [CrossRef] [PubMed]
- Gorchs, L.; Hellevik, T.; Bruun, J.-A.; Camilio, K.-A.; Al-Saad, S.; Stuge, T.-B.; Martinez-Zubiaurre, I. Cancer-Associated Fibroblasts from Lung Tumors Maintain Their Immunosuppressive Abilities after High-Dose Irradiation. Front. Oncol. 2015, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.P.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Efferth, T. Cancer combination therapy of the sesquiterpenoid artesunate and the selective EGFR-tyrosine kinase inhibitor erlotinib. Phytomedicine 2017, 37, 58–61. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, L.; Xiang, J.-D.; Jin, C.-S.; Li, M.-Q.; He, Y.-Y. Artesunate-induced ATG5-related autophagy enhances the cytotoxicity of NK92 cells on endometrial cancer cells via interactions between CD155 and CD226/TIGIT. Int. Immunopharmacol. 2021, 97, 107705. [Google Scholar] [CrossRef]
- Buoncervello, M.; Romagnoli, G.; Buccarelli, M.; Fragale, A.; Toschi, E.; Parlato, S.; Lucchetti, D.; Macchia, D.; Spada, M.; Canini, I.; et al. IFN-α potentiates the direct and immune-mediated antitumor effects of epigenetic drugs on both metastatic and stem cells of colorectal cancer. Oncotarget 2016, 7, 26361–26373. [Google Scholar] [CrossRef]
- Fragale, A.; Romagnoli, G.; Licursi, V.; Buoncervello, M.; Del Vecchio, G.; Giuliani, C.; Parlato, S.; Leone, C.; De Angelis, M.; Canini, I.; et al. Antitumor Effects of Epidrug/IFNα Combination Driven by Modulated Gene Signatures in Both Colorectal Cancer and Dendritic Cells. Cancer Immunol. Res. 2017, 5, 604–616. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Z.; Feng, W.; Gao, H.; Xu, Z.; Miao, Y.; Li, W.; Chen, F.; Lv, Z.; Huo, J.; et al. Small molecule inhibitors from organoid-based drug screen induce concurrent apoptosis and gasdermin E-dependent pyroptosis in colorectal cancer. Clin. Transl. Med. 2022, 12, e812. [Google Scholar] [CrossRef] [PubMed]
- Long, K.; Gu, L.; Li, L.; Zhang, Z.; Li, E.; Zhang, Y.; He, L.; Pan, F.; Guo, Z.; Hu, Z. Small-molecule inhibition of APE1 induces apoptosis, pyroptosis, and necroptosis in non-small cell lung cancer. Cell Death Dis. 2021, 12, 503. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| NCT | ICB | Trial Arms | Population | Size | Results | Ref. |
|---|---|---|---|---|---|---|
| NCT00861614 | Ipilimumab (CTLA-4) | Ipilimumab vs. placebo | Castration resistant prostate cancer with previous treatment with docetaxel | 988 | Median OS was 11.2 months (95% CI 9.5–12.7) with ipilimumab and 10.0 months (8.3–11) with placebo (hazard ratio [HR] 0.85, 0.72–1.00; p = 0.053) | [13] |
| NCT01057810 | Ipilimumab (CTLA-4) | Ipilimumab vs. placebo | Castration-resistant prostate cancer—asymptomatic or minimally symptomatic with metastatic chemotherapy-naive | 400 | Median OS was 28.7 months (95% CI, 24.5 to 32.5 months) in the ipilimumab arm versus 29.7 months (95% CI, 26.1 to 34.2 months) in the placebo arm (hazard ratio, 1.11; 95.87% CI, 0.88 to 1.39; p = 0.3667) | [14] |
| NCT02617589 | Nivolumab (PD-1) | Nivolumab vs. temozolomide | Newly diagnosed MGMT-unmethylated Glioblastoma | 560 | Press release—did not meet primary end points of OS or PFS. | [15] |
