
Prognosis-Related Molecular Subtypes and Immune Features Associated with Hepatocellular Carcinoma
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
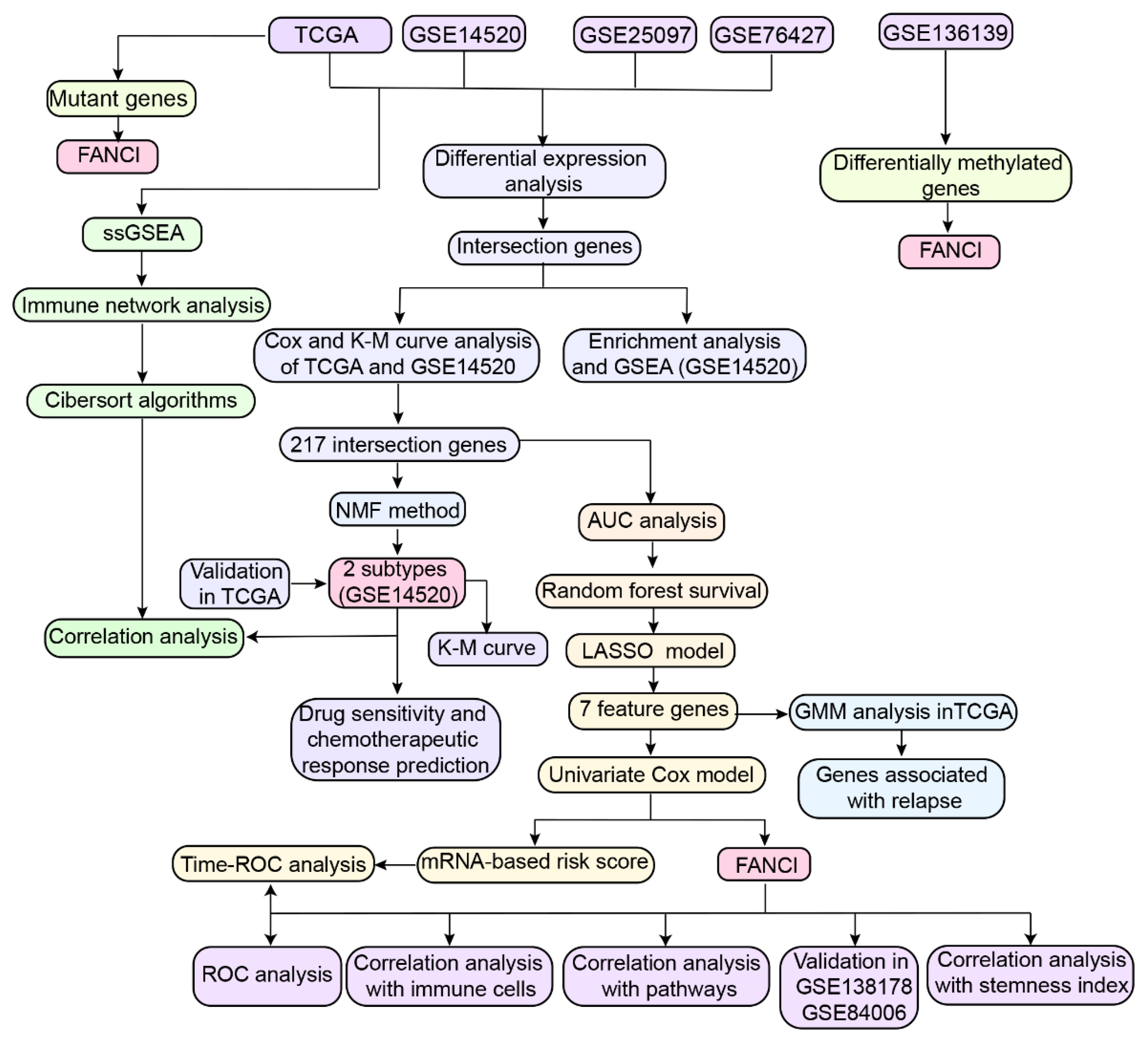
2. Material and Methods
2.1. Human Subject
2.2. Data Collection
2.3. Screening for Differentially Expressed Genes (DEGs) and Enrichment Analysis
2.4. Cox Regression and Kaplan-Meier Analyses
2.5. Construction of Random Survival Forest and Least Absolute Shrinkage and Selection Operator Regression Models
2.6. Construction of the Gaussian Finite Mixture Model
2.7. Construction of the Feature Gene-Based Risk Score Prognostic Model
2.8. Non-Negative Matrix Factorization
2.9. Subtype-Related Drug Sensitivity and Chemotherapeutic Response
2.10. Gene Expression-Related Stemness Index and Key Gene Expression
2.11. ssGSEA
2.12. Mutant Genes and DNA Methylation Analysis in HCC
2.13. Transcriptome Sequencing
2.14. Statistical Analysis
3. Results
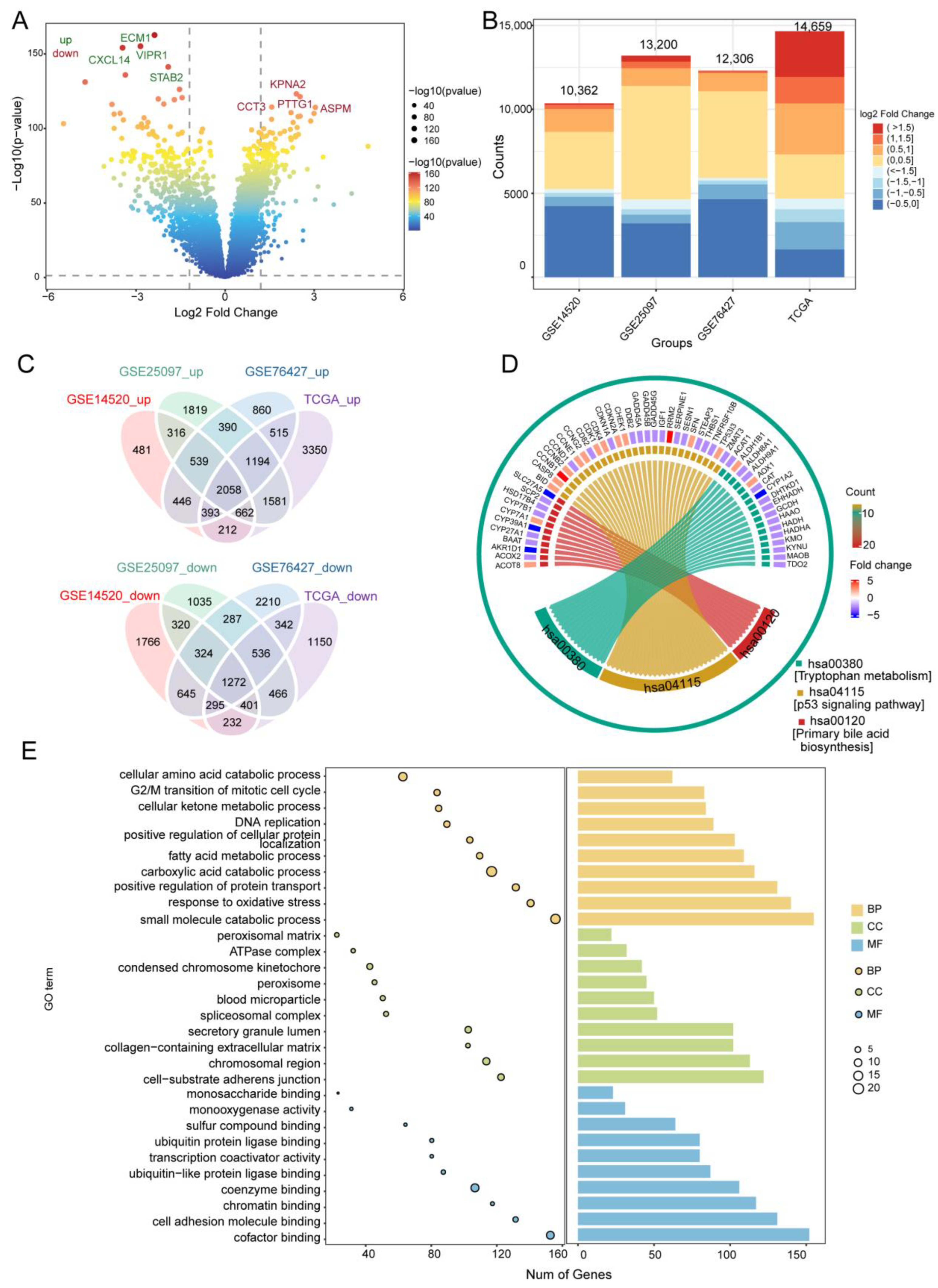
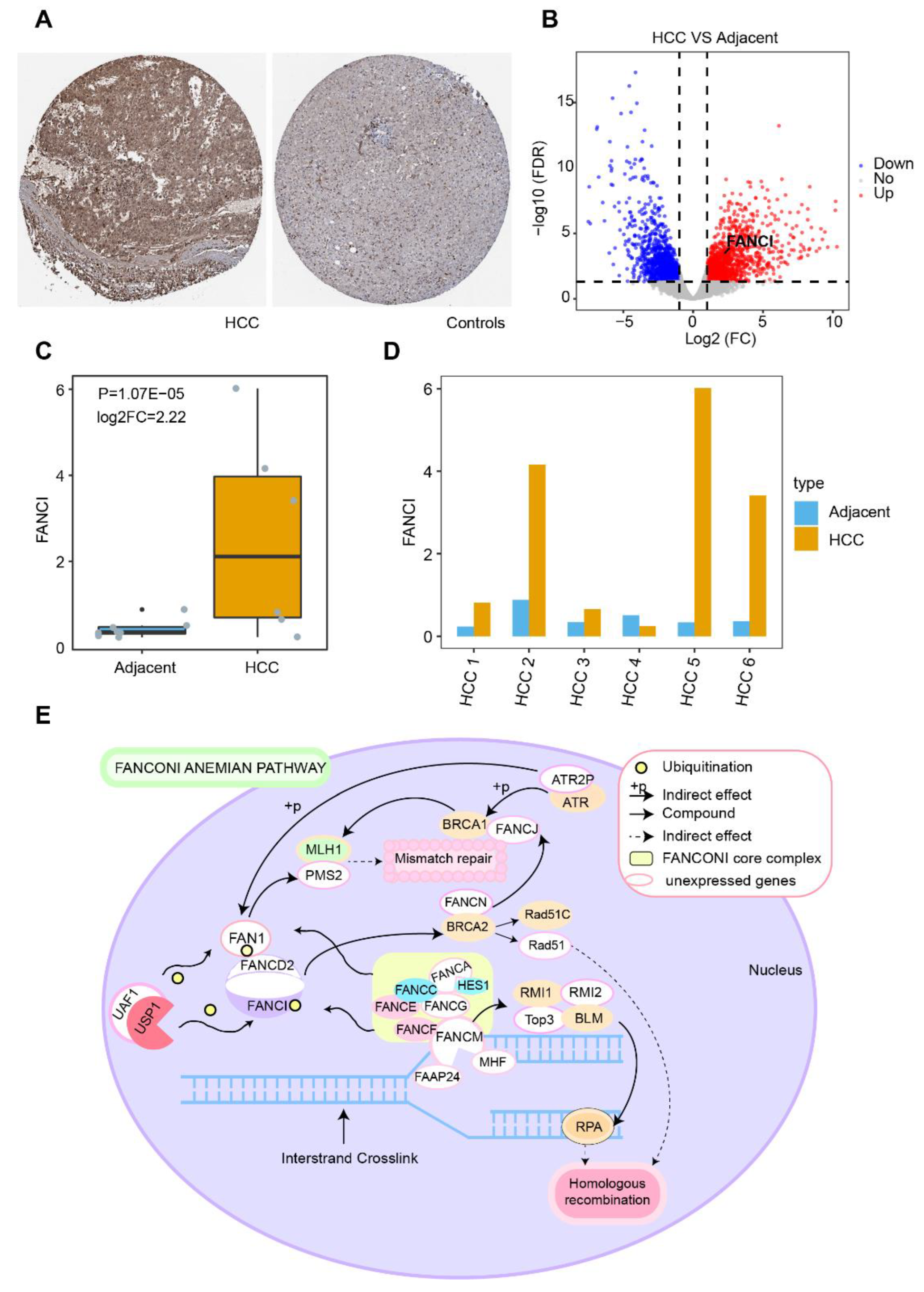
3.1. DEGs in HCC and Their Functional Enrichment
