
Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer


 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
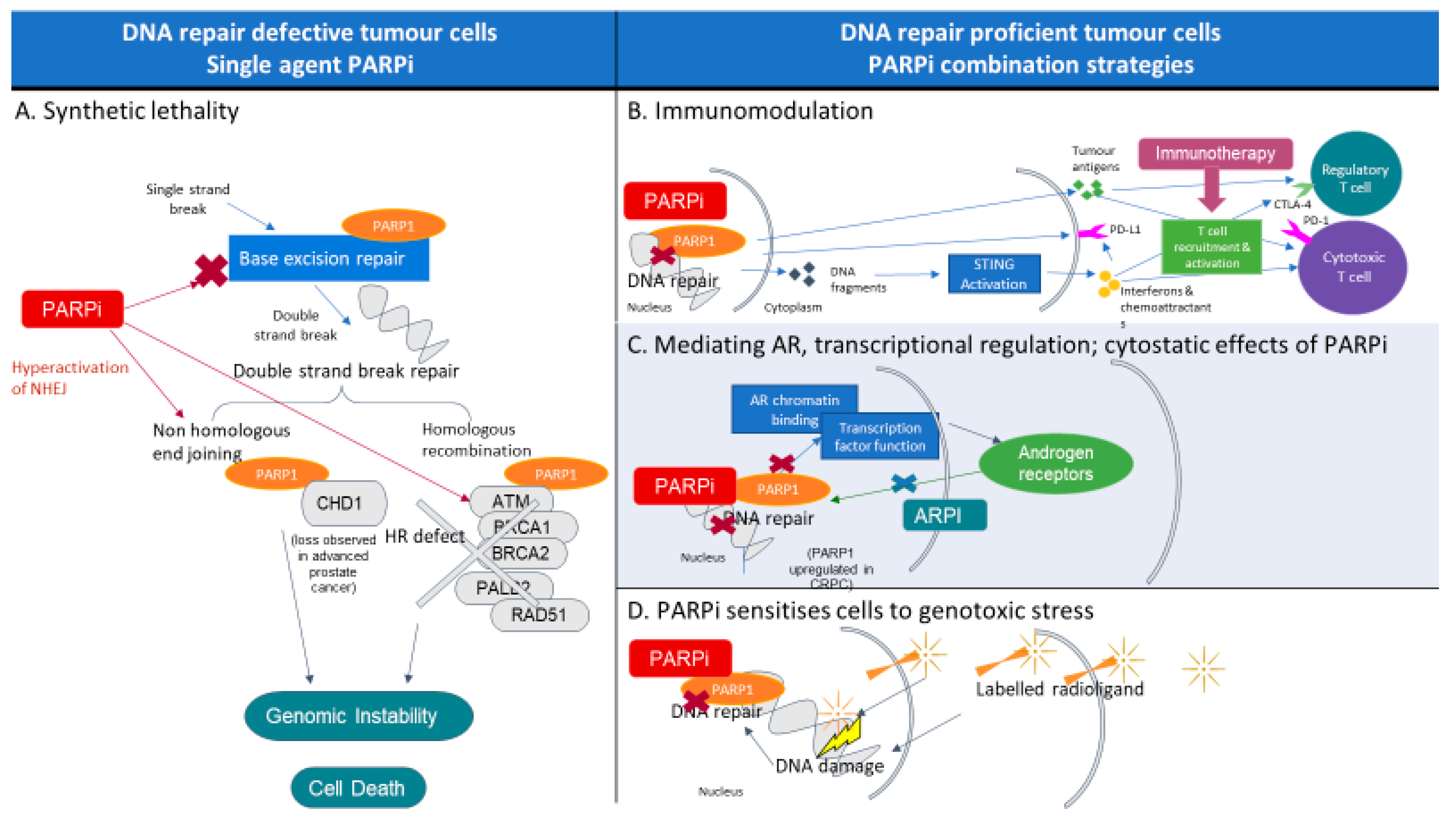
2. PARP and PARP Inhibitors
3. PARP Inhibitors in Prostate Cancer
4. Phase II Clinical Trials of PARP Inhibitors in Prostate Cancer

{kind=link}
| Trial Name | Phase of Trial (n = Number of Patients) | Treatment Arms | Patient Population | HRR Biomarker Inclusion | Key Endpoints | Key Efficacy Results | Responses in Molecular Subtypes |
|---|---|---|---|---|---|---|---|
| Phase II PARP inhibitor single agent trials in metastatic castrate resistant prostate cancer after taxane therapy | |||||||
| TOPARP-A [3] | Phase II (n = 50) | Olaparib 400 mg BD. | mCRPC, progressed after 1 or 2 taxane-based regimes. | Unselected; subsequent molecular characterization for HRR alterations. | PE: Composite response rate; radiological, PSA50 or CTC reduction. SE: rPFS, OS. | All patients: Composite response 33% (95% CI 20–48), PSA50 22%. | 88% composite response in HRR altered (BM+) cohort vs. 6% without HRR alterations (BM-) rPFS 9.8 (BM+) vs. 2.7 mo (BM-) (p < 0.001). Median OS 13.8 (BM+) vs. 7.5 mo (BM-) (p = 0.05). Composite response BRCA2: 100%, ATM: 80%. |
| TOPARP-B [6] | Phase II (n = 98) | Olaparib 300 mg BD or 400 mg BD. | mCRPC progressed after 1 or 2 taxane-based regimes. | Biallelic deleterious HRR alterations. | PE: Composite response rate (defined as TOPARP-A) SE: rPFS, OS. | Composite response 400 mg: 54.3% (95% CI 39.0–69.1), 300 mg: 39.1% (25.1–54.6). Median rPFS 5.5 mo (95% CI 4.4–8.3) 400 mg vs. 5.6 mo (3.7–7.7) 300 mg. Median OS 14.3 mo (9.7–18.9) 400 mg vs. 10.1 mo (9.0–17.7) 300 mg. | Composite response BRCA1/2 83.3% (65.3–94.4), PALB2 57.1% (18.4–90.1), ATM 36.8% (16.3–61.6), CDK12 25% (8.7–49.1), Other 20% (5.7–43.7). |
| TRITON 2 [8,9,34] | Phase II (n = 115 BRCA 78 non-BRCA) | Rucaparib 600 mg BD. | mCRPC, progressed after at least 1 taxane-based regimen and 1 ARPI. | Bi- or monoallelic, germline, or somatic deleterious BRCA1/2 mutation or other prespecified HRR gene. | PE: ORR SE: DOR, PSA50. | BRCA1/2 ORR 43.5% (95% CI 31.0–56.7), PSA50 54.8% (45.2–64.1). Median DOR not reached (NR; 95% CI 6.4 mo-NR), PSA50 54.8% (95% CI 45.2–64.1). | PSA50 gBRCA 61.7% (95% CI 46.4–75.5), sBRCA 50% (37.6–62.4). ORR gBRCA 45.8% (95% CI 25.6–67.2), sBRCA 42.1% (26.3–59.2). ATM: ORR 10.5%, PSA50 4.1%. CDK12: ORR 0%, PSA50 6.7%. CHEK12: ORR 11.1%, PSA50 16.7%. |
| GALAHAD [5] | Phase II (n = 289) | Niraparib 300 mg OD. | mCRPC progressed after at least 1 taxane-based regimen and 1 ARPI. | Biallelic HRR or germline pathogenic BRCA1/2 alterations (BRCA cohort) or biallelic alterations in other prespecified DDR genes (non-BRCA cohort)—included ATM, BRIP1, CHEK2, FANCA, HDAC2, PALB2. | PE: ORR (trial amended PE) in BRCA cohort. SE: ORR in non-BRCA, OS, rPFS, composite response (radiological, PSA50 or CTC reduction). | ORR BRCA: 34.2% (95% CI 23.7–46.0). Non-BRCA: 10.6% (3.5–23.1). OS BRCA 13.01, non-BRCA 9.63 mo. rPFS BRCA 8.08, non-BRCA 3.71 mo. Composite response BRCA 58%, non-BRCA 15%. | Results for BRCA1 and BRCA2 cohorts reported together. |
| TALAPRO 1 [35] | Phase II (n = 128) | Talazoparib 1 mg OD. | mCRPC progressed after at least 1 taxane-based regimen and 1 ARPI. | Mono- or biallelic HRR alterations (CDK12 excluded). | PE: ORR, SE: OS rPFS, PSA50. | ORR 29.8% (95% CI 21.2–39.6). Median OS 16.4 mo (95% CI 12.2–19.9). Median rPFS 5.6 mo (95% CI 3.7–8.8). PSA50 42% . | ORR: BRCA1/2 46%, BRCA2 46%, PALB2 25%, ATM 12%, Other 9%. |
