Neurological Complications of Conventional and Novel Anticancer Treatments

 ,
, 
 ,
,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Chemotherapy-Induced Peripheral Neurotoxicity (CIPN)
2.1. Definition and Clinical Presentation

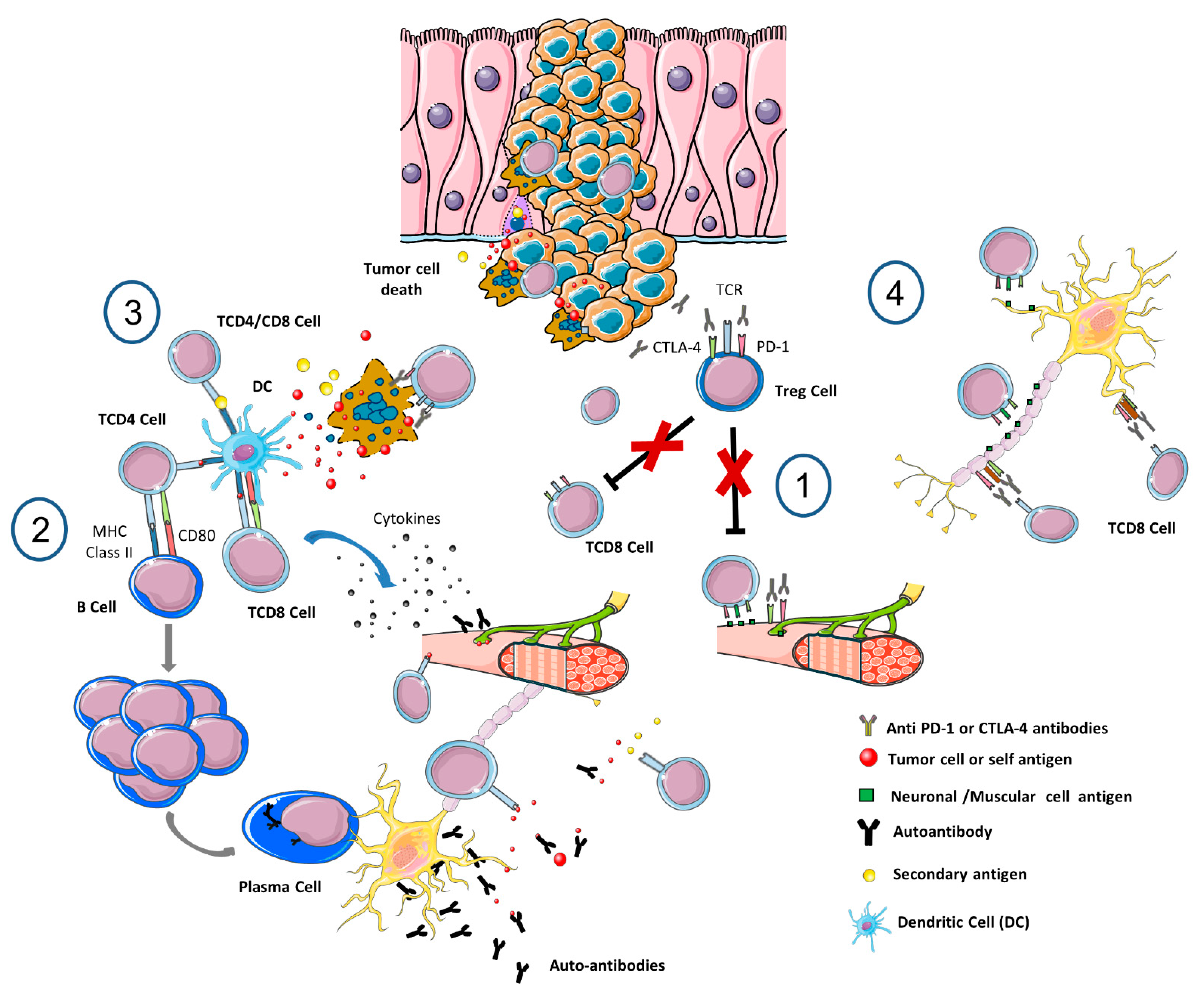{kind=link}
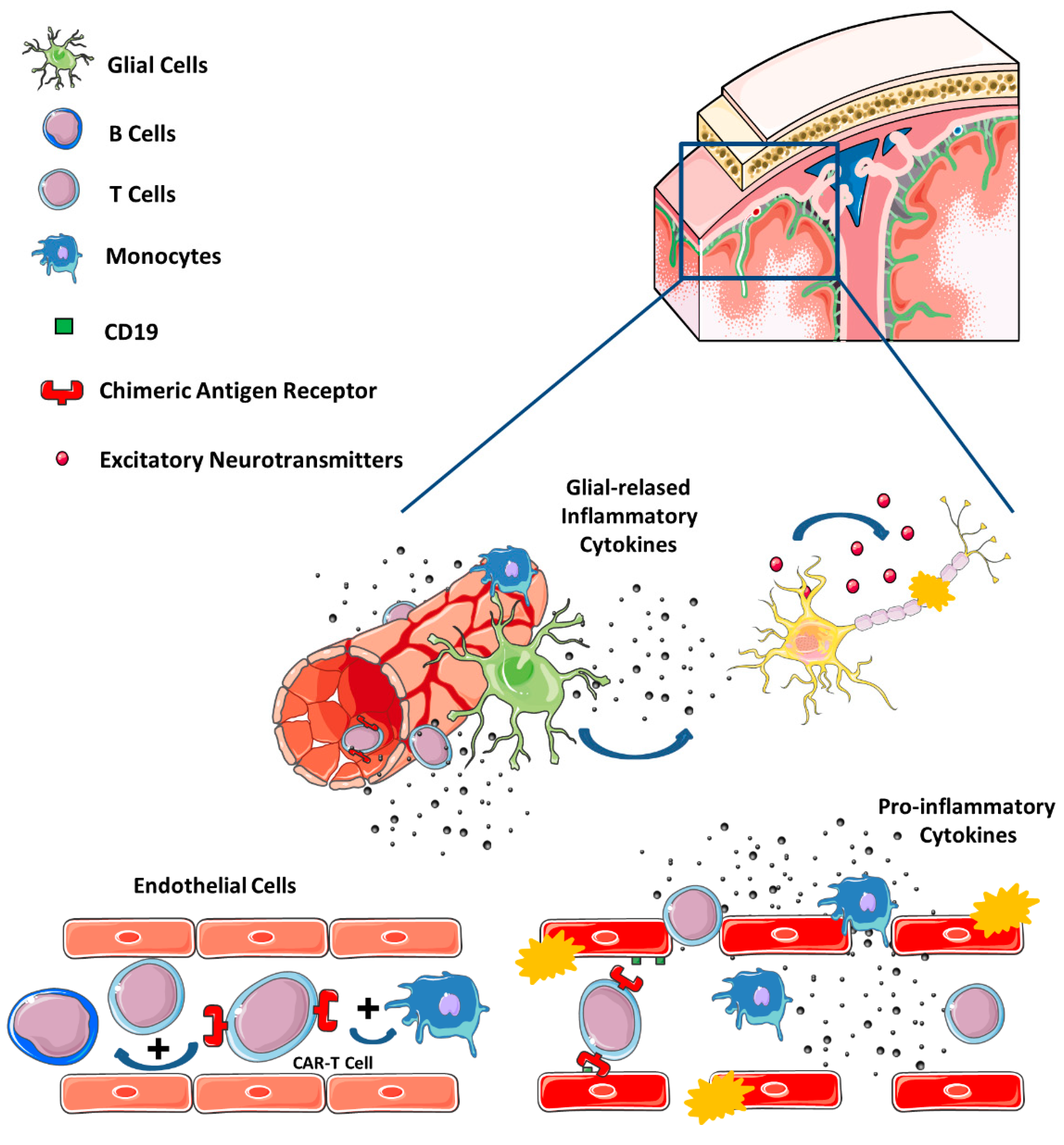{kind=link}
| Drug Class | Clinical Presentation |
|---|---|
| Platinum drugs—cisplatin and carboplatin |
|
| Platinum drugs—oxaliplatin (acute) |
|
| Platinum drugs—oxaliplatin (chronic) |
|
| Taxanes |
|
| Epothilones |
|
| Vinca Alkaloids |
|
| Bortezomib |
|
| Thalidomide and analogues |
|
2.2. Mechanisms
2.3. Assessment and Therapeutic Approach/Strategy
3. Novel Anticancer Drugs and Peripheral Neurotoxicity
3.1. Conjugated Monoclonal Antibodies
3.2. Small Molecule Inhibitors
3.3. Immune Checkpoint Inhibitors (ICI)
4. Immune-Mediated Toxicities
4.1. Common Forms of Central Nervous System (CNS) Neurotoxicity after ICI Therapy
4.2. Common Forms of PNS Neurotoxicity after ICI Therapy
4.3. Neurotoxicity after CAR T Cell Therapies
4.4. Possible Mechanisms of Novel Immune-Mediated Neurotoxicity
4.5. Treatment or Potential Treatment Strategies
5. Chemotherapy-Related Cognitive Impairment
5.1. Definition and Clinical Presentation
5.2. Mechanisms of Chemotherapy-Related Cognitive Impairment (CRCI)
5.3. Assessment and Therapeutic Approach/Strategy
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sinha, M.K.; Barman, A.; Sahu, S.; Jha, A.K.; Asharaf, A.A. Tamoxifen in Mastalgia: A Meta-Analysis. J. Obstet. Gynaecol. Can. 2022, 44, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Lavanya, K.J.; Nandini; Kaur, K.; Jaitak, V. Effectiveness of Selective Estrogen Receptor Modulators in Breast Cancer Therapy: An Update. Curr. Med. Chem. 2022. [Google Scholar] [CrossRef]
- Briani, C.; Argyriou, A.A.; Izquierdo, C.; Velasco, R.; Campagnolo, M.; Alberti, P.; Frigeni, B.; Cacciavillani, M.; Bergamo, F.; Cortinovis, D.; et al. Long-term course of oxaliplatin-induced polyneuropathy: A prospective 2-year follow-up study. J. Peripher. Nerv. Syst. 2014, 19, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Pike, C.T.; Birnbaum, H.G.; Muehlenbein, C.E.; Pohl, G.M.; Natale, R.B. Healthcare costs and workloss burden of patients with chemotherapy-associated peripheral neuropathy in breast, ovarian, head and neck, and nonsmall cell lung cancer. Chemother. Res. Pract. 2012, 2012, 913848. [Google Scholar] [CrossRef]
- Cavaletti, G.; Marmiroli, P. Pharmacotherapy options for managing chemotherapy-induced peripheral neurotoxicity. Expert Opin. Pharmacother. 2018, 19, 113–121. [Google Scholar] [CrossRef]
- Islam, B.; Lustberg, M.; Staff, N.P.; Kolb, N.; Alberti, P.; Argyriou, A.A. Vinca alkaloids, thalidomide and eribulin-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24 (Suppl. S2), S63–S73. [Google Scholar] [CrossRef]
- Dorsey, S.G.; Kleckner, I.R.; Barton, D.; Mustian, K.; O’Mara, A.; St Germain, D.; Cavaletti, G.; Danhauer, S.C.; Hershman, D.; Hohmann, A.G.; et al. NCI Clinical Trials Planning Meeting for prevention and treatment of chemotherapy-induced peripheral neuropathy. J. Natl. Cancer Inst. 2019, 111, 531–537. [Google Scholar] [CrossRef]
- Gewandter, J.S.; Brell, J.; Cavaletti, G.; Dougherty, P.M.; Evans, S.; Howie, L.; McDermott, M.P.; O’Mara, A.; Smith, A.G.; Dastros-Pitei, D.; et al. Trial designs for chemotherapy-induced peripheral neuropathy prevention: ACTTION recommendations. Neurology 2018, 91, 403–413. [Google Scholar] [CrossRef]
- Hu, B.; Zhou, Q.; Wu, T.; Zhuang, L.; Yi, L.; Cao, J.; Yang, X.; Wang, J. Efficacy and safety of subcutaneous versus intravenous bortezomib in multiple myeloma: A meta-analysis. Int. J. Clin. Pharmacol. Ther. 2017, 55, 329–338. [Google Scholar] [CrossRef]
- Alberti, P. Platinum-drugs induced peripheral neurotoxicity: Clinical course and preclinical evidence. Expert Opin. Drug Metab. Toxicol. 2019, 15, 487–497. [Google Scholar] [CrossRef]
- Grolleau, F.; Gamelin, L.; Boisdron-Celle, M.; Lapied, B.; Pelhate, M.; Gamelin, E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. J. Neurophysiol. 2001, 85, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Park, S.B.; Lin, C.S.; Krishnan, A.V.; Goldstein, D.; Friedlander, M.L.; Kiernan, M.C. Oxaliplatin-induced neurotoxicity: Changes in axonal excitability precede development of neuropathy. Brain 2009, 132, 2712–2723. [Google Scholar] [CrossRef] [PubMed]
- Lucchetta, M.; Lonardi, S.; Bergamo, F.; Alberti, P.; Velasco, R.; Argyriou, A.A.; Briani, C.; Bruna, J.; Cazzaniga, M.; Cortinovis, D.; et al. Incidence of atypical acute nerve hyperexcitability symptoms in oxaliplatin-treated patients with colorectal cancer. Cancer Chemother. Pharmacol. 2012, 70, 899–902. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, A.A.; Velasco, R.; Briani, C.; Cavaletti, G.; Bruna, J.; Alberti, P.; Cacciavillani, M.; Lonardi, S.; Santos, C.; Cortinovis, D.; et al. Peripheral neurotoxicity of oxaliplatin in combination with 5-fluorouracil (FOLFOX) or capecitabine (XELOX): A prospective evaluation of 150 colorectal cancer patients. Ann. Oncol. 2012, 23, 3116–3122. [Google Scholar] [CrossRef] [PubMed]
- Nozza, A.; Terenghi, F.; Gallia, F.; Adami, F.; Briani, C.; Merlini, G.; Giordano, L.; Santoro, A.; Nobile-Orazio, E. Lenalidomide and dexamethasone in patients with POEMS syndrome: Results of a prospective, open-label trial. Br. J. Haematol. 2017, 179, 748–755. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Palumbo, A.; Corradini, P.; Cavo, M.; Delforge, M.; Di Raimondo, F.; Weisel, K.C.; Oriol, A.; Hansson, M.; Vacca, A.; et al. Safety and efficacy of pomalidomide plus low-dose dexamethasone in STRATUS (MM-010): A phase 3b study in refractory multiple myeloma. Blood 2016, 128, 497–503. [Google Scholar] [CrossRef]
- Yuki, E.F.N.; Soares, R.; Kupa, L.V.K.; Heise, C.O.; Aikawa, N.E.; Arnone, M.; Romiti, R.; Pedrosa, T.D.N.; Silva, C.A.A.D.; Bonfa, E.; et al. One-year prospective nerve conduction study of thalidomide neuropathy in lupus erythematosus: Incidence, coasting effect and drug plasma levels. Lupus 2021, 30, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Zara, G.; Rondinone, R.; Della Libera, S.; Ermani, M.; Ruggero, S.; Ghirardello, A.; Zampieri, S.; Doria, A. Thalidomide neurotoxicity: Prospective study in patients with lupus erythematosus. Neurology 2004, 62, 2288–2290. [Google Scholar] [CrossRef]
- Tamburin, S.; Park, S.B.; Alberti, P.; Demichelis, C.; Schenone, A.; Argyriou, A.A. Taxane and epothilone-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24 (Suppl. S2), S40–S51. [Google Scholar] [CrossRef]