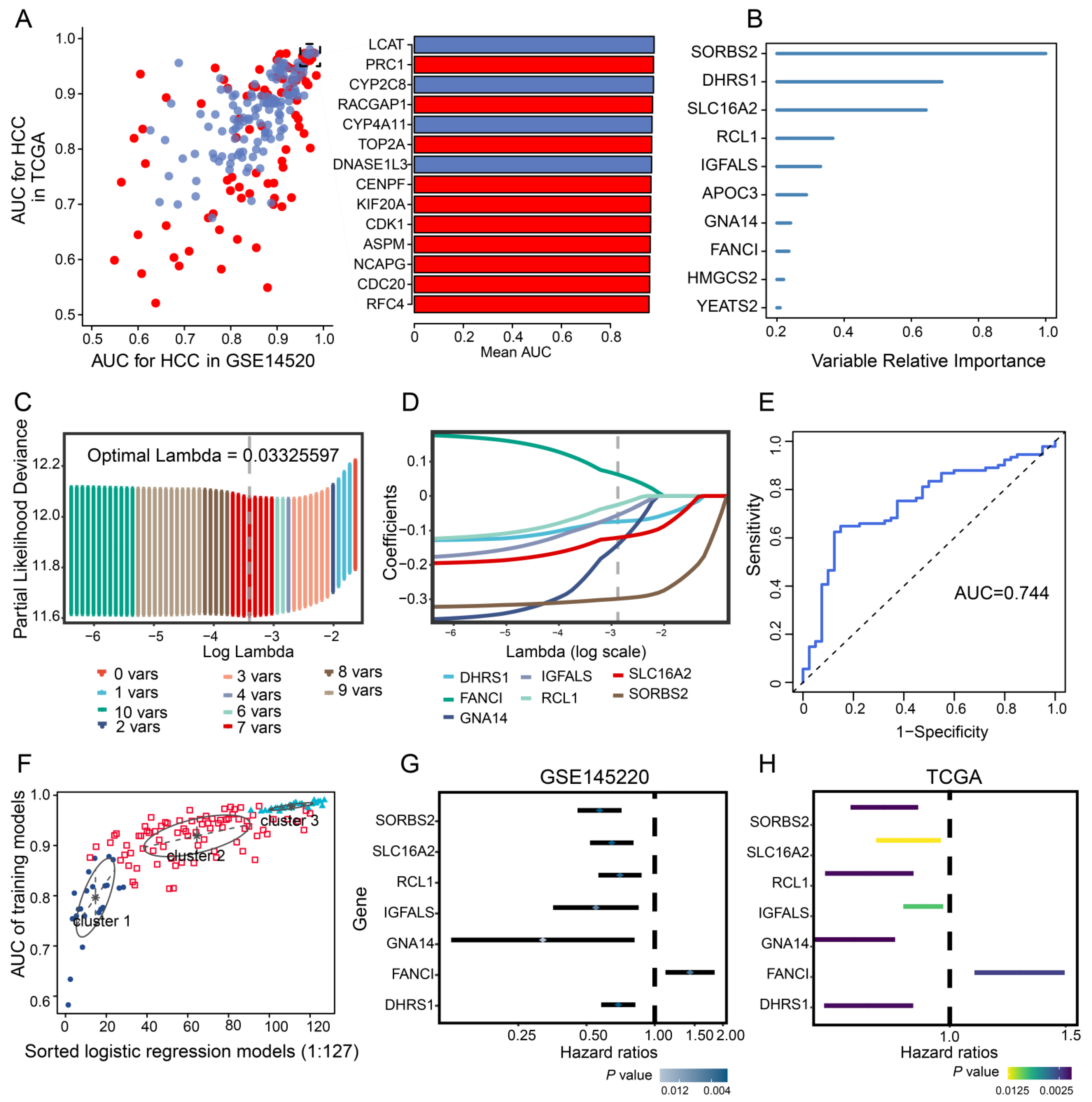
3.2. Identification of Diagnostic Genes in HCC
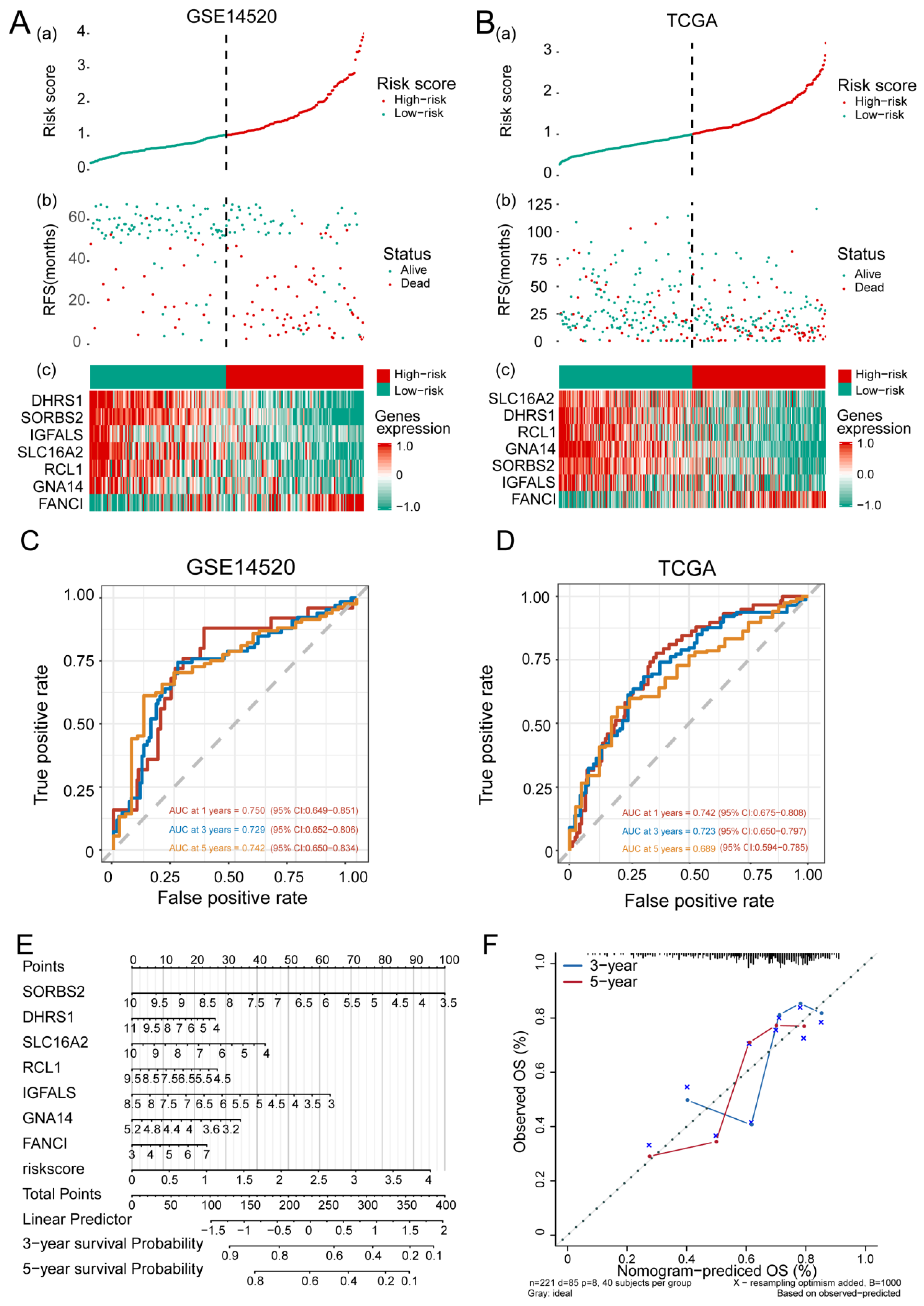
3.3. Feature Gene-Based Prognostic Risk Score as a Prognostic Tool in HCC
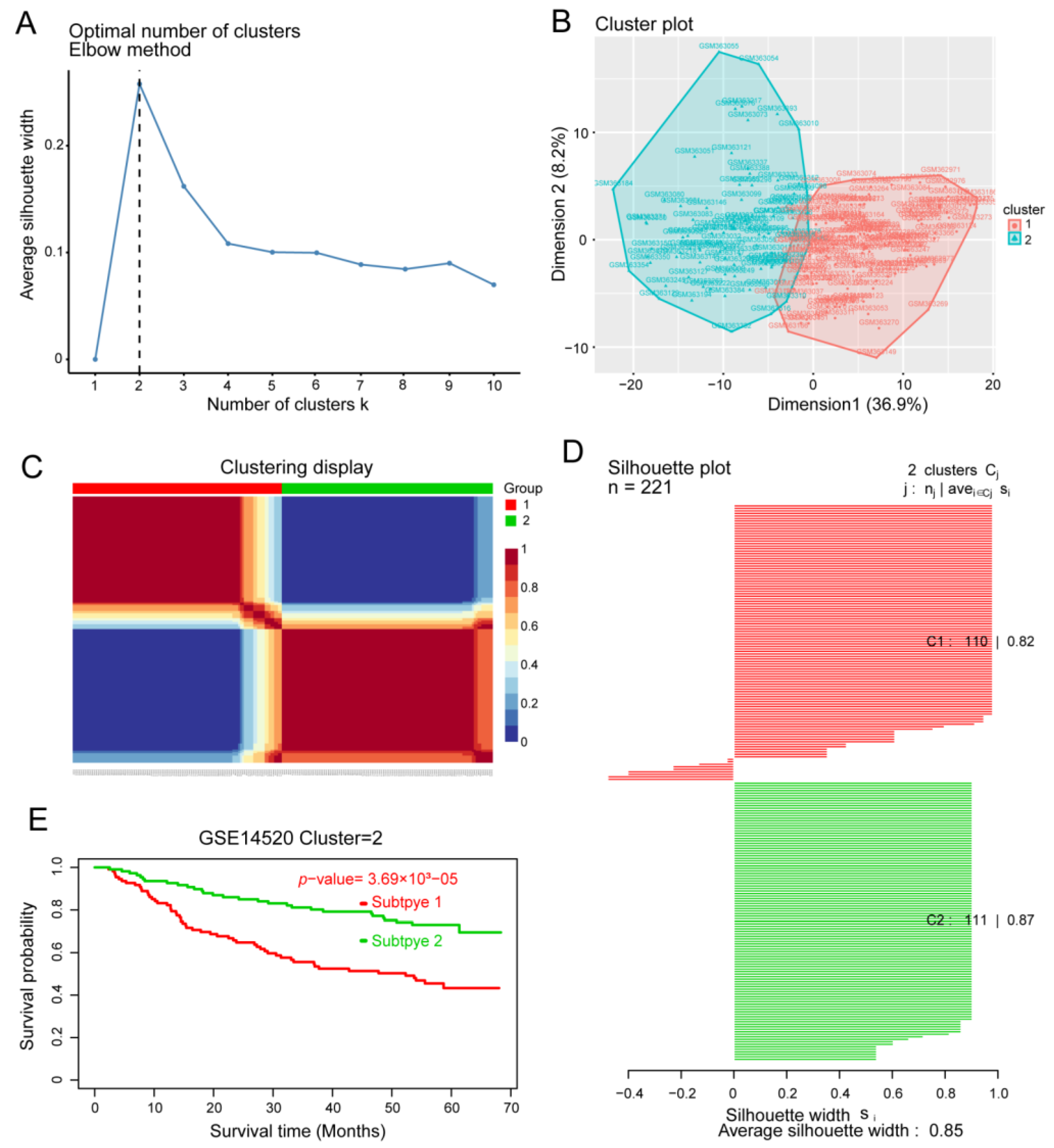
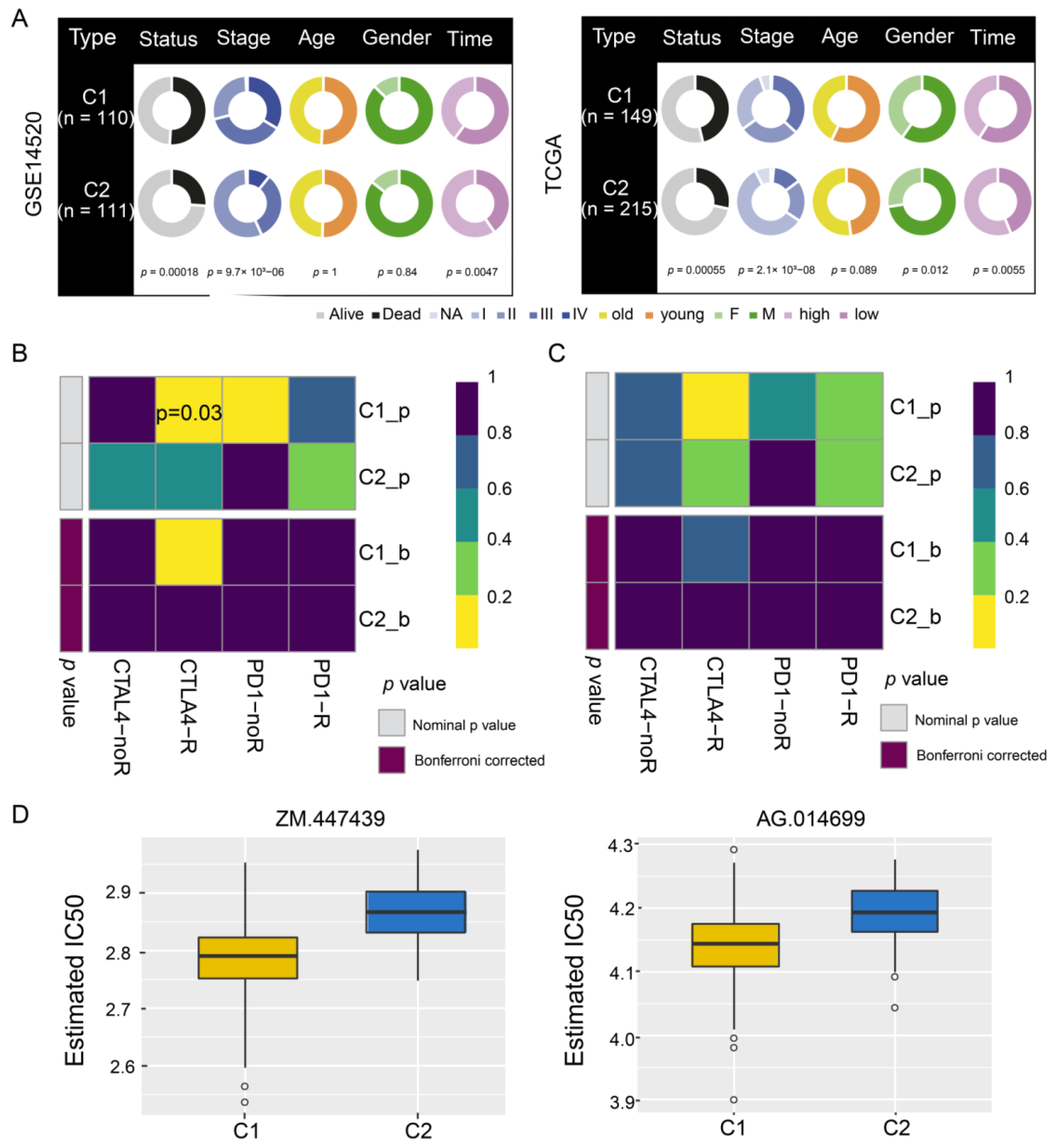
3.4. Identification of HCC Subtypes by NMF of Prognostic Genes
3.5. Sensitivity of HCC Subtypes to Immunotherapy and Chemotherapeutic Drugs
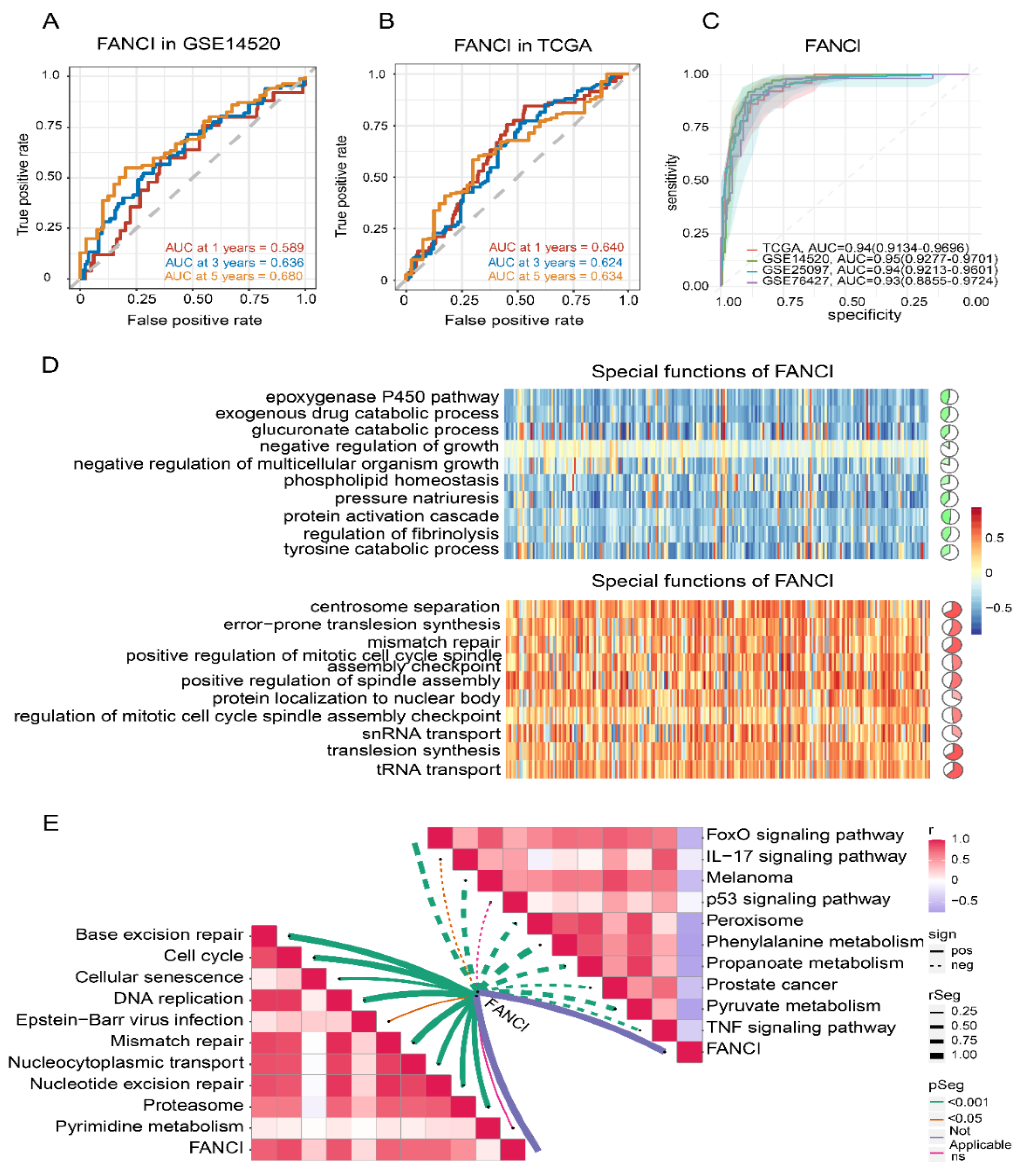
3.6. Stemness Index and FANCI Expression
3.7. Enrichment of FANCI in Biological Pathways
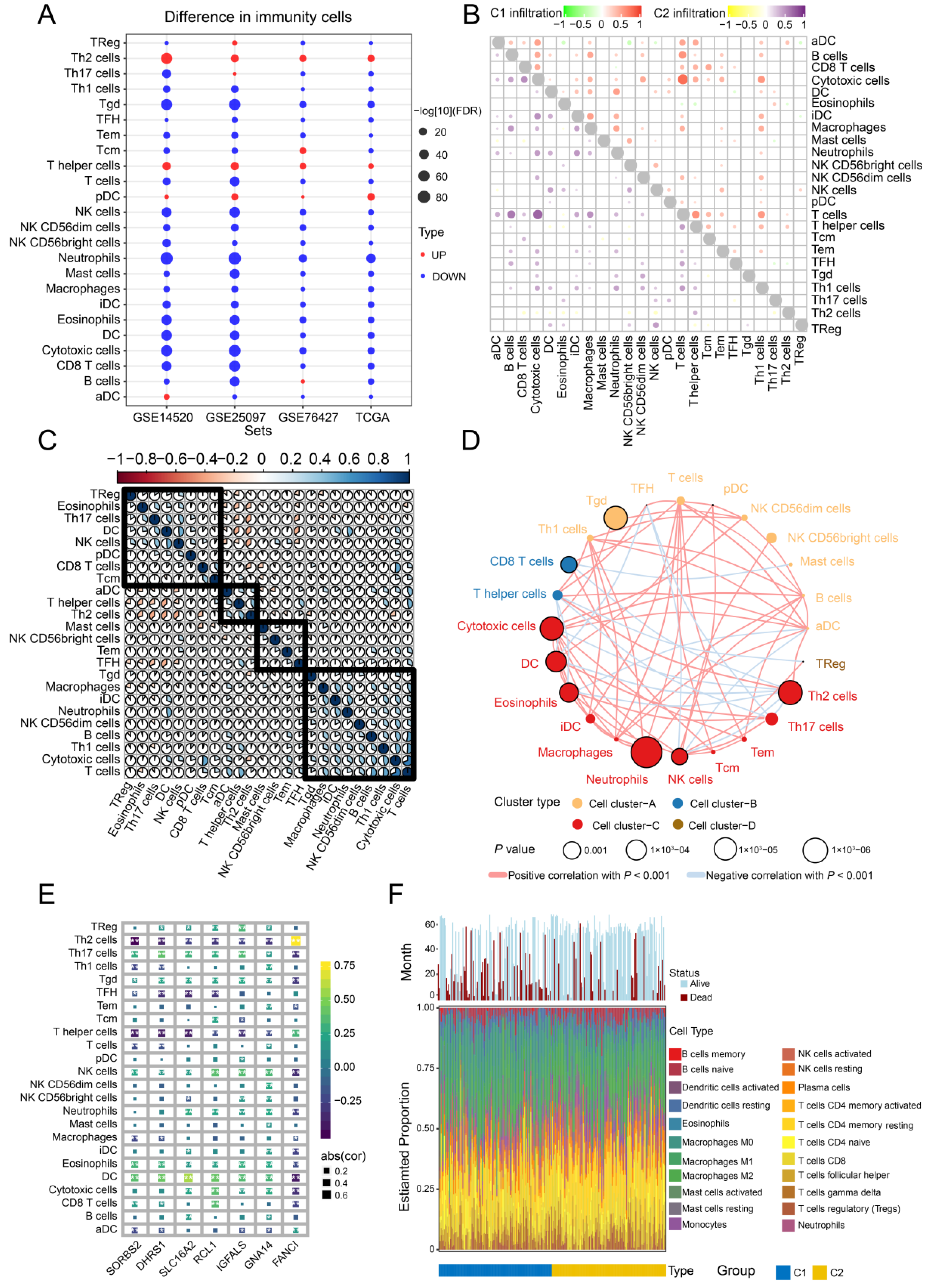
3.8. Immune Cell Infiltration
3.9. Somatic Mutations and DNA Methylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the receiver operating characteristic curve |
| BP | Biological process |
| CC | Cellular component |
| CS | Conditional survival |
| DEGs | Differentially expressed genes |
| GEO | Gene Expression Omnibus |
| GMM | Gaussian mixture model |
| GO | Gene Ontology |
| GSEA | Gene set enrichment analysis |
| HCC | Hepatocellular carcinoma |