| NCT02017717 | Nivolumab (PD-1) | Nivolumab vs. bevacizumab | Grade IV Glioblastoma | 529 | median OS (mOS) was comparable between groups: nivolumab, 9.8 months (95% CI, 8.2–11.8); bevacizumab, 10.0 months (95% CI, 9.0–11.8); HR, 1.04 (95% CI, 0.83–1.30); p = 0.76. | [16] |
| NCT02991482 | Pembrolizumab (PD-1) | Pembrolizumab vs. SOC | Advanced malignant mesothelioma previously treated with platinum-based chemotherapy | 144 | No difference in OS was detected between groups (HR = 1.12, 95% CI: 0.74–1.69; p = 0.59) | [17] |
| NCT02555657 | Pembrolizumab (PD-1) | Pembrolizumab vs. SOC | Metastatic triple negative breast cancer, previous treatment with two systemic therapies | 622 | In the overall population, median overall survival was 9.9 months (95% CI 8.3–11.4) for the pembrolizumab group and 10.8 months (9.1–12.6) for the chemotherapy group (HR 0.97 [95% CI 0.82–1.15]). | [18] |
| NCT02370498 | Pembrolizumab (PD-1) | Pembrolizumab vs. SOC | Advanced gastric/gastroesophageal junction adenocarcinoma progressive after platinum-based chemotherapy | 592 | Median overall survival was 9.1 months (95% CI 6.2–10.7) with pembrolizumab and 8.3 months (7.6–9.0) with paclitaxel (hazard ratio [HR] 0.82, 95% CI 0.66–1.03; one-sided p = 0.0421). | [19] |
| NCT02494583 | Pembrolizumab (PD-1) | Pembrolizumab vs. pembrolizumab plus SOC vs. SOC | Advanced Gastric or Gastroesophageal Junction Adenocarcinoma—first-Line Monotherapy and Combination Therapy | 763 | Pembrolizumab plus chemotherapy was not superior to chemotherapy for OS in patients with CPS of 1 or greater (12.5 vs. 11.1 months; HR, 0.85; 95% CI, 0.70–1.03; p = 0.05) or CPS of 10 or greater (12.3 vs. 10.8 months; HR, 0.85; 95% CI, 0.62–1.17; p = 0.16) | [20] |
| NCT02853305 | Pembrolizumab (PD-1) | Pembrolizumab vs. pembrolizumab plus SOC vs. SOC | Advanced or metastatic urothelial carcinoma with no previous systemic therapy | 1010 | Pembrolizumab plus chemotherapy versus chemotherapy did not significantly improve overall survival, with a median overall survival of 17.0 months (14.5–19.5) in the pembrolizumab plus chemotherapy group versus 14.3 months (12.3–16.7) in the chemotherapy group (0.86, 0.72–1.02; p = 0.0407). | [21] |
| NCT02702401 | Pembrolizumab (PD-1) | Pembrolizumab vs. placebo | Advanced hepatocellular carcinoma previously systemically treated | 413 | OS and PFS did not reach statistical significance per specified criteria. Median OS was 13.9 months (95% CI, 11.6 to 16.0 months) for pembrolizumab versus 10.6 months (95% CI, 8.3 to 13.5 months) for placebo (hazard ratio [HR], 0.781 | [22] |
| NCT02551159 | Durvalumab (PD-1) | Durvalumab vs. SOC | Recurrent/metastatic head neck squamous cell carcinoma—first line with high PD-1 expression | 823 | Press release—did not meet the primary endpoint of improving overall survival (OS) versus the EXTREME treatment regimen (chemotherapy plus cetuximab) | [23] |