- Gewandter, J.S.; Freeman, R.; Kitt, R.A.; Cavaletti, G.; Gauthier, L.R.; McDermott, M.P.; Mohile, N.A.; Mohlie, S.G.; Smith, A.G.; Tejani, M.A.; et al. Chemotherapy-induced peripheral neuropathy clinical trials: Review and recommendations. Neurology 2017, 89, 859–869. [Google Scholar] [CrossRef]
- Gewandter, J.S.; Dworkin, R.H.; Finnerup, N.B.; Mohile, N.A. Painful chemotherapy-induced peripheral neuropathy: Lack of treatment efficacy or the wrong clinical trial methodology? Pain 2017, 158, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lavoie Smith, E.M.; Haupt, R.; Kelly, J.P.; Lee, D.; Kanzawa-Lee, G.; Knoerl, R.; Bridges, C.; Alberti, P.; Prasertsri, N.; Donohoe, C. The Content Validity of a Chemotherapy-Induced Peripheral Neuropathy Patient-Reported Outcome Measure. Oncol. Nurs. Forum 2017, 44, 580–588. [Google Scholar] [CrossRef] [PubMed]
- Suchánková, T.; Kubíček, K.; Kašpárková, J.; Brabec, V.; Kozelka, J. Platinum-DNA interstrand crosslinks: Molecular determinants of bending and unwinding of the double helix. J. Inorg. Biochem. 2012, 108, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs--phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar] [PubMed]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Alberti, P.; Bruna, J.; Psimaras, D.; Argyriou, A.A. Bortezomib and other proteosome inhibitors-induced peripheral neurotoxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24 (Suppl. S2), S52–S62. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Li, Y.; Segal, R.A. A Mechanistic Understanding of Axon Degeneration in Chemotherapy-Induced Peripheral Neuropathy. Front. Neurosci. 2017, 11, 481. [Google Scholar] [CrossRef]
- Alé, A.; Bruna, J.; Herrando, M.; Navarro, X.; Udina, E. Toxic effects of bortezomib on primary sensory neurons and Schwann cells of adult mice. Neurotox. Res. 2015, 27, 430–440. [Google Scholar] [CrossRef]
- Staff, N.P.; Podratz, J.L.; Grassner, L.; Bader, M.; Paz, J.; Knight, A.M.; Loprinzi, C.L.; Trushina, E.; Windebank, A.J. Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. Neurotoxicology 2013, 39, 124–131. [Google Scholar] [CrossRef]
- Pero, M.E.; Meregalli, C.; Qu, X.; Shin, G.J.; Kumar, A.; Shorey, M.; Rolls, M.M.; Tanji, K.; Brannagan, T.H.; Alberti, P.; et al. Pathogenic role of delta 2 tubulin in bortezomib-induced peripheral neuropathy. Proc. Natl. Acad. Sci. USA 2021, 118, e2012685118. [Google Scholar] [CrossRef]
- Schellingerhout, D.; LeRoux, L.G.; Hobbs, B.P.; Bredow, S. Impairment of retrograde neuronal transport in oxaliplatin-induced neuropathy demonstrated by molecular imaging. PLoS ONE 2012, 7, e45776. [Google Scholar] [CrossRef]
- Calls, A.; Carozzi, V.; Navarro, X.; Monza, L.; Bruna, J. Pathogenesis of platinum-induced peripheral neurotoxicity: Insights from preclinical studies. Exp. Neurol. 2019, 325, 113141. [Google Scholar] [CrossRef]
- Canta, A.; Pozzi, E.; Carozzi, V.A. Mitochondrial Dysfunction in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Toxics 2015, 3, 198–223. [Google Scholar] [CrossRef]
- Alberti, P.; Semperboni, S.; Cavaletti, G.; Scuteri, A. Neurons: The Interplay between Cytoskeleton, Ion Channels/Transporters and Mitochondria. Cells 2022, 11, 2499. [Google Scholar] [CrossRef]
- Adelsberger, H.; Quasthoff, S.; Grosskreutz, J.; Lepier, A.; Eckel, F.; Lersch, C. The chemotherapeutic oxaliplatin alters voltage-gated Na(+) channel kinetics on rat sensory neurons. Eur. J. Pharmacol. 2000, 406, 25–32. [Google Scholar] [CrossRef]
- Webster, R.G.; Brain, K.L.; Wilson, R.H.; Grem, J.L.; Vincent, A. Oxaliplatin induces hyperexcitability at motor and autonomic neuromuscular junctions through effects on voltage-gated sodium channels. Br. J. Pharmacol. 2005, 146, 1027–1039. [Google Scholar] [CrossRef]
- Sittl, R.; Lampert, A.; Huth, T.; Schuy, E.T.; Link, A.S.; Fleckenstein, J.; Alzheimer, C.; Grafe, P.; Carr, R.W. Anticancer drug oxaliplatin induces acute cooling-aggravated neuropathy via sodium channel subtype Na(V)1.6-resurgent and persistent current. Proc. Natl. Acad. Sci. USA 2012, 109, 6704–6709. [Google Scholar] [CrossRef]
- Deuis, J.R.; Zimmermann, K.; Romanovsky, A.A.; Possani, L.D.; Cabot, P.J.; Lewis, R.J.; Vetter, I. An animal model of oxaliplatin-induced cold allodynia reveals a crucial role for Nav1.6 in peripheral pain pathways. Pain 2013, 154, 1749–1757. [Google Scholar] [CrossRef]
- Wu, S.N.; Chen, B.S.; Wu, Y.H.; Peng, H.; Chen, L.T. The mechanism of the actions of oxaliplatin on ion currents and action potentials in differentiated NG108-15 neuronal cells. Neurotoxicology 2009, 30, 677–685. [Google Scholar] [CrossRef]
- Benoit, E.; Brienza, S.; Dubois, J.M. Oxaliplatin, an anticancer agent that affects both Na+ and K+ channels in frog peripheral myelinated axons. Gen. Physiol. Biophys. 2006, 25, 263–276. [Google Scholar]
- Kagiava, A.; Tsingotjidou, A.; Emmanouilides, C.; Theophilidis, G. The effects of oxaliplatin, an anticancer drug, on potassium channels of the peripheral myelinated nerve fibres of the adult rat. Neurotoxicology 2008, 29, 1100–1106. [Google Scholar] [CrossRef]
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Cavaletti, G.; Antonacopoulou, A.; Genazzani, A.A.; Briani, C.; Bruna, J.; Terrazzino, S.; Velasco, R.; Alberti, P.; Campagnolo, M.; et al. Voltage-gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin-induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer 2013, 119, 3570–3577. [Google Scholar] [CrossRef]
- Ballarini, E.; Malacrida, A.; Rodriguez-Menendez, V.; Pozzi, E.; Canta, A.; Chiorazzi, A.; Monza, L.; Semperboni, S.; Meregalli, C.; Carozzi, V.A.; et al. Sodium-Calcium Exchanger 2: A Pivotal Role in Oxaliplatin Induced Peripheral Neurotoxicity and Axonal Damage? Int. J. Mol. Sci. 2022, 23, 63. [Google Scholar] [CrossRef]
- Alberti, P.; Canta, A.; Chiorazzi, A.; Fumagalli, G.; Meregalli, C.; Monza, L.; Pozzi, E.; Ballarini, E.; Rodriguez-Menendez, V.; Oggioni, N.; et al. Topiramate prevents oxaliplatin-related axonal hyperexcitability and oxaliplatin induced peripheral neurotoxicity. Neuropharmacology 2020, 164, 107905. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD. Neuron 2017, 93, 1334–1343.e5. [Google Scholar] [CrossRef]
- Gilley, J.; Orsomando, G.; Nascimento-Ferreira, I.; Coleman, M.P. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015, 10, 1974–1981. [Google Scholar] [CrossRef]
- Li, Y.; Pazyra-Murphy, M.F.; Avizonis, D.; de Sá Tavares Russo, M.; Tang, S.; Chen, C.Y.; Hsueh, Y.P.; Bergholz, J.S.; Jiang, T.; Zhao, J.J.; et al. Sarm1 activation produces cADPR to increase intra-axonal Ca++ and promote axon degeneration in PIPN. J. Cell Biol. 2022, 221, e202106080. [Google Scholar] [CrossRef]