| KM | Kaplan–Meier |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LIHC | Liver hepatocellular carcinoma |
| LASSO | Least absolute shrinkage and selection operator |
| MF | Molecular function |
| NMF | Nonnegative matrix factorization |
| OS | Overall survival |
| ssGSEA | Single-sample gene set enrichment analysis |
| TCGA | The Cancer Genome Atlas |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| TIMER | Tumor Immune Estimation Resource |
References
- Brunt, E.; Aishima, S.; Clavien, P.A.; Fowler, K.; Goodman, Z.; Gores, G.; Gouw, A.; Kagen, A.; Klimstra, D.; Komuta, M.; et al. cHCC-CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 2018, 68, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; Board, W.H.O.C.o.T.E. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global surveillance of cancer survival 1995-2009: Analysis of individual data for 25,676,887 patients from 279 population-based registries in 67 countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; Montella, M.; Polesel, J.; La Vecchia, C.; Crispo, A.; Dal Maso, L.; Casarin, P.; Izzo, F.; Tommasi, L.G.; Chemin, I.; et al. Hepatitis viruses, alcohol, and tobacco in the etiology of hepatocellular carcinoma in Italy. Cancer Epidemiol. Biomark. Prev. 2006, 15, 683–689. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Joliat, G.R.; Allemann, P.; Labgaa, I.; Demartines, N.; Halkic, N. Treatment and outcomes of recurrent hepatocellular carcinomas. Langenbecks Arch. Surg. 2017, 402, 737–744. [Google Scholar] [CrossRef]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Zhang, P.; Yang, Y.; Wen, F.; He, X.; Tang, R.; Du, Z.; Zhou, J.; Zhang, J.; Li, Q. Cost-effectiveness of sorafenib as a first-line treatment for advanced hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2015, 27, 853–859. [Google Scholar] [CrossRef]
- Moeini, A.; Sia, D.; Zhang, Z.; Camprecios, G.; Stueck, A.; Dong, H.; Montal, R.; Torrens, L.; Martinez-Quetglas, I.; Fiel, M.I.; et al. Mixed hepatocellular cholangiocarcinoma tumors: Cholangiolocellular carcinoma is a distinct molecular entity. J. Hepatol. 2017, 66, 952–961. [Google Scholar] [CrossRef]