| Phase III PARP inhibitor single agent trials in metastatic castrate resistant prostate cancer | |||||||
| PROfound [7] | Phase III (n = 387) | Olaparib 300 mg BD vs. physicians choice of enzalutamide or abiraterone. | mCRPC after at least 1 ARPI (previous taxane was allowed). | Bi- or monoallelic, somatic, or germline, deleterious HRR alterations. Cohort A: BRCA1/2 or ATM mutations. Cohort B: other 12 HRR genes mutations. | PE: rPFS in cohort A. SE: ORR, PSA50, OS. | Median rPFS Cohort A + B 5.8 olaparib vs. 3.5 mo control (HR 0.49 95% CI 0.38–0.63). ORR 22% olaparib vs. 4% control (OR 5.93; 95% CI 2.01–25.40). Median OS (prelim) 17.5 olaparib vs. 14.3 mo control (HR 0.67; 95% CI 0.49–0.93). PSA50 30% olaparib vs. 10% control. | Median PFS Cohort A: 7.4 olaparib vs. 3.6 mo control (HR 0.34; 95% CI 0.25–0.47). Cohort B: 5.8 mo vs. 3.5 mo (HR 0.49, 95% CI: 0.38–0.63). Cohort A: ORR 33% olaparib vs. 2% control (OR 20.86; 95% CI 4.18–379.18). Cohort A: PSA50 43% olaparib vs. 8% control. Cohort A: Median OS (prelim) 18.5 olaparib vs. 15.1 mo control (HR 0.64; 95% CI 0.43–0.97). |
| TRITON3 [41] | Phase III (n = 405) | Rucaparib vs. physicians choice of either enzalutamide, abiraterone or docetaxel. | mCRPC progressed after ARPI (no prior chemotherapy in castrate resistant setting). | BRCA1/2 or ATM alteration. | PE: rPFS. SE: OS, ORR. | Prelim: ITT population (ATM and BRCA) Median rPFS 10.2 rucaparib vs. 6.4 mo control:HR 0.50 (95% CI 0.36–0.69). OS data immature. | Prelim: BRCA: Median rPFS 11.2 rucaparib vs. 6.4 mo control (p < 0.0001). ATM: Median rPFS 8.1 rucaparib vs. 6.8 mo control (p = 0.8421). OS data immature. |
5. Phase III Clinical Trials of PARP Inhibitors in Prostate Cancer
6. Targeting Androgen Receptor (AR) and PARP Concurrently
7. Biomarkers of Response and Resistance
8. BRCA and HRR Testing in Advanced Prostate Cancer
9. Combination Therapeutics with PARP Inhibitors
9.1. PARP Inhibition and Immunotherapy
9.2. PARP and Other Targeted Therapies
9.3. PARP and Radioligand Therapy
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Smith, M.R.; Scher, H.I.; Sandhu, S.; Efstathiou, E.; Lara, P.N., Jr.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; Ståhl, O.; et al. Niraparib in patients with metastatic castration-resistant prostate cancer and DNA repair gene defects (GALAHAD): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2022, 23, 362–373. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Abida, W.; Campbell, D.; Patnaik, A.; Shapiro, J.D.; Sautois, B.; Vogelzang, N.J.; Voog, E.G.; Bryce, A.H.; McDermott, R.; Ricci, F.; et al. Non-BRCA DNA Damage Repair Gene Alterations and Response to the PARP Inhibitor Rucaparib in Metastatic Castration-Resistant Prostate Cancer: Analysis from the Phase II TRITON2 Study. Clin. Cancer Res. 2020, 26, 2487–2496. [Google Scholar] [CrossRef]
- Abida, W.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Rucaparib in Men With Metastatic Castration-Resistant Prostate Cancer Harboring a BRCA1 or BRCA2 Gene Alteration. J. Clin. Oncol. 2020, 38, 3763–3772. [Google Scholar] [CrossRef]
- Lozano Mejorada, R.; Castro Marcos, E.; Aragon, I.M.; Thorne, H.; Lopez Campos, F.; Sanz, A.; Alonso, C.; Anido, U.; Juan Fita, M.J.; Gutierrez Pecharromán, A.M.; et al. 612MO Clinical impact of somatic alterations in prostate cancer patients with and without previously known germline BRCA1/2 mutations: Results from PROREPAIR-A study. Ann. Oncol. 2020, 31, S509–S510. [Google Scholar] [CrossRef]