- Geisler, S.; Doan, R.A.; Strickland, A.; Huang, X.; Milbrandt, J.; DiAntonio, A. Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 2016, 139, 3092–3108. [Google Scholar] [CrossRef]
- Gould, S.A.; White, M.; Wilbrey, A.L.; Pór, E.; Coleman, M.P.; Adalbert, R. Protection against oxaliplatin-induced mechanical and thermal hypersensitivity in Sarm1. Exp. Neurol. 2021, 338, 113607. [Google Scholar] [CrossRef]
- Fumagalli, G.; Monza, L.; Cavaletti, G.; Rigolio, R.; Meregalli, C. Neuroinflammatory Process Involved in Different Preclinical Models of Chemotherapy-Induced Peripheral Neuropathy. Front. Immunol. 2020, 11, 626687. [Google Scholar] [CrossRef]
- Meregalli, C.; Monza, L.; Chiorazzi, A.; Scali, C.; Guarnieri, C.; Fumagalli, G.; Alberti, P.; Pozzi, E.; Canta, A.; Ballarini, E.; et al. Human Intravenous Immunoglobulin Alleviates Neuropathic Symptoms in a Rat Model of Paclitaxel-Induced Peripheral Neurotoxicity. Int. J. Mol. Sci. 2021, 22, 1058. [Google Scholar] [CrossRef]
- Meregalli, C.; Marjanovic, I.; Scali, C.; Monza, L.; Spinoni, N.; Galliani, C.; Brivio, R.; Chiorazzi, A.; Ballarini, E.; Rodriguez-Menendez, V.; et al. High-dose intravenous immunoglobulins reduce nerve macrophage infiltration and the severity of bortezomib-induced peripheral neurotoxicity in rats. J. Neuroinflamm. 2018, 15, 232. [Google Scholar] [CrossRef]
- Calls, A.; Torres-Espin, A.; Tormo, M.; Martínez-Escardó, L.; Bonet, N.; Casals, F.; Navarro, X.; Yuste, V.J.; Udina, E.; Bruna, J. A transient inflammatory response contributes to oxaliplatin neurotoxicity in mice. Ann. Clin. Transl. Neurol. 2022. [Google Scholar] [CrossRef]
- Cavaletti, G.; Cornblath, D.R.; Merkies, I.S.J.; Postma, T.J.; Rossi, E.; Alberti, P.; Bruna, J.; Argyriou, A.A.; Briani, C.; Velasco, R.; et al. Patients’ and physicians’ interpretation of chemotherapy-induced peripheral neurotoxicity. J. Peripher. Nerv. Syst. 2019, 24, 111–119. [Google Scholar] [CrossRef]
- Cavaletti, G.; Cornblath, D.R.; Merkies, I.S.; Postma, T.J.; Rossi, E.; Frigeni, B.; Alberti, P.; Bruna, J.; Velasco, R.; Argyriou, A.A.; et al. The chemotherapy-induced peripheral neuropathy outcome measures standardization study: From consensus to the first validity and reliability findings. Ann. Oncol. 2013, 24, 454–462. [Google Scholar] [CrossRef]
- Cavaletti, G.; Frigeni, B.; Lanzani, F.; Mattavelli, L.; Susani, E.; Alberti, P.; Cortinovis, D.; Bidoli, P. Chemotherapy-Induced Peripheral Neurotoxicity assessment: A critical revision of the currently available tools. Eur. J. Cancer 2010, 46, 479–494. [Google Scholar] [CrossRef]
- Alberti, P.; Bernasconi, D.P.; Cornblath, D.R.; Merkies, I.S.J.; Park, S.B.; Velasco, R.; Bruna, J.; Psimaras, D.; Koeppen, S.; Pace, A.; et al. Prospective Evaluation of Health Care Provider and Patient Assessments in Chemotherapy-Induced Peripheral Neurotoxicity. Neurology 2021, 97, e660–e672. [Google Scholar] [CrossRef]
- Alberti, P. Role of neurophysiology in Chemotherapy-Induced Peripheral Neuropathy (CIPN). Clin. Neurophysiol. 2020, 131, 1964–1965. [Google Scholar] [CrossRef]
- Alberti, P.; Rossi, E.; Argyriou, A.A.; Kalofonos, H.P.; Briani, C.; Cacciavillani, M.; Campagnolo, M.; Bruna, J.; Velasco, R.; Cazzaniga, M.E.; et al. Risk stratification of oxaliplatin induced peripheral neurotoxicity applying electrophysiological testing of dorsal sural nerve. Support. Care Cancer 2018, 26, 3143–3151. [Google Scholar] [CrossRef]
- Alberti, P. Chemotherapy-induced peripheral neurotoxicity—Outcome measures: The issue. Expert Opin. Drug Metab. Toxicol. 2017, 13, 241–243. [Google Scholar] [CrossRef][Green Version]
- Alberti, P. A review of novel biomarkers and imaging techniques for assessing the severity of chemotherapy-induced peripheral neuropathy. Expert Opin. Drug Metab. Toxicol. 2020, 16, 1147–1158. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Park, S.B.; Islam, B.; Tamburin, S.; Velasco, R.; Alberti, P.; Bruna, J.; Psimaras, D.; Cavaletti, G.; Cornblath, D.R.; et al. Neurophysiological, nerve imaging and other techniques to assess chemotherapy-induced peripheral neurotoxicity in the clinical and research settings. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1361–1369. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Rohrer, J.D.; Woollacott, I.O.; Dick, K.M.; Brotherhood, E.; Gordon, E.; Fellows, A.; Toombs, J.; Druyeh, R.; Cardoso, M.J.; Ourselin, S.; et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016, 87, 1329–1336. [Google Scholar] [CrossRef]
- Mariotto, S.; Farinazzo, A.; Magliozzi, R.; Alberti, D.; Monaco, S.; Ferrari, S. Serum and cerebrospinal neurofilament light chain levels in patients with acquired peripheral neuropathies. J. Peripher. Nerv. Syst. 2018, 23, 174–177. [Google Scholar] [CrossRef]
- Sandelius, Å.; Zetterberg, H.; Blennow, K.; Adiutori, R.; Malaspina, A.; Laura, M.; Reilly, M.M.; Rossor, A.M. Plasma neurofilament light chain concentration in the inherited peripheral neuropathies. Neurology 2018, 90, e518–e524. [Google Scholar] [CrossRef]
- Meregalli, C.; Fumagalli, G.; Alberti, P.; Canta, A.; Carozzi, V.A.; Chiorazzi, A.; Monza, L.; Pozzi, E.; Sandelius, Å.; Blennow, K.; et al. Neurofilament light chain as disease biomarker in a rodent model of chemotherapy induced peripheral neuropathy. Exp. Neurol. 2018, 307, 129–132. [Google Scholar] [CrossRef]
- Kim, S.H.; Choi, M.K.; Park, N.Y.; Hyun, J.W.; Lee, M.Y.; Kim, H.J.; Jung, S.K.; Cha, Y. Serum neurofilament light chain levels as a biomarker of neuroaxonal injury and severity of oxaliplatin-induced peripheral neuropathy. Sci. Rep. 2020, 10, 7995. [Google Scholar] [CrossRef]
- Meregalli, C.; Fumagalli, G.; Alberti, P.; Canta, A.; Chiorazzi, A.; Monza, L.; Pozzi, E.; Carozzi, V.A.; Blennow, K.; Zetterberg, H.; et al. Neurofilament light chain: A specific serum biomarker of axonal damage severity in rat models of Chemotherapy-Induced Peripheral Neurotoxicity. Arch. Toxicol. 2020, 94, 2517–2522. [Google Scholar] [CrossRef]
- Bendstrup, N.; Hejl, A.M.; Salvesen, L. Neurofilament Light Chain Levels in Frontotemporal Dementia and Progressive Supranuclear Palsy: A Systematic Review. J. Alzheimers Dis. 2022, 87, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Wendel, E.M.; Bertolini, A.; Kousoulos, L.; Rauchenzauner, M.; Schanda, K.; Wegener-Panzer, A.; Baumann, M.; Reindl, M.; Otto, M.; Rostásy, K. Serum neurofilament light-chain levels in children with monophasic myelin oligodendrocyte glycoprotein-associated disease, multiple sclerosis, and other acquired demyelinating syndrome. Mult. Scler. 2022, 10, 13524585221081090. [Google Scholar] [CrossRef] [PubMed]