| NCT02952586 | Avelumab (PD-L1) | Avelumab + SOC vs. SOC | Locally advanced head neck squamous cell carcinoma | 697 | Median progression-free survival was not reached (95% CI 16.9 months–not estimable) in the avelumab group and not reached (23.0 months–not estimable) in the placebo group (stratified hazard ratio 1.21 [95% CI 0.93–1.57] favouring the placebo group; one-sided p = 0.92). | [24] |
| NCT02542293 | Durvalumab (PD-1) + Tremelimumab (CTLA-4) | Combination immunotherapy vs. SOC | Metastatic NSCLC—first line | 953 | Press release—did not meet primary endpoints | [25] |
| NCT02538666 | Nivolumab (PD-1) and Ipilimumab (CTLA-4) | Combination immunotherapy vs. placebo | Extensive disease NSCLC—as maintenance therapy post platinum-based chemotherapy | 1212 | OS was not significantly prolonged with nivolumab plus ipilimumab versus placebo (hazard ratio [HR], 0.92; 95% CI, 0.75 to 1.12; p = 0.37; median, 9.2 v 9.6 months) | [26] |
| NCT02279732 | Ipilimumab (CTLA-4) | Ipilimumab + SOC vs. placebo + SOC | Metastatic or recurrent squamous NSCLC | 342 | ClinicalTrail.Gov result posted. Recruitment stopped at 204 patients and primary end point not analysed. | [27] |
| NCT01285609 | Ipilimumab (CTLA-4) | Ipilimumab + SOC vs. placebo + SOC | Metastatic or recurrent squamous NSCLC | 1289 | Median OS was 13.4 months for chemotherapy plus ipilimumab and 12.4 months for chemotherapy plus placebo (hazard ratio, 0.91; 95% CI, 0.77 to 1.07; p = 0.25). | [28] |
| NCT01450761 | Ipilimumab (CTLA-4) | Ipilimumab + SOC vs. placebo + SOC | Newly diagnosed extensive-stage SCLC | 1351 | Median OS was 11.0 months for chemotherapy plus ipilimumab versus 10.9 months for chemotherapy plus placebo (hazard ratio, 0.94; 95% CI, 0.81 to 1.09; p = 0.3775). | [29] |
| NCT | Trial Name | ICB | SMI | Trial Arms | Population | Size | Status | Outcomes | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| Published/Completed Trials | |||||||||
| NCT03361865 | ECHO-007 | Pembrolizumab (PD-1) | Epacadostat (IDO1) | 1. Pembrolizumab + Epacadostat 2. Pembrolizumab | Cisplatin-ineligible advanced or metastatic urothelial Carcinoma | 93 | Completed, not published | Source—ClinicalTrials.Gov ORR 31.8 (22.46 to 55.24) vs. 24.5 (15.33 to 43.67) | [46] |
| NCT02752074 | ECHO-301 | Pembrolizumab (PD-1) | Epacadostat (IDO1) | 1. Pembrolizumab + Epacadostat 2. Pembrolizumab | Unresectable or metastatic melanoma | 706 | Completed | No significant difference in PFS or OS | [47] |
| NCT03829332 | LEAP-007 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + lenvatinib + SOC 2. Pembrolizumab + SOC | Treatment-naïve, Metastatic NSCLC | 623 | Completed, not published | ClinicalTrials.Gov PFS 6.6 months (Combination) vs. 4.2 months (Pembrolizumab monotherapy) HR 0.78 (p = 0.006). No benefit to overall survival. | [48] |
| NCT03517449 | KEYNOTE-775 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + lenvatinib 2. SOC | Advanced, recurrent or metastatic endometrial cancer. | 827 | Completed | PFS combo 7.2 vs. SOC 3.8 months; hazard ratio, 0.56; 95% CI, 0.47 to 0.66; p < 0.001. OS 8.3 vs. 11.4 months; hazard ratio, 0.62; 95% CI, 0.51 to 0.75; p < 0.001 | [49] |