- Trigos, A.S.; Pasam, A.; Banks, P.; Wallace, R.; Guo, C.; Keam, S.; Thorne, H.; Mitchell, C.; Lade, S.; Clouston, D.; et al. Tumor immune microenvironment of primary prostate cancer with and without germline mutations in homologous recombination repair genes. J. ImmunoTherapy Cancer 2022, 10, e003744. [Google Scholar] [CrossRef] [PubMed]
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef]
- Tutt, A.; Bertwistle, D.; Valentine, J.; Gabriel, A.; Swift, S.; Ross, G.; Griffin, C.; Thacker, J.; Ashworth, A. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 2001, 20, 4704–4716. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- Panzarino, N.J.; Krais, J.J.; Cong, K.; Peng, M.; Mosqueda, M.; Nayak, S.U.; Bond, S.M.; Calvo, J.A.; Doshi, M.B.; Bere, M.; et al. Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res. 2021, 81, 1388–1397. [Google Scholar] [CrossRef]
- Cong, K.; Peng, M.; Kousholt, A.N.; Lee, W.T.C.; Lee, S.; Nayak, S.; Krais, J.; VanderVere-Carozza, P.S.; Pawelczak, K.S.; Calvo, J.; et al. Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell 2021, 81, 3128–3144.e7. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of Poly(ADP-Ribose) Polymerase in Tumors from BRCA Mutation Carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 2014, 12, 1069–1080. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Kraus, W.L. The PARP side of the nucleus: Molecular actions, physiological outcomes, and clinical targets. Mol. Cell 2010, 39, 8–24. [Google Scholar] [CrossRef]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef]
- Lang, S.H.; Swift, S.L.; White, H.; Misso, K.; Kleijnen, J.; Quek, R.G.W. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int. J. Oncol. 2019, 55, 597–616. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Sandhu, S.K.; Carden, C.P.; de Bono, J.S. Poly(ADP-ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA Cancer J. Clin. 2011, 61, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Loehr, A.; Patnaik, A.; Campbell, D.; Shapiro, J.; Bryce, A.H.; McDermott, R.; Sautois, B.; Vogelzang, N.J.; Bambury, R.M.; Voog, E.; et al. Response to Rucaparib in BRCA-Mutant Metastatic Castration-Resistant Prostate Cancer Identified by Genomic Testing in the TRITON2 Study. Clin. Cancer Res. 2021, 27, 6677–6686. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Mehra, N.; Scagliotti, G.V.; Castro, E.; Dorff, T.; Stirling, A.; Stenzl, A.; Fleming, M.T.; Higano, C.S.; Saad, F.; et al. Talazoparib monotherapy in metastatic castration-resistant prostate cancer with DNA repair alterations (TALAPRO-1): An open-label, phase 2 trial. Lancet Oncol. 2021, 22, 1250–1264. [Google Scholar] [CrossRef] [PubMed]
- Teyssonneau, D.; Margot, H.; Cabart, M.; Anonnay, M.; Sargos, P.; Vuong, N.S.; Soubeyran, I.; Sevenet, N.; Roubaud, G. Prostate cancer and PARP inhibitors: Progress and challenges. J. Hematol. Oncol. 2021, 14, 51. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Gomella, L.G.; Petrylak, D.P. When and How to Use PARP Inhibitors in Prostate Cancer: A Systematic Review of the Literature with an Update on On-Going Trials. Eur. Urol. Oncol. 2020, 3, 594–611. [Google Scholar] [CrossRef]
- Risdon, E.N.; Chau, C.H.; Price, D.K.; Sartor, O.; Figg, W.D. PARP Inhibitors and Prostate Cancer: To Infinity and Beyond BRCA. Oncologist 2021, 26, e115–e129. [Google Scholar] [CrossRef]