- Van den Bosch, A.; Fransen, N.; Mason, M.; Rozemuller, A.J.; Teunissen, C.; Smolders, J.; Huitinga, I. Neurofilament Light Chain Levels in Multiple Sclerosis Correlate with Lesions Containing Foamy Macrophages and with Acute Axonal Damage. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1154. [Google Scholar] [CrossRef] [PubMed]
- Karteri, S.; Bruna, J.; Argyriou, A.A.; Mariotto, S.; Velasco, R.; Alemany, M.; Kalofonou, F.; Alberti, P.; Dinoto, A.; Velissaris, D.; et al. Prospectively Assessing Serum Neurofilament Light Chain Levels as A Biomarker of Paclitaxel-Induced Peripheral Neurotoxicity in Breast Cancer Patients. J. Peripher. Nerv. Syst. 2022, 27, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Huehnchen, P.; Schinke, C.; Bangemann, N.; Dordevic, A.D.; Kern, J.; Maierhof, S.K.; Hew, L.; Nolte, L.; Körtvelyessy, P.; Göpfert, J.C.; et al. Neurofilament proteins as a potential biomarker in chemotherapy-induced polyneuropathy. JCI Insight 2022, 7, e154395. [Google Scholar] [CrossRef]
- Loprinzi, C.L.; Lacchetti, C.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Hertz, D.L.; Kelley, M.R.; Lavino, A.; Lustberg, M.B.; Paice, J.A.; et al. Prevention and Management of Chemotherapy-Induced Peripheral Neuropathy in Survivors of Adult Cancers: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, JCO2001399. [Google Scholar] [CrossRef]
- Mariotto, S.; Tecchio, C.; Sorio, M.; Bertolasi, L.; Turatti, M.; Tozzi, M.C.; Benedetti, F.; Cavaletti, G.; Monaco, S.; Ferrari, S. Clinical and neurophysiological serial assessments of brentuximab vedotin-associated peripheral neuropathy. Leuk. Lymphoma 2019, 60, 2806–2809. [Google Scholar] [CrossRef]
- Mariotto, S.; Ferrari, S.; Sorio, M.; Benedetti, F.; Tridente, G.; Cavallaro, T.; Gajofatto, A.; Monaco, S. Brentuximab vedotin: Axonal microtubule’s Apollyon. Blood Cancer J. 2015, 5, e343. [Google Scholar] [CrossRef]
- Mariotto, S.; Ferrari, S.; Monaco, S. Brentuximab vedotin-induced peripheral neuropathy: Looking at microtubules. J. Neurooncol. 2018, 137, 665–666. [Google Scholar] [CrossRef]
- Chen, R.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Connors, J.M.; Engert, A.; Larsen, E.K.; Huebner, D.; et al. Five-year survival and durability results of brentuximab vedotin in patients with relapsed or refractory Hodgkin lymphoma. Blood 2016, 128, 1562–1566. [Google Scholar] [CrossRef]
- Straus, D.J.; Długosz-Danecka, M.; Connors, J.M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; et al. Brentuximab vedotin with chemotherapy for stage III or IV classical Hodgkin lymphoma (ECHELON-1): 5-year update of an international, open-label, randomised, phase 3 trial. Lancet Haematol. 2021, 8, e410–e421. [Google Scholar] [CrossRef] [PubMed]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Visentin, A.; Salvalaggio, A.; Imbergamo, S.; Piazza, F.; Cacciavillani, M.; Campagnolo, M.; Frezzato, F.; Semenzato, G.; Trentin, L. Peripheral neuropathies in chronic lymphocytic leukemia: A single center experience on 816 patients. Haematologica 2017, 102, e140–e143. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Visentin, A.; Cavallaro, T.; Cacciavillani, M.; Cabrini, I.; Ferrari, S.; Zambello, R.; Trentin, L. Primary neurolymphomatosis as clinical onset of chronic lymphocytic leukemia. Ann. Hematol. 2017, 96, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.M.; Nichelatti, M.; Deodato, M.; Zamprogna, G.; Minga, P.; Pioltelli, M.L.; Cairoli, R.; Tedeschi, A. Health-related quality of life in Waldenstrom Macroglobulinemia and IgM-related disorders: A single institution experience. Hematol. Oncol. 2020, 38, 111–113. [Google Scholar] [CrossRef]
- Treon, S.P.; Meid, K.; Gustine, J.; Yang, G.; Xu, L.; Liu, X.; Patterson, C.J.; Hunter, Z.R.; Branagan, A.R.; Laubach, J.P.; et al. Long-Term Follow-Up of Ibrutinib Monotherapy in Symptomatic, Previously Treated Patients with Waldenström Macroglobulinemia. J. Clin. Oncol. 2021, 39, 565–575. [Google Scholar] [CrossRef]
- Castellani, F.; Visentin, A.; Campagnolo, M.; Salvalaggio, A.; Cacciavillani, M.; Candiotto, C.; Bertorelle, R.; Trentin, L.; Briani, C. The Bruton tyrosine kinase inhibitor ibrutinib improves anti-MAG antibody polyneuropathy. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e720. [Google Scholar] [CrossRef]
- Furman, R.R.; Byrd, J.C.; Owen, R.G.; O’Brien, S.M.; Brown, J.R.; Hillmen, P.; Stephens, D.M.; Chernyukhin, N.; Lezhava, T.; Hamdy, A.M.; et al. Pooled analysis of safety data from clinical trials evaluating acalabrutinib monotherapy in mature B-cell malignancies. Leukemia 2021, 35, 3201–3211. [Google Scholar] [CrossRef]
- Tam, C.S.; Dimopoulos, M.; Garcia-Sanz, R.; Trotman, J.; Opat, S.; Roberts, A.W.; Owen, R.; Song, Y.; Xu, W.; Zhu, J.; et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2022, 6, 1296–1308. [Google Scholar] [CrossRef]
- Visentin, A.; Frezzato, F.; Severin, F.; Imbergamo, S.; Pravato, S.; Romano Gargarella, L.; Manni, S.; Pizzo, S.; Ruggieri, E.; Facco, M.; et al. Lights and Shade of Next-Generation Pi3k Inhibitors in Chronic Lymphocytic Leukemia. OncoTargets Ther 2020, 13, 9679–9688. [Google Scholar] [CrossRef]
- Davids, M.S.; Hallek, M.; Wierda, W.; Roberts, A.W.; Stilgenbauer, S.; Jones, J.A.; Gerecitano, J.F.; Kim, S.Y.; Potluri, J.; Busman, T.; et al. Comprehensive Safety Analysis of Venetoclax Monotherapy for Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2018, 24, 4371–4379. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Visentin, A.; Castellani, F.; Cacciavillani, M.; Trentin, L. The BCL2 Inhibitor Venetoclax Plus Rituximab Is Active in MYD88 Wild-Type Polyneuropathy With Anti-MAG Antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2022, 9, e1181. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. Immune effector cell associated neurotoxicity syndrome in chimeric antigen receptor-T cell therapy. Front. Immunol. 2022, 13, 879608. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chmielowski, B.; Lao, C.D.; Hodi, F.S.; Sharfman, W.; Weber, J.; Suijkerbuijk, K.P.M.; Azevedo, S.; Li, H.; Reshef, D.; et al. Neurologic Serious Adverse Events Associated with Nivolumab Plus Ipilimumab or Nivolumab Alone in Advanced Melanoma, Including a Case Series of Encephalitis. Oncologist 2017, 22, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Velasco, R.; Villagrán, M.; Jové, M.; Simó, M.; Vilariño, N.; Alemany, M.; Palmero, R.; Martínez-Villacampa, M.M.; Nadal, E.; Bruna, J. Encephalitis Induced by Immune Checkpoint Inhibitors: A Systematic Review. JAMA Neurol. 2021, 78, 864–873. [Google Scholar] [CrossRef]
- Yshii, L.M.; Hohlfeld, R.; Liblau, R.S. Inflammatory CNS disease caused by immune checkpoint inhibitors: Status and perspectives. Nat. Rev. Neurol. 2017, 13, 755–763. [Google Scholar] [CrossRef]