| NCT02853331 | KEYNOTE-426 | Pembrolizumab (PD-1) | Axitinib (TKR) | 1. Pembrolizumab + Axitinib 2. Sunitinib | First-line in Locally Advanced or Metastatic Renal Cell Carcinoma | 861 | Completed | PFS -15.1 months pembrolizumab + axitinib group vs. 11.1—month sunitinib group (HR for disease progression or death, 0.69; 95% CI, 0.57 to 0.84; p < 0.001 | [50,51] |
| NCT02684006 | JAVELIN Renal 101 | Avelumab (PD-L1) | Sunitinib (TKR) | 1. Avelumab + axitinib 2. Sunitinib | First-line in Locally Advanced Renal Cell Carcinoma | 888 | Completed | Median PFS l 13.8 months combination vs. 8.4 months monotherapy (hazard ratio, 0.69; 95% CI, 0.56 to 0.84; p < 0.001 | [52] |
| NCT02788279 | IMblaze370 | Atezolizumab (PD-L1) | Cobimetinib (MEK) | 1. Atezolizumab 2. Cobimetinib + Atezolizumab 3. Regorafenib | Previously Treated Unresectable Locally Advanced or Metastatic Colorectal Adenocarcinoma | 363 | Completed | Not significant difference. Median overall survival was 8.87 months with atezolizumab plus cobimetinib, 7, 10 months with atezolizumab, and 8.51 months with regorafenib; HR 1.00 for the combination versus regorafenib and HR 1.19 (p = 0.34) for atezolizumab versus regorafenib | [53] |
| NCT03141177 | CheckMate 9ER | Nivolumab (PD-1) | Cabozantinib (TKR) | 1. Nivolumab and Cabozantinib 2. Sunitinib 3. Nivolumab, Ipilimumab, Cabozantinib (discontinued) | First line Advanced or Metastatic Renal Cell Carcinoma | 701 | Completed | PFS 16.6 months (95% CI, 12.5 to 24.9) with nivolumab + cabozantinib vs. 8.3 months (95% CI, 7.0 to 9.7) sunitinib (HR 0.51; 95% CI, 0.41 to 0.64; p < 0.001). OS at 12 months 85.7% (95% CI, 81.3 to 89.1) with nivolumab + cabozantinib vs. 75.6% (95% CI, 70.5 to 80.0) with sunitinib (HR 0.60; 98.89% CI, 0.40 to 0.89; p = 0.001). | [54] |
| NCT03937219 | COSMIC-313 | Nivolumab (PD-1_ and Ipilimumab (CTLA-4) | Cabozantinib (TKR) | 1. Cabozantinib + nivolumab + ipilimumab followed by cabozantinib + nivolumab 2. nivolumab + ipilimumab followed by nivolumab | First line Advanced or Metastatic Renal Cell Carcinoma of Intermediate or Poor Risk | 840 | Completed. Collecting OS data | press release/meeting abstract. Primary PFS endpoint (HR 0.73, 95% CI, 0.57–0.94; p = 0.013) in favour of combination | [55] |
| NCT03713593 | LEAP-002 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. lenvatinib plus pembrolizumab 2. Lenvatinib + placebo | First-line Therapy for Advanced HCC | 794 | Completed | Press release—did not meet primary outcome measures | [56] |
| In Progress Trials (by Tumor Type) | |||||||||
| NCT04335006 | Carelizumab (PD-1) | Apatinib (TKR) | 1. Carelizumab + Nab-paclitaxel + Apatinib 2. Carelizumab + Nab-paclitaxel 3. Nab-paclitaxel | Advanced or metastatic Triple Negative Breast Cancer | 780 | Recruiting | PFS | ||
| NCT04177108 | Atezolizumab (PD-L1) | Ipatasertib (AKT) | 1. Paclitaxel, Atezolizumab and Ipatasertib 2. Paclitaxel, ipatasertib b 3. Paclitaxel | Locally Advanced Unresectable or Metastatic Triple-Negative Breast Cancer. | 242 | Active, not recruiting | PFS, OS | ||