- Snyder, L.A.; Damle, R.; Patel, S.; Bohrer, J.; Fiorella, A.; Driscoll, J.; Hawkins, R.; Stratton, C.F.; Manning, C.D.; Tatikola, K.; et al. Niraparib Shows Superior Tissue Distribution and Efficacy in a Prostate Cancer Bone Metastasis Model Compared with Other PARP Inhibitors. Mol. Cancer Ther. 2022, 21, 1115–1124. [Google Scholar] [CrossRef]
- Wang, C.; Li, J. Haematologic toxicities with PARP inhibitors in cancer patients: An up-to-date meta-analysis of 29 randomized controlled trials. J. Clin. Pharm. 2021, 46, 571–584. [Google Scholar] [CrossRef]
- A Study of Rucaparib Versus Physician’s Choice of Therapy in Patients with Metastatic Castration-Resistant Prostate Cancer and Homologous Recombination Gene Deficiency (TRITON3): ClinicalTrials.gov Identifier: NCT02975934. 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02975934 (accessed on 21 November 2022).
- Ratta, R.; Guida, A.; Scotté, F.; Neuzillet, Y.; Teillet, A.B.; Lebret, T.; Beuzeboc, P. PARP inhibitors as a new therapeutic option in metastatic prostate cancer: A systematic review. Prostate Cancer Prost. Dis. 2020, 23, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef] [PubMed]
- TRITON3 Phase 3 Trial of Rubraca® (rucaparib) Achieves Primary Endpoint in Men with Metastatic Castration-Resistant Prostate Cancer with BRCA or ATM Mutations: Clovis Oncology. 2022. Available online: https://ir.clovisoncology.com/investors-and-news/news-releases/press-release-details/2022/TRITON3-Phase-3-Trial-of-Rubraca-rucaparib-Achieves-Primary-Endpoint-in-Men-with-Metastatic-Castration-Resistant-Prostate-Cancer-with-BRCA-or-ATM-Mutations/default.aspx (accessed on 21 November 2022).
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib combined with abiraterone in patients with metastatic castration-resistant prostate cancer: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.W.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.; Loredo, E.; Procopio, G.; Menezes, J.D.; Girotto, G.; Arslan, C.; et al. Abiraterone and Olaparib for Metastatic Castration-Resistant Prostate Cancer. NEJM Evid. 2022, 1, EVIDoa2200043. [Google Scholar] [CrossRef]
- Saad, F.; Armstrong, A.J.; Thiery-Vuillemin, A.; Oya, M.; Shore, N.D.; Procopio, G.; Arslan, C.; Mehra, N.; Parnis, F.; Brown, E.; et al. 1357O—Biomarker analysis and updated results from the Phase III PROpel trial of abiraterone (abi) and olaparib (ola) vs abi and placebo (pbo) as first-line (1L) therapy for patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). Ann. Oncol. 2022, 33, S616–S652. [Google Scholar] [CrossRef]
- Chi, K.N.; Rathkopf, D.E.; Smith, M.R.; Efstathiou, E.; Attard, G.; Olmos, D.; Lee, J.Y.; Small, E.J.; Gomes, A.J.; Roubaud, G.; et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J. Clin. Oncol. 2022, 40, 12. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Shore, N.D.; Carles, J.; Fay, A.P.; Dunshee, C.; Karsh, L.I.; Paccagnella, M.L.; Santo, N.D.; Elmeliegy, M.; et al. TALAPRO-2: A phase 3 randomized study of enzalutamide (ENZA) plus talazoparib (TALA) versus placebo in patients with new metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, TPS5089. [Google Scholar] [CrossRef]
- Pfizer Announces Positive Topline Results from Phase 3 TALAPRO-2 Trial: Pfizer. 2022. Available online: https://www.pfizer.com/news/press-release/press-release-detail/pfizer-announces-positive-topline-results-phase-3-talapro-2 (accessed on 21 November 2022).