- Daxini, A.; Cronin, K.; Sreih, A.G. Vasculitis associated with immune checkpoint inhibitors-a systematic review. Clin. Rheumatol. 2018, 37, 2579–2584. [Google Scholar] [CrossRef]
- Agarwal, A.; Rübsam, A.; Zur Bonsen, L.; Pichi, F.; Neri, P.; Pleyer, U. A Comprehensive Update on Retinal Vasculitis: Etiologies, Manifestations and Treatments. J. Clin. Med. 2022, 11, 2525. [Google Scholar] [CrossRef]
- Narumi, Y.; Yoshida, R.; Minami, Y.; Yamamoto, Y.; Takeguchi, S.; Kano, K.; Takahashi, K.; Saito, T.; Sawada, J.; Terui, H.; et al. Neuromyelitis optica spectrum disorder secondary to treatment with anti-PD-1 antibody nivolumab: The first report. BMC Cancer 2018, 18, 95. [Google Scholar] [CrossRef]
- Gerdes, L.A.; Held, K.; Beltrán, E.; Berking, C.; Prinz, J.C.; Junker, A.; Tietze, J.K.; Ertl-Wagner, B.; Straube, A.; Kümpfel, T.; et al. CTLA4 as Immunological Checkpoint in the Development of Multiple Sclerosis. Ann. Neurol. 2016, 80, 294–300. [Google Scholar] [CrossRef]
- Yeh, O.L.; Francis, C.E. Ipilimumab-associated bilateral optic neuropathy. J. Neuroophthalmol. 2015, 35, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.A.; Guld, K.; Galetta, S.; Walsh, R.D.; Kharlip, J.; Tamhankar, M.; McGettigan, S.; Schuchter, L.M.; Fecher, L.A. Acute visual loss after ipilimumab treatment for metastatic melanoma. J. Immunother. Cancer 2016, 4, 66. [Google Scholar] [CrossRef]
- Makri, O.E.; Dimitrakopoulos, F.I.; Tsapardoni, F.; Tsekouras, I.; Argyriou, A.A.; Kalofonos, H.; Georgakopoulos, C.D. Isolated optic neuritis after pembrolizumab administration for non-small-cell lung carcinoma. Int. J. Neurosci. 2022, 132, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Kartal, Ö.; Ataş, E. Bilateral Optic Neuritis Secondary to Nivolumab Therapy: A Case Report. Medicina 2018, 54, 82. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef]
- Mori, S.; Kurimoto, T.; Ueda, K.; Enomoto, H.; Sakamoto, M.; Keshi, Y.; Yamada, Y.; Nakamura, M. Optic Neuritis Possibly Induced by Anti-PD-L1 Antibody Treatment in a Patient with Non-Small Cell Lung Carcinoma. Case Rep. Ophthalmol. 2018, 9, 348–356. [Google Scholar] [CrossRef]
- Noble, C.W.; Gangaputra, S.S.; Thompson, I.A.; Yuan, A.; Apolo, A.B.; Lee, J.M.; Papaliodis, G.N.; Kodati, S.; Bishop, R.; Magone, M.T.; et al. Ocular Adverse Events following Use of Immune Checkpoint Inhibitors for Metastatic Malignancies. Ocul. Immunol. Inflamm. 2020, 28, 854–859. [Google Scholar] [CrossRef]
- Jaben, K.A.; Francis, J.H.; Shoushtari, A.N.; Abramson, D.H. Isolated Abducens Nerve Palsy Following Pembrolizumab. Neuroophthalmology 2020, 44, 182–185. [Google Scholar] [CrossRef]
- Jono, M.; Kinehara, Y.; Utsu, Y.; Tamura, Y.; Koseto, M.; Murakami, T.; Uota, A.; Ninomiya, R.; Komo, S.; Sumitani, S.; et al. Neuropsychiatric Immune-related Adverse Events Induced by Pembrolizumab in a Patient with Lung Adenocarcinoma and Systemic Lupus Erythematosus. Intern. Med. 2020, 59, 569–572. [Google Scholar] [CrossRef]
- Shah, M.; Tayar, J.H.; Abdel-Wahab, N.; Suarez-Almazor, M.E. Myositis as an adverse event of immune checkpoint blockade for cancer therapy. Semin. Arthritis Rheum. 2019, 48, 736–740. [Google Scholar] [CrossRef]
- Psimaras, D.; Velasco, R.; Birzu, C.; Tamburin, S.; Lustberg, M.; Bruna, J.; Argyriou, A.A. Immune checkpoint inhibitors-induced neuromuscular toxicity: From pathogenesis to treatment. J. Peripher. Nerv. Syst. 2019, 24 (Suppl. S2), S74–S85. [Google Scholar] [CrossRef] [PubMed]
- Aldrich, J.; Pundole, X.; Tummala, S.; Palaskas, N.; Andersen, C.R.; Shoukier, M.; Abdel-Wahab, N.; Deswal, A.; Suarez-Almazor, M.E. Inflammatory Myositis in Cancer Patients Receiving Immune Checkpoint Inhibitors. Arthritis Rheumatol. 2021, 73, 866–874. [Google Scholar] [CrossRef]
- Liewluck, T.; Kao, J.C.; Mauermann, M.L. PD-1 Inhibitor-associated Myopathies: Emerging Immune-mediated Myopathies. J. Immunother. 2018, 41, 208–211. [Google Scholar] [CrossRef]
- Bruna, J.; Argyriou, A.A.; Anastopoulou, G.G.; Alemany, M.; Nadal, E.; Kalofonou, F.; Piulats, J.M.; Simó, M.; Velasco, R.; Kalofonos, H.P. Incidence and characteristics of neurotoxicity in immune checkpoint inhibitors with focus on neuromuscular events: Experience beyond the clinical trials. J. Peripher. Nerv. Syst. 2020, 25, 171–177. [Google Scholar] [CrossRef]
- Touat, M.; Maisonobe, T.; Knauss, S.; Ben Hadj Salem, O.; Hervier, B.; Auré, K.; Szwebel, T.A.; Kramkimel, N.; Lethrosne, C.; Bruch, J.F.; et al. Immune checkpoint inhibitor-related myositis and myocarditis in patients with cancer. Neurology 2018, 91, e985–e994. [Google Scholar] [CrossRef]
- Kao, J.C.; Brickshawana, A.; Liewluck, T. Neuromuscular Complications of Programmed Cell Death-1 (PD-1) Inhibitors. Curr. Neurol. NeuroSci. Rep. 2018, 18, 63. [Google Scholar] [CrossRef]
- Makarious, D.; Horwood, K.; Coward, J.I.G. Myasthenia gravis: An emerging toxicity of immune checkpoint inhibitors. Eur. J. Cancer 2017, 82, 128–136. [Google Scholar] [CrossRef]
- Man, J.; Ritchie, G.; Links, M.; Lord, S.; Lee, C.K. Treatment-related toxicities of immune checkpoint inhibitors in advanced cancers: A meta-analysis. Asia Pac. J. Clin. Oncol. 2018, 14, 141–152. [Google Scholar] [CrossRef]
- Touat, M.; Talmasov, D.; Ricard, D.; Psimaras, D. Neurological toxicities associated with immune-checkpoint inhibitors. Curr. Opin. Neurol. 2017, 30, 659–668. [Google Scholar] [CrossRef]
- Holtzman, N.G.; Xie, H.; Bentzen, S.; Kesari, V.; Bukhari, A.; El Chaer, F.; Lutfi, F.; Siglin, J.; Hutnick, E.; Gahres, N.; et al. Immune effector cell-associated neurotoxicity syndrome after chimeric antigen receptor T-cell therapy for lymphoma: Predictive biomarkers and clinical outcomes. Neuro-Oncology 2021, 23, 112–121. [Google Scholar] [CrossRef]
- Karschnia, P.; Jordan, J.T.; Forst, D.A.; Arrillaga-Romany, I.C.; Batchelor, T.T.; Baehring, J.M.; Clement, N.F.; Gonzalez Castro, L.N.; Herlopian, A.; Maus, M.V.; et al. Clinical presentation, management, and biomarkers of neurotoxicity after adoptive immunotherapy with CAR T cells. Blood 2019, 133, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Guha-Thakurta, N.; Wierda, W.G. Cerebral edema secondary to chimeric antigen receptor T-cell immunotherapy. Neurology 2018, 91, 843. [Google Scholar] [CrossRef]
- Rubin, D.B.; Al Jarrah, A.; Li, K.; LaRose, S.; Monk, A.D.; Ali, A.B.; Spendley, L.N.; Nikiforow, S.; Jacobson, C.; Vaitkevicius, H. Clinical Predictors of Neurotoxicity after Chimeric Antigen Receptor T-Cell Therapy. JAMA Neurol. 2020, 77, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.; Nagle, S.; Randall, J.; Hinson, H.E. Chimeric Antigen Receptor T Cell-Related Neurotoxicity: Mechanisms, Clinical Presentation, and Approach to Treatment. Curr. Treat. Options Neurol. 2019, 21, 40. [Google Scholar] [CrossRef]