- Bidkhori, G.; Benfeitas, R.; Klevstig, M.; Zhang, C.; Nielsen, J.; Uhlen, M.; Boren, J.; Mardinoglu, A. Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes. Proc. Natl. Acad. Sci. USA 2018, 115, E11874–E11883. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lou, Y.; Yang, J.; Wang, J.; Feng, J.; Zhao, Y.; Wang, L.; Huang, X.; Fu, Q.; Ye, M.; et al. Integrated multiomic analysis reveals comprehensive tumour heterogeneity and novel immunophenotypic classification in hepatocellular carcinomas. Gut 2019, 68, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Angell, H.; Galon, J. From the immune contexture to the Immunoscore: The role of prognostic and predictive immune markers in cancer. Curr. Opin. Immunol. 2013, 25, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. (Pozn.) 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets--update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Shah, M.M.; Meyer, B.I.; Rhee, K.; NeMoyer, R.E.; Lin, Y.; Tzeng, C.D.; Jabbour, S.K.; Kennedy, T.J.; Nosher, J.L.; Kooby, D.A.; et al. Conditional survival analysis of hepatocellular carcinoma. J. Surg. Oncol. 2020. [Google Scholar] [CrossRef]
- Cucchetti, A.; Piscaglia, F.; Cescon, M.; Ercolani, G.; Terzi, E.; Bolondi, L.; Zanello, M.; Pinna, A.D. Conditional survival after hepatic resection for hepatocellular carcinoma in cirrhotic patients. Clin. Cancer Res. 2012, 18, 4397–4405. [Google Scholar] [CrossRef]
- Nathan, H.; de Jong, M.C.; Pulitano, C.; Ribero, D.; Strub, J.; Mentha, G.; Gigot, J.F.; Schulick, R.D.; Choti, M.A.; Aldrighetti, L.; et al. Conditional survival after surgical resection of colorectal liver metastasis: An international multi-institutional analysis of 949 patients. J. Am. Coll. Surg. 2010, 210, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Blanche, P.; Dartigues, J.F.; Jacqmin-Gadda, H. Estimating and comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. [Google Scholar] [CrossRef] [PubMed]
- Barakat, A.; Mittal, A.; Ricketts, D.; Rogers, B.A. Understanding survival analysis: Actuarial life tables and the Kaplan-Meier plot. Br. J. Hosp. Med. (Lond.) 2019, 80, 642–646. [Google Scholar] [CrossRef]