- Rao, A.; Ryan, C.J.; VanderWeele, D.J.; Heller, G.; Lewis, L.D.; Watt, C.; Chen, R.C.; Grubb, R.; Hahn, O.M.; Beltran, H.; et al. CASPAR (Alliance A031902): A randomized, phase III trial of enzalutamide (ENZ) with rucaparib (RUCA)/placebo (PBO) as a novel therapy in first-line metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, TPS181. [Google Scholar] [CrossRef]
- Agarwal, N.; Azad, A.; Fizazi, K.; Mateo, J.; Matsubara, N.; Shore, N.D.; Chakrabarti, J.; Chen, H.-C.; Lanzalone, S.; Niyazov, A.; et al. Talapro-3: A phase 3, double-blind, randomized study of enzalutamide (ENZA) plus talazoparib (TALA) versus placebo plus enza in patients with DDR gene mutated metastatic castration-sensitive prostate cancer (mCSPC). J. Clin. Oncol. 2022, 40, TPS221. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Chi, K.N.; Olmos, D.; Cheng, H.H.; Agarwal, N.; Graff, J.N.; Sandhu, S.K.; Hayreh, V.; Lopez-Gitlitz, A.; Francis, P.S.J.; et al. AMPLITUDE: A study of niraparib in combination with abiraterone acetate plus prednisone (AAP) versus AAP for the treatment of patients with deleterious germline or somatic homologous recombination repair (HRR) gene-altered metastatic castration-sensitive prostate cancer (mCSPC). J. Clin. Oncol. 2021, 39, TPS176. [Google Scholar] [CrossRef]
- Markowski, M.C.; Antonarakis, E.S. BRCA1 Versus BRCA2 and PARP Inhibitor Sensitivity in Prostate Cancer: More Different Than Alike? J. Clin. Oncol. 2020, 38, 3735–3739. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Porta, N.; Arce-Gallego, S.; Seed, G.; Llop-Guevara, A.; Bianchini, D.; Rescigno, P.; Paschalis, A.; Bertan, C.; Baker, C.; et al. Biomarkers Associating with PARP Inhibitor Benefit in Prostate Cancer in the TOPARP-B Trial. Cancer Discov. 2021, 11, 2812–2827. [Google Scholar] [CrossRef]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Goodall, J.; Mateo, J.; Yuan, W.; Mossop, H.; Porta, N.; Miranda, S.; Perez-Lopez, R.; Dolling, D.; Robinson, D.R.; Sandhu, S.; et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Discov. 2017, 7, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Mejía, N.; García-Illescas, D.; Morales-Barrera, R.; Suarez, C.; Planas, J.; Maldonado, X.; Carles, J.; Mateo, J. PARP inhibitors in advanced prostate cancer: When to use them? Endocr. Relat. Cancer 2021, 28, T79–T93. [Google Scholar] [CrossRef]
- Lozano, R.; Castro, E.; Aragon, I.; Thorne, H.; López-Campos, F.; Sanz, A.; Alonso, C.; Anido, U.; Jose, J.m.; Pecharroman, A.G.; et al. PROREPAIR-A: Clinical and molecular characterization study of prostate cancer (PC) patients with and without previously known germline BRCA1/2 mutations. J. Clin. Oncol. 2020, 38, 5511. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Knudsen, K.E. DNA Damage Response in Prostate Cancer. Cold Spring Harb. Perspect. Med. 2019, 9, a030486. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. Prostate Cancer Version 4. 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate.pdf (accessed on 21 November 2022).
- Cheng, H.H.; Sokolova, A.O.; Schaeffer, E.M.; Small, E.J.; Higano, C.S. Germline and Somatic Mutations in Prostate Cancer for the Clinician. J. Natl. Compr. Cancer Netw. 2019, 17, 515–521. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Investig. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Hussain, M.; Corcoran, C.; Sibilla, C.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Mateo, J.; Olmos, D.; Mehra, N.; et al. Tumor Genomic Testing for >4000 Men with Metastatic Castration-resistant Prostate Cancer in the Phase III Trial PROfound (Olaparib). Clin. Cancer Res. 2022, 28, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Barnicle, A.; Sibilla, C.; Lai, Z.; Corcoran, C.; Williams, J.A.; Barrett, J.C.; Adelman, C.A.; Qiu, P.; Easter, A.; et al. Concordance of BRCA1, BRCA2 (BRCA), and ATM mutations identified in matched tumor tissue and circulating tumor DNA (ctDNA) in men with metastatic castration-resistant prostate cancer (mCRPC) screened in the PROfound study. J. Clin. Oncol. 2021, 39, 26. [Google Scholar] [CrossRef]
- Vandekerkhove, G.; Struss, W.J.; Annala, M.; Kallio, H.M.L.; Khalaf, D.; Warner, E.W.; Herberts, C.; Ritch, E.; Beja, K.; Loktionova, Y.; et al. Circulating Tumor DNA Abundance and Potential Utility in De Novo Metastatic Prostate Cancer. Eur. Urol. 2019, 75, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Niimi, A.; Yasuhara, T.; Permata, T.B.M.; Hagiwara, Y.; Isono, M.; Nuryadi, E.; Sekine, R.; Oike, T.; Kakoti, S.; et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 2017, 8, 1751. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Kim, H.J.; Wang, Q.; Kearns, M.; Jiang, T.; Ohlson, C.E.; Li, B.B.; Xie, S.; Liu, J.F.; Stover, E.H.; et al. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018, 25, 2972–2980.e5. [Google Scholar] [CrossRef] [PubMed]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L.; et al. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Yu, E.Y.; Piulats, J.M.; Gravis, G.; Laguerre, B.; Arija, J.A.A.; Oudard, S.; Fong, P.C.C.; Kolinsky, M.P.; Augustin, M.; Feyerabend, S.; et al. KEYNOTE-365 cohort A updated results: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 100. [Google Scholar] [CrossRef]
- Yu, E.Y.; Park, S.H.; Goh, J.C.H.; Shin, S.J.; Mehra, N.; McDermott, R.; Gonzalez, M.S.; Fong, P.C.; Greil, R.; Retz, M.; et al. 1362MO—Pembrolizumab + olaparib vs abiraterone (abi) or enzalutamide (enza) for patients (pts) with previously treated metastatic castration-resistant prostate cancer (mCRPC): Randomized open-label phase III KEYLYNK-010 study. Ann. Oncol. 2022, 33, S1163–S1164. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Retz, M.; Goh, J.C.; Burotto, M.; Gravis, G.; Castellano, D.; Flechon, A.; Zschaebitz, S.; Shaffer, D.R.; Limon, J.C.V.; et al. CheckMate 9KD cohort A1 final analysis: Nivolumab (NIVO) + rucaparib for post-chemotherapy (CT) metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2021, 39, 5044. [Google Scholar] [CrossRef]
- A Study of Niraparib Combination Therapies for the Treatment of Metastatic Castration-Resistant Prostate Cancer (QUEST): ClinicalTrials.gov Identifier: NCT03431350. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03431350 (accessed on 21 November 2022).