- Strati, P.; Nastoupil, L.J.; Westin, J.; Fayad, L.E.; Ahmed, S.; Fowler, N.H.; Hagemeister, F.B.; Lee, H.J.; Iyer, S.P.; Nair, R.; et al. Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B-cell lymphoma. Blood Adv. 2020, 4, 3943–3951. [Google Scholar] [CrossRef]
- Vilariño, N.; Bruna, J.; Kalofonou, F.; Anastopoulou, G.G.; Argyriou, A.A. Immune-Driven Pathogenesis of Neurotoxicity after Exposure of Cancer Patients to Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 5774. [Google Scholar] [CrossRef]
- Selby, M.J.; Engelhardt, J.J.; Quigley, M.; Henning, K.A.; Chen, T.; Srinivasan, M.; Korman, A.J. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 2013, 1, 32–42. [Google Scholar] [CrossRef]
- Lim, S.Y.; Lee, J.H.; Gide, T.N.; Menzies, A.M.; Guminski, A.; Carlino, M.S.; Breen, E.J.; Yang, J.Y.H.; Ghazanfar, S.; Kefford, R.F.; et al. Circulating Cytokines Predict Immune-Related Toxicity in Melanoma Patients Receiving Anti-PD-1-Based Immunotherapy. Clin. Cancer Res. 2019, 25, 1557–1563. [Google Scholar] [CrossRef]
- De Moel, E.C.; Rozeman, E.A.; Kapiteijn, E.H.; Verdegaal, E.M.E.; Grummels, A.; Bakker, J.A.; Huizinga, T.W.J.; Haanen, J.B.; Toes, R.E.M.; van der Woude, D. Autoantibody Development under Treatment with Immune-Checkpoint Inhibitors. Cancer Immunol. Res. 2019, 7, 6–11. [Google Scholar] [CrossRef]
- Kohm, A.P.; Carpentier, P.A.; Anger, H.A.; Miller, S.D. Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J. Immunol. 2002, 169, 4712–4716. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Dalmau, J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2019, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Vogrig, A.; Muñiz-Castrillo, S.; Antoine, J.G.; Desestret, V.; Dubey, D.; Giometto, B.; Irani, S.R.; Joubert, B.; Leypoldt, F.; et al. Updated Diagnostic Criteria for Paraneoplastic Neurologic Syndromes. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1014. [Google Scholar] [CrossRef] [PubMed]
- Kwek, S.S.; Dao, V.; Roy, R.; Hou, Y.; Alajajian, D.; Simko, J.P.; Small, E.J.; Fong, L. Diversity of antigen-specific responses induced in vivo with CTLA-4 blockade in prostate cancer patients. J. Immunol. 2012, 189, 3759–3766. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Deng, Q.; Han, G.; Puebla-Osorio, N.; Ma, M.C.J.; Strati, P.; Chasen, B.; Dai, E.; Dang, M.; Jain, N.; Yang, H.; et al. Characteristics of anti-CD19 CAR T cell infusion products associated with efficacy and toxicity in patients with large B cell lymphomas. Nat. Med. 2020, 26, 1878–1887. [Google Scholar] [CrossRef]
- Caruso, H.G.; Hurton, L.V.; Najjar, A.; Rushworth, D.; Ang, S.; Olivares, S.; Mi, T.; Switzer, K.; Singh, H.; Huls, H.; et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015, 75, 3505–3518. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hill, T.; Shamah, S.M.; Salter, A.I.; Chen, Y.; Mohler, K.M.; Riddell, S.R. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia 2017, 31, 2191–2199. [Google Scholar] [CrossRef]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Salter, A.I.; Ivey, R.G.; Kennedy, J.J.; Voillet, V.; Rajan, A.; Alderman, E.J.; Voytovich, U.J.; Lin, C.; Sommermeyer, D.; Liu, L.; et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal. 2018, 11, eaat6753. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 3978–3992. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.J.; Naidoo, J.; Santomasso, B.D.; Lacchetti, C.; Adkins, S.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: ASCO Guideline Update. J. Clin. Oncol. 2021, 39, 4073–4126. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Beattie, J.A.; Fuentes, P.; Rizvi, H.; Egger, J.V.; Kern, J.A.; Leung, D.Y.M.; Lacouture, M.E.; Kris, M.G.; Gambarin, M.; et al. Beyond Steroids: Immunosuppressants in Steroid-Refractory or Resistant Immune-Related Adverse Events. J. Thorac. Oncol. 2021, 16, 1759–1764. [Google Scholar] [CrossRef]
- Ferreros, P.; Trapero, I. Interleukin Inhibitors in Cytokine Release Syndrome and Neurotoxicity Secondary to CAR-T Therapy. Diseases 2022, 10, 41. [Google Scholar] [CrossRef]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights Into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Gardner, R.A.; Ceppi, F.; Rivers, J.; Annesley, C.; Summers, C.; Taraseviciute, A.; Gust, J.; Leger, K.J.; Tarlock, K.; Cooper, T.M.; et al. Preemptive mitigation of CD19 CAR T-cell cytokine release syndrome without attenuation of antileukemic efficacy. Blood 2019, 134, 2149–2158. [Google Scholar] [CrossRef]
- Whittaker, A.L.; George, R.P.; O’Malley, L. Prevalence of cognitive impairment following chemotherapy treatment for breast cancer: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 2135. [Google Scholar] [CrossRef]
- Cruzado, J.A.; López-Santiago, S.; Martínez-Marín, V.; José-Moreno, G.; Custodio, A.B.; Feliu, J. Longitudinal study of cognitive dysfunctions induced by adjuvant chemotherapy in colon cancer patients. Support. Care Cancer 2014, 22, 1815–1823. [Google Scholar] [CrossRef]
- Vardy, J.L.; Dhillon, H.M.; Pond, G.R.; Rourke, S.B.; Bekele, T.; Renton, C.; Dodd, A.; Zhang, H.; Beale, P.; Clarke, S.; et al. Cognitive Function in Patients with Colorectal Cancer Who Do and Do Not Receive Chemotherapy: A Prospective, Longitudinal, Controlled Study. J. Clin. Oncol. 2015, 33, 4085–4092. [Google Scholar] [CrossRef]
- Hwang, S.Y.; Kim, K.; Ha, B.; Lee, D.; Kim, S.; Ryu, S.; Yang, J.; Jung, S.J. Neurocognitive Effects of Chemotherapy for Colorectal Cancer: A Systematic Review and a Meta-Analysis of 11 Studies. Cancer Res. Treat. 2021, 53, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Duijts, S.F.; van der Beek, A.J.; Boelhouwer, I.G.; Schagen, S.B. Cancer-related cognitive impairment and patients’ ability to work: A current perspective. Curr. Opin. Supportive Palliat. Care 2017, 11, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Bolton, G.; Isaacs, A. Women’s experiences of cancer-related cognitive impairment, its impact on daily life and care received for it following treatment for breast cancer. Psychol. Health Med. 2018, 23, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Vardy, J. Cognitive function in breast cancer survivors. Cancer Treat. Res. 2009, 151, 387–419. [Google Scholar] [CrossRef] [PubMed]