| NCT03740165 | KEYLYNK-001 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. Pembrolizumab + Olaparib + SOC 2. Pembrolizumab + SOC 3. SOC | BRCA Non-mutated Advanced Epithelial Ovarian Cancer | 1284 | Active, not recruiting | PFS | |
| NCT05145218 | TQB2450 (PD-L1) | Anlotinib (TKR) | 1. TQB2450 + Anlotinib 2. Paclitaxel | Recurrent platinum-resistant ovarian cancer | 405 | Recruiting | PFS, OS | ||
| NCT03651206 | ROCSAN | Dostarlimab (PD-1) | Niraparib (PARP) | 1. Niraparib 2. Niraparib + TSR-042 (Dostarlimab) 3. SOC | Metastatic or Recurrent Endometrial or Ovarian Carcinosarcoma | 196 | Recruiting | RR, OS | |
| NCT03598270 | Atezolizumab (PD-L1) | Niraparib (PARP) | 1. SOC 2. SOC + Atezolizumab with maintaince atezolizumab + niraparib | Recurrent ovarian cancer | 414 | Active, not recruiting | PFS | ||
| NCT03793166 | PDGREEI | Nivolumab (PD-1) | Cabozantinib (TKR) | 1. Nivolumab 2. Nivolumab + Cabozantinib | Metastatic clear cell renal cancer | 1046 | Recruiting | OS | |
| NCT04523272 | TQB2450 (PD-L1) | Anlotinib (TKR) | 1. TQB2450 + Anlotinib 2. Sunitinib | Locally advanced clear cell renal cancer | 418 | Recruiting | PFS | ||
| NCT05219318 | SPICI | PD-1/PD-L1 ICI | VEGFR-Tyrosine Kinase Inhibitor | 1. Treatment pause post-12 months of therapy. 2. PD-1/PD-L1 inhibitor + TKI | Good or Intermediate Risk Metastatic Renal Cell Carcinoma | 372 | Not yet recruiting | PFS | |
| NCT04338269 | CONTACT-03 | Atezolizumab (PD-L1) | Cabozantinib (TKR) | 1. Atezolizumab + cabazntinib 2. cabazantinib | Inoperable, Locally Advanced, or Metastatic Renal Cell Carcinoma | 523 | Active, not recruiting | PFS, OS | |
| NCT04987203 | Nivolumab (PD-1) | Tivozanib (TKR) | 1. Nivolumab + Tivozanib 2. Tivozanib | Locally advanced or metastatic Renal cell carcinoma-with progression following at least 6 weeks of treatment with ICI | 326 | Recruiting | PFS | ||
| NCT03898180 | LEAP-011 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + Lenvatinib 2. Pembrolizumab monotherapy 3. Placebo + pembrolizumab | First-line Cisplatin-ineligible Participants with PDL1 expression. Ineligible for Platinum-containing Chemotherapy Urothelial Carcinoma | 487 | Active, not recruiting | PFS, OS | |
| NCT03834519 | KEYLYNK-010 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. Pembrolizumab + Olaparib 2. Abiraterone + Prednisone or Enzalutamide | Metastatic Castration-resistant Prostate Cancer | 793 | Active, not recruiting | PFS, OS | |
| NCT03976375 | LEAP-008 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + Lenvatinib 2. Docetaxel 3. Lenvatinib monotherapy | Metastatic NSCLC | 405 | Active, not recruiting | OS, PFS | |
| NCT03178552 | Atezolizumab (PD-L1) | Cobimetinib (MEK), Alectinib (ALK), Entrectinib (ROS1), Vemurafenib (BRAF), GDC-6036 (KRAS) | Multiple trial arms including different combinations | Advanced or metastatic NSCLC | 1000 | Recruiting | ORR | ||
| NCT04471428 | Atezolizumab (PD-L1) | Cabozantinib (TKR) | 1. Atezolizuman + cabozantinib 2. Docetaxel | Metastatic NSCLC | 366 | Active, not recruiting | OS | ||