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR Inhibitor AZD6738 (Ceralasertib) Exerts Antitumor Activity as a Monotherapy and in Combination with Chemotherapy and the PARP Inhibitor Olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Targeting Resistant Prostate Cancer with ATR and PARP Inhibition (TRAP Trial): ClinicalTrials.gov Identifier: NCT03787680. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT03787680 (accessed on 21 November 2022).
- Reichert, Z.R.; Devitt, M.E.; Alumkal, J.J.; Smith, D.C.; Caram, M.V.; Palmbos, P.; Vaishampayan, U.N.; Alva, A.S.; Braun, T.; Yentz, S.E.; et al. Targeting resistant prostate cancer, with or without DNA repair defects, using the combination of ceralasertib (ATR inhibitor) and olaparib (the TRAP trial). J. Clin. Oncol. 2022, 40, 88. [Google Scholar] [CrossRef]
- Phase II Trial of AZD6738 Alone and in Combination with Olaparib: ClinicalTrials.gov Identifier: NCT03682289. Available online: https://clinicaltrials.gov/ct2/show/NCT03682289 (accessed on 21 November 2022).
- Gallyas, F., Jr.; Sumegi, B.; Szabo, C. Role of Akt Activation in PARP Inhibitor Resistance in Cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer—The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef]
- Pook, D.W.; Geynisman, D.M.; Carles, J.; Braud, F.G.D.; Joshua, A.M.; Perez-Gracia, J.L.; Pérez, C.L.; Shin, S.J.; Fang, B.; Barve, M.A.; et al. A phase Ib, open-label study evaluating the safety and efficacy of ipatasertib + rucaparib in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2022, 40, 95. [Google Scholar] [CrossRef]
- Kim, J.W.; Cardin, D.B.; Vaishampayan, U.N.; Kato, S.; Grossman, S.R.; Glazer, P.M.; Shyr, Y.; Ivy, S.P.; LoRusso, P.M. Clinical Activity and Safety of Cediranib and Olaparib Combination in Patients with Metastatic Pancreatic Ductal Adenocarcinoma without BRCA Mutation. Oncologist 2021, 26, e1104–e1109. [Google Scholar] [CrossRef]
- Kim, J.W.; McKay, R.R.; Radke, M.R.; Zhao, S.; Taplin, M.-E.; Davis, N.B.; Monk, P.; Appleman, L.J.; Jr, P.N.L.; Vaishampayan, U.N.; et al. Randomized Trial of Olaparib with or without Cediranib for Metastatic Castration-Resistant Prostate Cancer: The Results From National Cancer Institute 9984. J. Clin. Oncol. 2022. [Google Scholar] [CrossRef]
- Phase I/II Study of the Anti-Programmed Death Ligand-1 Durvalumab Antibody (MEDI4736) in Combination with Olaparib and/or Cediranib for Advanced Solid Tumors and Advanced or Recurrent Ovarian, Triple Negative Breast, Lung, Prostate and Colorectal Can: ClinicalTrials.gov Identifier: NCT02484404. 2015. Available online: https://clinicaltrials.gov/ct2/show/NCT02484404 (accessed on 21 November 2022).
- Pidnarulex and Talazoparib in Patients with Metastatic Castration Resistant Prostate Cancer (REPAIR): ClinicalTrials.gov Identifier: NCT05425862. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05425862 (accessed on 21 November 2022).
- Marchetti, A.; Rosellini, M.; Nuvola, G.; Tassinari, E.; Mollica, V.; Rizzo, A.; Santoni, M.; Cimadamore, A.; Farolfi, A.; Montironi, R.; et al. PARP Inhibitors and Radiometabolic Approaches in Metastatic Castration-Resistant Prostate Cancer: What’s Now, What’s New, and What’s Coming? Cancers 2022, 14, 907. [Google Scholar] [CrossRef]
- 177Lu-PSMA-617 Therapy and Olaparib in Patients with Metastatic Castration Resistant Prostate Cancer (LuPARP): ClinicalTrials.gov. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT03874884 (accessed on 21 November 2022).