- Selamat, M.H.; Loh, S.Y.; Mackenzie, L.; Vardy, J. Chemobrain experienced by breast cancer survivors: A meta-ethnography study investigating research and care implications. PLoS ONE 2014, 9, e108002. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, A.D.; Hosking, J.R.; Kichenadasse, G.; Mattiske, J.K.; Wilson, C. Objective and subjective cognitive impairment following chemotherapy for cancer: A systematic review. Cancer Treat. Rev. 2012, 38, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Cerulla Torrente, N.; Navarro Pastor, J.B.; de la Osa Chaparro, N. Systematic review of cognitive sequelae of non-central nervous system cancer and cancer therapy. J. Cancer Surviv. 2020, 14, 464–482. [Google Scholar] [CrossRef]
- Simó, M.; Root, J.C.; Vaquero, L.; Ripollés, P.; Jové, J.; Ahles, T.; Navarro, A.; Cardenal, F.; Bruna, J.; Rodríguez-Fornells, A. Cognitive and brain structural changes in a lung cancer population. J. Thorac. Oncol. 2015, 10, 38–45. [Google Scholar] [CrossRef]
- Vannorsdall, T.D. Cognitive Changes Related to Cancer Therapy. Med. Clin. N. Am. 2017, 101, 1115–1134. [Google Scholar] [CrossRef]
- Myers, J.S. Cancer- and chemotherapy-related cognitive changes: The patient experience. Semin. Oncol. Nurs. 2013, 29, 300–307. [Google Scholar] [CrossRef]
- Vega, J.N.; Dumas, J.; Newhouse, P.A. Cognitive Effects of Chemotherapy and Cancer-Related Treatments in Older Adults. Am. J. Geriatr. Psychiatry 2017, 25, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Ahles, T.A.; Root, J.C. Cognitive Effects of Cancer and Cancer Treatments. Annu. Rev. Clin. Psychol. 2018, 14, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Koppelmans, V.; Breteler, M.M.; Boogerd, W.; Seynaeve, C.; Gundy, C.; Schagen, S.B. Neuropsychological performance in survivors of breast cancer more than 20 years after adjuvant chemotherapy. J. Clin. Oncol. 2012, 30, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Wefel, J.S.; Saleeba, A.K.; Buzdar, A.U.; Meyers, C.A. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer 2010, 116, 3348–3356. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, E.Y.; Domenicano, I.; Nyhan, K.; Elfil, M.; Mougalian, S.S.; Cartmel, B.; Ehrlich, B.E. Cognitive Effects and Depression Associated With Taxane-Based Chemotherapy in Breast Cancer Survivors: A Meta-Analysis. Front. Oncol. 2021, 11, 642382. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.L.; Gong, X.; Di, K.; Bota, D.A. Low-doses of cisplatin injure hippocampal synapses: A mechanism for ‘chemo’ brain? Exp. Neurol. 2014, 255, 137–144. [Google Scholar] [CrossRef]
- Moruno-Manchon, J.F.; Uzor, N.E.; Kesler, S.R.; Wefel, J.S.; Townley, D.M.; Nagaraja, A.S.; Pradeep, S.; Mangala, L.S.; Sood, A.K.; Tsvetkov, A.S. TFEB ameliorates the impairment of the autophagy-lysosome pathway in neurons induced by doxorubicin. Aging (Albany NY) 2016, 8, 3507–3519. [Google Scholar] [CrossRef]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Cleeland, C.S.; Bennett, G.J.; Dantzer, R.; Dougherty, P.M.; Dunn, A.J.; Meyers, C.A.; Miller, A.H.; Payne, R.; Reuben, J.M.; Wang, X.S.; et al. Are the symptoms of cancer and cancer treatment due to a shared biologic mechanism? A cytokine-immunologic model of cancer symptoms. Cancer 2003, 97, 2919–2925. [Google Scholar] [CrossRef]
- Capuron, L.; Ravaud, A.; Dantzer, R. Timing and specificity of the cognitive changes induced by interleukin-2 and interferon-alpha treatments in cancer patients. Psychosom. Med. 2001, 63, 376–386. [Google Scholar] [CrossRef]
- Wang, X.M.; Walitt, B.; Saligan, L.; Tiwari, A.F.; Cheung, C.W.; Zhang, Z.J. Chemobrain: A critical review and causal hypothesis of link between cytokines and epigenetic reprogramming associated with chemotherapy. Cytokine 2015, 72, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Joly, F.; Vardy, J.; Ahles, T.; Dubois, M.; Tron, L.; Winocur, G.; De Ruiter, M.B.; Castel, H. Cancer-related cognitive impairment: An update on state of the art, detection, and management strategies in cancer survivors. Ann. Oncol. 2019, 30, 1925–1940. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.A.; Acharya, M.M.; Parihar, V.K.; Nguyen, A.; Martirosian, V.; Limoli, C.L. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin. Cancer Res. 2012, 18, 1954–1965. [Google Scholar] [CrossRef] [PubMed]
- Pomykala, K.L.; Ganz, P.A.; Bower, J.E.; Kwan, L.; Castellon, S.A.; Mallam, S.; Cheng, I.; Ahn, R.; Breen, E.C.; Irwin, M.R.; et al. The association between pro-inflammatory cytokines, regional cerebral metabolism, and cognitive complaints following adjuvant chemotherapy for breast cancer. Brain Imaging Behav. 2013, 7, 511–523. [Google Scholar] [CrossRef]
- Seigers, R.; Loos, M.; Van Tellingen, O.; Boogerd, W.; Smit, A.B.; Schagen, S.B. Neurobiological changes by cytotoxic agents in mice. Behav. Brain Res. 2016, 299, 19–26. [Google Scholar] [CrossRef]
- Lomeli, N.; Di, K.; Czerniawski, J.; Guzowski, J.F.; Bota, D.A. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic. Biol. Med. 2017, 102, 274–286. [Google Scholar] [CrossRef]
- McGinnis, G.J.; Friedman, D.; Young, K.H.; Torres, E.R.; Thomas, C.R.; Gough, M.J.; Raber, J. Neuroinflammatory and cognitive consequences of combined radiation and immunotherapy in a novel preclinical model. Oncotarget 2017, 8, 9155–9173. [Google Scholar] [CrossRef]
- Bianchi, M.; Heidbreder, C.; Crespi, F. Cytoskeletal changes in the hippocampus following restraint stress: Role of serotonin and microtubules. Synapse 2003, 49, 188–194. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Ehrlich, B.E. Cellular mechanisms and treatments for chemobrain: Insight from aging and neurodegenerative diseases. EMBO Mol. Med. 2020, 12, e12075. [Google Scholar] [CrossRef]
- You, Z.; Zhang, S.; Shen, S.; Yang, J.; Ding, W.; Yang, L.; Lim, G.; Doheny, J.T.; Tate, S.; Chen, L.; et al. Cognitive impairment in a rat model of neuropathic pain: Role of hippocampal microtubule stability. Pain 2018, 159, 1518–1528. [Google Scholar] [CrossRef]
- Deprez, S.; Amant, F.; Smeets, A.; Peeters, R.; Leemans, A.; Van Hecke, W.; Verhoeven, J.S.; Christiaens, M.R.; Vandenberghe, J.; Vandenbulcke, M.; et al. Longitudinal assessment of chemotherapy-induced structural changes in cerebral white matter and its correlation with impaired cognitive functioning. J. Clin. Oncol. 2012, 30, 274–281. [Google Scholar] [CrossRef]
- Koppelmans, V.; de Ruiter, M.B.; van der Lijn, F.; Boogerd, W.; Seynaeve, C.; van der Lugt, A.; Vrooman, H.; Niessen, W.J.; Breteler, M.M.; Schagen, S.B. Global and focal brain volume in long-term breast cancer survivors exposed to adjuvant chemotherapy. Breast Cancer Res. Treat. 2012, 132, 1099–1106. [Google Scholar] [CrossRef]
- Kesler, S.R.; Blayney, D.W. Neurotoxic Effects of Anthracycline- vs Nonanthracycline-Based Chemotherapy on Cognition in Breast Cancer Survivors. JAMA Oncol. 2016, 2, 185–192. [Google Scholar] [CrossRef]