| NCT04921358 | SAFFRON-301: | Tislelizumab (PD-1) | Sitravatinib (TKR) | 1. Tislelizumab + Sitravatinib 2. Docetaxel | Metastatic NSCLC | 420 | Recruiting | OS, PFS | |
| NCT03348904 | Nivolumab (PD-1) | Epacadostat (IDO1) | 1. Nivolumab + epacadostat + platnium 2. Platinum chemotherapy 3. Platinum + Nivolumab | Metastatic or recurrent NSCLC | 2 | Terminated early | |||
| NCT04380636 | KEYLYNK-012 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. pembrolizumab + chemoradiation → pembrolizumab + olaparib placebo 2. pembrolizumab + chemoradiation → pembrolizumab + olaparib 3. chemoradiation → durvalumab | Unresectable, locally advanced NSCLC | 870 | Recruiting | PFS, OS | |
| NCT03906071 | SAPPHIRE | Nivolumab (PD-1) | Sitravatinib (TKR) | 1. Nivolumab and Sitravatinib 2. Docetaxel | Advanced or metastatic NSCLC | 532 | Active, not recruiting | OS | |
| NCT03976362 | KEYLYNK-008 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. Pembrolizumab + Carboplatin + Taxane + Maintenance Olaparib 2. Pembrolizumab + Carboplatin + Taxane + Maintenance placebo | First-line Metastatic NSCLC | 857 | Active, not recruiting | PFS, OS | |
| NCT03976323 | KEYLYNK-006 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. Pembrolizumab + Pemetrexed + Platinum Therapy + Maintenance Olaparib 2. Pembrolizumab + Pemetrexed + Platinum Therapy + Maintenance Pemetrexed | First-line Metastatic NSCLC | 1005 | Active, not recruiting | PFS, OS | |
| NCT03829319 | LEAP-006 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + lenvatinib + SOC 2. Pembrolizumab + SOC | Metastatic Nonsquamous NSCLC | 726 | Active, not recruiting | Safety, PFS, OS | |
| NCT05042375 | Camrelizumab (PD-1) | Famitinib (TKR) | 1. camrelizumab + famitinib 2. pembrolizumab 3. camrelizumab | PD-L1-Positive Recurrent or Metastatic NSCLC | 450 | Not yet recruiting | PFS | ||
| NCT05346952 | TQB2450 (PD-L1) | Anlotinib (TKR) | 1. TQB2450 + carboplatin + pemetrexed 2. TQB2450 + Anlotinib + Pemetrexed | First-line Treatment on Patient with Advanced Non-squamous NSCLC | 390 | Recruiting | PFS, OS | ||
| NCT05106335 | Camrelizumab (PD-1) | Famitinib (TKR) | 1. Camerlizumab + famitinib 2. famitinib 3. docetaxel | Advanced NSCLC | 524 | Recruiting | OS | ||
| NCT04234607 | ETER701 | TQB2450 (PD-L1) | Anlotinib (TKR) | 1. TQB2450 + Anlotinib + etoposide + carboplatin 2. Anlotinib + etoposide + carboplatin 3. etoposide + carboplatin | Extensive SCLC | 738 | Not yet recruiting | PFS, OS | |
| NCT04624204 | KEYLYNK-013 | Pembrolizumab (PD-1) | Olaparib (PARP) | 1. Pembrolizumab + SOC 2. Pembrolizumab + Olaparib + SOC 3. SOC | Newly Diagnosed Treatment-Naïve Limited-Stage SCLC | 672 | Recruiting | PFS, OS | |
| NCT04674683 | Nivolumab (PD-1) | HBI-8000 (HDAC) | 1. HBI-8000 + nivolumab 2. Placebo + nivolumab | Unresectable or metastatic melanoma | 480 | Recruiting | ORR, PFS | ||
| NCT03820986 | LEAP-003 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Lenvatinib + pembrolizumab 2. Pembrolizumab + placebo | First-line in adults With Advance Melanoma | 660 | Active, not recruiting | PFS, OS | |