- Shaya, J.; Xie, W.; Saraiya, B.; Parikh, M.; Folefac, E.; Olson, A.C.; Choudhury, A.D.; Einstein, D.J.; Heath, E.I.; Parikh, R.A.; et al. A phase I/II study of combination olaparib and radium-223 in men with metastatic castration-resistant prostate cancer with bone metastases (COMRADE): A trial in progress. J. Clin. Oncol. 2021, 39, TPS182. [Google Scholar] [CrossRef]
- Kelly, W.K.; Leiby, B.; Einstein, D.J.; Szmulewitz, R.Z.; Sartor, A.O.; Yang, E.S.-H.; Sonpavde, G. Radium-223 (Rad) and niraparib (Nira) treatment (tx) in castrate-resistant prostate cancer (CRPC) patients (pts) with and without prior chemotherapy (chemo). J. Clin. Oncol. 2020, 38, 5540. [Google Scholar] [CrossRef]
- A Study of Olaparib and Durvalumab in Prostate Cancer: ClinicalTrials.gov. 2019. Available online: https://clinicaltrials.gov/ct2/show/NCT03810105 (accessed on 21 November 2022).

| Trial Name | Phase of Trial (n = Number of Patients) | Treatment Arms | Patient Population | HRR Biomarker Inclusion | Key Endpoints | Key Efficacy Results |
|---|---|---|---|---|---|---|
| NCT01972217 [46] | Phase II (n = 142) | Olaparib plus abiraterone vs. placebo and abiraterone. | mCRPC following prior docetaxel. | Unselected. | PE: rPFS. SE: PFS 2, OS. | rPFS 13.8 olaparib vs. 8.2 mo placebo (HR 0.65; 95% CI 0.44–0.97; p = 0.034). Median PFS2 23.3 (95% CI 17.4—not reached) olaparib vs. 18.5 mo (16.1–23.8) placebo (HR 0.79; 95% CI 0.51–1.21; p = 0.28). Median OS 22.7 (95% CI 17.4–29.4) olaparib vs. 20.9 mo (17.6–26.3) placebo (HR 0.91; 95% CI 0.60–1.38; p = 0.66). |
| TALAPRO-2 [49] | Phase III (n = 1095) | Talazoparib plus enzalutamide vs. placebo and enzalutamide. | mCRPC, first-line (ARPI allowed in the castration sensitive setting). | Cohort 1: HRR unselected. Cohort 2: selected for HRR gene alterations. | PE: rPFS. SE: OS. | Prelim: rPFS benefit with talazoparib combination exceeded prespecified HR 0.696. |
| PROpel [47] | Phase III (n = 796) | Olapaib plus abiraterone and prednisolone vs. placebo abiraterone and prednisolone. | mCRPC, first-line (study allowed for prior treatment with docetaxel in the first-line hormone sensitive setting). | Unselected | PE: rPFS. SE: OS. | Prelim: rPFS 24.8 olaparib and abiraterone vs. 16.6 mo (HR 0.66, 0.95% CI 0.54–0.81; p < 0.0001). HRRm cohort: median rPFS 28.8 olaparib and abiraterone vs. 13.8 mo (HR 0.45; 95% CI 0.31–0.65). Non-HRRm cohort: median rPFS 27.6 olaparib and abiraterone vs. 19.1 mo (HR 0.72; 95% CI 0.56–0.93). Cohort with BRCA alterations: median rPFS NR olaparib and abiraterone vs. 8.4 mo (HR 0.18; 95% CI 0.09–0.34). Cohort without BRCA alterations: median rPFS 27.6 olaparib and abiraterone vs. 16.6 mo (HR 0.72; 95% CI 0.58–0.90). OS data immature. |
| CASPAR [51] | Phase III (n = 984) | Rucaparib plus enzalutamide vs. placebo and enzalutamide. | mCRPC treatment-naïve. | Unselected. | PE: rPFS, OS. SE: rPFS in BRCA1/2 and PALB2. | Ongoing. |
| MAGNITUDE [48] | Phase III (n = 670) | Niraparib plus abiraterone vs. placebo and abiraterone. | mCRPC treatment-naïve (allowed for patients who had received less than four months prior abiraterone therapy). | Cohort without HRR alterations and HRR altered cohort (BM+): ATM, BRCA1, BRCA2, BRIP1, CDK12, CHEK2, FANCA, HDAC2, PALB2. | PE: rPFS in BRCA1/2 alterations and BM+ cohort. SE: OS. | Prelim: rPFS in BRCA1/2: 16.6 vs. 10.9 mo (HR 0.53 95%CI 0.36–0.79); p = 0.0014). rPFS in BM+ cohort 16.5 vs. 13.7 mo (HR 0.73 95% CI 0.56–0.96); p = 0.0217). |
| TALAPRO-3 [52] | Phase III (n = 550) | Talazoparib plus enzalutamide vs. placebo and enzalutamide. | mHSPC. | Selected for alterations in ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, RAD51C. | PE: rPFS. SE: OS. | Ongoing. |
| AMPLITUDE [53] | Phase III (n = 788) | Niraparib plus abiraterone vs. placebo and abiraterone. | mHSPC (allowed less than 6 months of ADT prior to randomisation and <3 y total). | Selected for HRR gene alteration. | PE: rPFS. SE: OS. | Ongoing. |