- Nelson, C.J.; Nandy, N.; Roth, A.J. Chemotherapy and cognitive deficits: Mechanisms, findings, and potential interventions. Palliat. Support. Care 2007, 5, 273–280. [Google Scholar] [CrossRef]
- Almey, A.; Milner, T.A.; Brake, W.G. Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm. Behav. 2015, 74, 125–138. [Google Scholar] [CrossRef]
- Wagner, L.I.; Gray, R.J.; Sparano, J.A.; Whelan, T.J.; Garcia, S.F.; Yanez, B.; Tevaarwerk, A.J.; Carlos, R.C.; Albain, K.S.; Olson, J.A.; et al. Patient-Reported Cognitive Impairment Among Women With Early Breast Cancer Randomly Assigned to Endocrine Therapy Alone Versus Chemoendocrine Therapy: Results From TAILORx. J. Clin. Oncol. 2020, 38, 1875–1886. [Google Scholar] [CrossRef]
- Barton, D.; Loprinzi, C. Novel approaches to preventing chemotherapy-induced cognitive dysfunction in breast cancer: The art of the possible. Clin. Breast Cancer 2002, 3 (Suppl. S3), S121–S127. [Google Scholar] [CrossRef]
- Ahles, T.A.; Saykin, A.J.; Noll, W.W.; Furstenberg, C.T.; Guerin, S.; Cole, B.; Mott, L.A. The relationship of APOE genotype to neuropsychological performance in long-term cancer survivors treated with standard dose chemotherapy. Psychooncology 2003, 12, 612–619. [Google Scholar] [CrossRef]
- Stern, Y. Cognitive reserve in ageing and Alzheimer’s disease. Lancet Neurol. 2012, 11, 1006–1012. [Google Scholar] [CrossRef]
- Mandelblatt, J.S.; Stern, R.A.; Luta, G.; McGuckin, M.; Clapp, J.D.; Hurria, A.; Jacobsen, P.B.; Faul, L.A.; Isaacs, C.; Denduluri, N.; et al. Cognitive impairment in older patients with breast cancer before systemic therapy: Is there an interaction between cancer and comorbidity? J. Clin. Oncol. 2014, 32, 1909–1918. [Google Scholar] [CrossRef]
- Wefel, J.S.; Vardy, J.; Ahles, T.; Schagen, S.B. International Cognition and Cancer Task Force recommendations to harmonise studies of cognitive function in patients with cancer. Lancet Neurol. 2011, 12, 703–708. [Google Scholar] [CrossRef]
- Dhillon, H.M.; Tannock, I.F.; Pond, G.R.; Renton, C.; Rourke, S.B.; Vardy, J.L. Perceived cognitive impairment in people with colorectal cancer who do and do not receive chemotherapy. J. Cancer Surviv. 2018, 12, 178–185. [Google Scholar] [CrossRef]
- Bray, V.J.; Dhillon, H.M.; Vardy, J.L. Systematic review of self-reported cognitive function in cancer patients following chemotherapy treatment. J. Cancer Surviv. 2018, 12, 537–559. [Google Scholar] [CrossRef]
- Brailean, A.; Steptoe, A.; Batty, G.D.; Zaninotto, P.; Llewellyn, D.J. Are subjective memory complaints indicative of objective cognitive decline or depressive symptoms? Findings from the English Longitudinal Study of Ageing. J. Psychiatr. Res. 2019, 110, 143–151. [Google Scholar] [CrossRef]
- Apple, A.C.; Schroeder, M.P.; Ryals, A.J.; Wagner, L.I.; Cella, D.; Shih, P.A.; Reilly, J.; Penedo, F.J.; Voss, J.L.; Wang, L. Hippocampal functional connectivity is related to self-reported cognitive concerns in breast cancer patients undergoing adjuvant therapy. Neuroimage Clin. 2018, 20, 110–118. [Google Scholar] [CrossRef]
- Argyriou, A.A.; Karteri, S.; Bruna, J.; Mariotto, S.; Simo, M.; Velissaris, D.; Kalofonou, F.; Cavaletti, G.; Ferrari, S.; Kalofonos, H.P. Serum neurofilament light chain levels as biomarker of paclitaxel-induced cognitive impairment in patients with breast cancer: A prospective study. Support. Care Cancer 2022, 30, 1807–1814. [Google Scholar] [CrossRef]
- Winocur, G.; Wojtowicz, J.M.; Huang, J.; Tannock, I.F. Physical exercise prevents suppression of hippocampal neurogenesis and reduces cognitive impairment in chemotherapy-treated rats. Psychopharmacology 2014, 231, 2311–2320. [Google Scholar] [CrossRef]
- Bedillion, M.F.; Ansell, E.B.; Thomas, G.A. Cancer treatment effects on cognition and depression: The moderating role of physical activity. Breast 2019, 44, 73–80. [Google Scholar] [CrossRef]
- Bray, V.J.; Dhillon, H.M.; Bell, M.L.; Kabourakis, M.; Fiero, M.H.; Yip, D.; Boyle, F.; Price, M.A.; Vardy, J.L. Evaluation of a Web-Based Cognitive Rehabilitation Program in Cancer Survivors Reporting Cognitive Symptoms After Chemotherapy. J. Clin. Oncol. 2017, 35, 217–225. [Google Scholar] [CrossRef]
- Ercoli, L.M.; Petersen, L.; Hunter, A.M.; Castellon, S.A.; Kwan, L.; Kahn-Mills, B.A.; Embree, L.M.; Cernin, P.A.; Leuchter, A.F.; Ganz, P.A. Cognitive rehabilitation group intervention for breast cancer survivors: Results of a randomized clinical trial. Psychooncology 2015, 24, 1360–1367. [Google Scholar] [CrossRef]
- Schagen, S.B.; Tsvetkov, A.S.; Compter, A.; Wefel, J.S. Cognitive adverse effects of chemotherapy and immunotherapy: Are interventions within reach? Nat. Rev. Neurol. 2022, 18, 173–185. [Google Scholar] [CrossRef]
- Nekhlyudov, L.; Aziz, N.M.; Lerro, C.; Virgo, K.S. Oncologists’ and primary care physicians’ awareness of late and long-term effects of chemotherapy: Implications for care of the growing population of survivors. J. Oncol. Pract. 2014, 10, e29–e36. [Google Scholar] [CrossRef]
- Hershman, D.L.; Lacchetti, C.; Dworkin, R.H.; Lavoie Smith, E.M.; Bleeker, J.; Cavaletti, G.; Chauhan, C.; Gavin, P.; Lavino, A.; Lustberg, M.B.; et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J. Clin. Oncol. 2014, 32, 1941–1967. [Google Scholar] [CrossRef]
- Bruna, J.; Videla, S.; Argyriou, A.A.; Velasco, R.; Villoria, J.; Santos, C.; Nadal, C.; Cavaletti, G.; Alberti, P.; Briani, C.; et al. Efficacy of a Novel Sigma-1 Receptor Antagonist for Oxaliplatin-Induced Neuropathy: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Clinical Trial. Neurotherapeutics 2018, 15, 178–189. [Google Scholar] [CrossRef]
- Available online: https://www.pnsociety.com/ (accessed on 15 November 2022).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alberti, P.; Salvalaggio, A.; Argyriou, A.A.; Bruna, J.; Visentin, A.; Cavaletti, G.; Briani, C. Neurological Complications of Conventional and Novel Anticancer Treatments. Cancers 2022, 14, 6088. https://doi.org/10.3390/cancers14246088
Alberti P, Salvalaggio A, Argyriou AA, Bruna J, Visentin A, Cavaletti G, Briani C. Neurological Complications of Conventional and Novel Anticancer Treatments. Cancers. 2022; 14(24):6088. https://doi.org/10.3390/cancers14246088
Chicago/Turabian StyleAlberti, Paola, Alessandro Salvalaggio, Andreas A. Argyriou, Jordi Bruna, Andrea Visentin, Guido Cavaletti, and Chiara Briani. 2022. "Neurological Complications of Conventional and Novel Anticancer Treatments" Cancers 14, no. 24: 6088. https://doi.org/10.3390/cancers14246088
APA StyleAlberti, P., Salvalaggio, A., Argyriou, A. A., Bruna, J., Visentin, A., Cavaletti, G., & Briani, C. (2022). Neurological Complications of Conventional and Novel Anticancer Treatments. Cancers, 14(24), 6088. https://doi.org/10.3390/cancers14246088