| NCT03813784 | SHR-1210 (PD-1) | Apatinib (TKR) | 1. SHR-1210 + Apatinib + SOC 2. SOC 3. SOC + SHR-1210 | Advanced or metastatic gastric cancer | 887 | Active, not recruiting | OS | ||
| NCT04949256 | LEAP-014 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Pembrolizumab + Lenvatinib + Chemotherapy 2. Pembrolizumab + Chemotherapy | First-line Metastatic Esophageal Carcinoma | 862 | Recruiting | Safety, PFS, OS | |
| NCT04662710 | LEAP-015 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Lenvatinib + Pembrolizumab + SOC 2. SOC | First-line in Advanced/Metastatic Gastroesophageal Adenocarcinoma | 790 | Recruiting | PFS, OS | |
| NCT04879368 | INTEGRATEIIb | Nivolumab (PD-1) | Regorafenib (TKR) | 1. Nivolumab + regorafenib 2. SOC | Refractory Advanced Gastro-Oesophageal Cancer | 450 | Recruiting | OS | |
| NCT05049681 | SHR-1210 (PD-1) | Apatinib (TKR) | 1. SHR-1210 + Apatinib 2. SHR-1210 | Locally advanced/unresectable, recurrence or metastatic esophegeal SCC | 234 | Not yet recruiting | OS | ||
| NCT04776148 | LEAP-17 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. lenvatinib + pembrolizumab 2. SOC | Metastatic Colorectal Cancer | 424 | Active, not recruiting | OS | |
| NCT04669496 | Toripalimab (PD-1) | Lenvatinib (TKR) | 1. Neoadjuvant GEMOX + Lenvatinib + Toripalimab 2. No neoadjuvant therapy | Resectable Intrahepatic Cholangiocarcinoma with High-risk Recurrence Factors | 178 | Recruiting | PFS | ||
| NCT04246177 | LEAP-012 | Pembrolizumab (PD-1) | Lenvatinib (TKR) | 1. Lenvatinib plus Pembrolizumab plus TACE 2. Oral Placebo plus IV Placebo plus TACE | Incurable Locally Advanced HCC | 950 | Recruiting | PFS, OS | |
| NCT04523493 | Toripalimab (PD-1) | Lenvatinib (TKR) | 1. Toripalimab + Lenvatinib 2. Lenvatinib | First-line Therapy for Advanced HCC | 519 | Recruiting | PFS, OS | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, D.; Moseley, P.; Lu, X.; Wright, Q.; Gabrielli, B.; Frazer, I.H.; Cruz, J.L.G. Repurposing of Commercially Existing Molecular Target Therapies to Boost the Clinical Efficacy of Immune Checkpoint Blockade. Cancers 2022, 14, 6150. https://doi.org/10.3390/cancers14246150
Sinha D, Moseley P, Lu X, Wright Q, Gabrielli B, Frazer IH, Cruz JLG. Repurposing of Commercially Existing Molecular Target Therapies to Boost the Clinical Efficacy of Immune Checkpoint Blockade. Cancers. 2022; 14(24):6150. https://doi.org/10.3390/cancers14246150
Chicago/Turabian StyleSinha, Debottam, Philip Moseley, Xuehan Lu, Quentin Wright, Brian Gabrielli, Ian H. Frazer, and Jazmina L. G. Cruz. 2022. "Repurposing of Commercially Existing Molecular Target Therapies to Boost the Clinical Efficacy of Immune Checkpoint Blockade" Cancers 14, no. 24: 6150. https://doi.org/10.3390/cancers14246150
APA StyleSinha, D., Moseley, P., Lu, X., Wright, Q., Gabrielli, B., Frazer, I. H., & Cruz, J. L. G. (2022). Repurposing of Commercially Existing Molecular Target Therapies to Boost the Clinical Efficacy of Immune Checkpoint Blockade. Cancers, 14(24), 6150. https://doi.org/10.3390/cancers14246150