| TRAP [78] | Phase II (n = 47) | Ceralasertib (AZD 6738; ATR inhibitor) plus olaparib. | Multiple solid organ malignancies, includes mCRPC cohort with previous ARPI (n = 10). | Cohort 1 with and Cohort 2 without HRR alterations. | PE: Composite response (radiological or PSA50) SE: PFS. | Prelim: Cohort 1 (HRR alterations): PSA50 40%. |
| NCT03840200 [82] | Phase Ib (n = 51) | Ipatasertib (AKT inhibitor) plus rucaparib. | Breast, ovarian, or prostate cancer: including progressive mCRPC with prior ARPI. | Unselected. | Key PE: PSA50, dose-limiting toxicities, recommended phase II dose. SE: ORR, rPFS. | Prelim: PSA50 22% all mCRPC patients, 50% in HRR alterations, 25% without HRR alterations. ORR 4% all patients. |
| NCT02893917 [84] | Phase II (n = 90) | Cediranib plus olaparib (Arm A) or olaparib alone (Arm B). | mCRPC ≥ 1 prior line of therapy. | Unselected. | PE: rPFS. SE: ORR, PSA50. | Median rPFS 8.5 Arm A vs. 4.0 mo (HR 0.617, 95% CI 0.392–0.969 = 0.0359). ORR 19% Arm A vs. 12%. |
| NCT02484404 [70] | Phase I/II (n = 384) | Durvalumab plus olaparib and/or cediranib. | mCRPC previously treated with ARPI. | Unselected. | PE: clinical efficacy. SE: ORR, PSA50. | Radiographic and/or PSA response in 53%. All patients: 12-month rPFS 51.5%. Median rPFS for cohort with HRR alterations: 16.1 mo. |
| REPAIR [86] | Phase I (n = 48) | Pidnarulex (CX-5461) plus talazoparib. | mCRPC prior taxane, and ARPI. | Unselected. | PE: maximum tolerated dose. SE: PSA50, rPFS, ORR, OS. | Ongoing. |
| NCT03810105 [91] | Phase II (n = 5) | Olaparib plus durvalumab. | Castration sensitive biochemically recurrent nonmetastatic prostate cancer. | HRR deleterious mutations. | PE: undetectable PSA. | Ongoing. |
| KEYNOTE-365 [71] | Phase Ib/2 (Cohort A n = 84) | Olaparib with pembrolizumab. | Cohort A: mCRPC post docetaxel, up to 2 prior ARPI. | Unselected. | Key PE: PSA50 ORR. SE: OS, rPFS. | Prelim: PSA50 9%. ORR 8%. OS 14 mo. |
| KEYLYNK-010 [72] | Phase III (n = 793) | Olaparib with pembrolizumab vs. ARPI. | mCRPC progressed with prior ARPI and docetaxel. | Unselected. | PE: rPFS and OS. SE: ORR. | Stopped for futility in 2022. Median rPFS 4.4 pembrolizumab and olaparib vs. 4.2 mo ARPI (HR 1.02; 95% CI 0.82–1.25). OS 15.8 pembrolizumab and olaparib vs. 14.6 mo (HR 0.94; 95% CI 0.77–1.14) ORR 17% vs. 6% ARPI (p = 0.002). |
| CheckMate 9KD [73] | Phase II (n = 81 cohort A1) | Nivolumab combined with rucaparib (Arm A), docetaxel (Arm B) or enzalutamide (Arm C). | mCRPC (Arm A1: post 1–2 prior taxane regimes ≤ 2 ARPIs). | Unselected. | PE: ORR, PSA50. SE: rPFS, OS. | Cohort A1: ORR 10.3%, all, 17.2% HRR alterations, 3.4% without. PSA50 11.9% all, 18.2% HRR alterations, 5% without. All patients in cohort A1: Median rPFS 4.9 mo. Median OS 13.9 mo. |
| QUEST [74] | Phase II (n = 136) | Niraparib with abiraterone plus cetrelimab. | mCRPC. | Unselected. | Key PE: ORR, composite response (radiological, CTC, or PSA50 response). SE: OS. | Ongoing. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inderjeeth, A.-J.; Topp, M.; Sanij, E.; Castro, E.; Sandhu, S. Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer. Cancers 2022, 14, 5922. https://doi.org/10.3390/cancers14235922
Inderjeeth A-J, Topp M, Sanij E, Castro E, Sandhu S. Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer. Cancers. 2022; 14(23):5922. https://doi.org/10.3390/cancers14235922
Chicago/Turabian StyleInderjeeth, Andrisha-Jade, Monique Topp, Elaine Sanij, Elena Castro, and Shahneen Sandhu. 2022. "Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer" Cancers 14, no. 23: 5922. https://doi.org/10.3390/cancers14235922
APA StyleInderjeeth, A.-J., Topp, M., Sanij, E., Castro, E., & Sandhu, S. (2022). Clinical Application of Poly(ADP-ribose) Polymerase (PARP) Inhibitors in Prostate Cancer. Cancers, 14(23), 5922. https://doi.org/10.3390/cancers14235922