- Reich, M.; Liefeld, T.; Gould, J.; Lerner, J.; Tamayo, P.; Mesirov, J.P. GenePattern 2.0. Nat. Genet. 2006, 38, 500–501. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e315. [Google Scholar] [CrossRef]
- Rhodes, D.R.; Kalyana-Sundaram, S.; Mahavisno, V.; Varambally, R.; Yu, J.; Briggs, B.B.; Barrette, T.R.; Anstet, M.J.; Kincead-Beal, C.; Kulkarni, P.; et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 2007, 9, 166–180. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS One 2016, 11, e0160519. [Google Scholar] [CrossRef]
- Koch, A.; Jeschke, J.; Van Criekinge, W.; van Engeland, M.; De Meyer, T. MEXPRESS update 2019. Nucleic Acids Res. 2019, 47, W561–W565. [Google Scholar] [CrossRef]
- Chen, J.; Rajasekaran, M.; Xia, H.; Zhang, X.; Kong, S.N.; Sekar, K.; Seshachalam, V.P.; Deivasigamani, A.; Goh, B.K.; Ooi, L.L.; et al. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/beta-catenin signalling pathway. Gut 2016, 65, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Goh, M.J.; Kang, W.; Jeong, W.K.; Sinn, D.H.; Gwak, G.Y.; Paik, Y.H.; Choi, M.S.; Lee, J.H.; Koh, K.C.; Paik, S.W. Prognostic significance of cachexia index in patients with advanced hepatocellular carcinoma treated with systemic chemotherapy. Sci. Rep. 2022, 12, 7647. [Google Scholar] [CrossRef] [PubMed]
- Rich, N.E.; Phen, S.; Desai, N.; Mittal, S.; Yopp, A.C.; Yang, J.D.; Marrero, J.A.; Iyengar, P.; Infante, R.E.; Singal, A.G. Cachexia is Prevalent in Patients With Hepatocellular Carcinoma and Associated With Worse Prognosis. Clin. Gastroenterol. Hepatol. 2022, 20, e1157–e1169. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wang, Z.; Ren, S.; Wang, W.; Duan, L.; Zhu, D.; Zhang, C.; Duan, Y. Prognostic biomarker MITD1 and its correlation with immune infiltrates in hepatocellular carcinoma (HCC). Int. Immunopharmacol. 2020, 81, 106222. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lei, Q.; Zhang, S.; Kong, L.; Qin, B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol. Rep. 2017, 38, 2607–2618. [Google Scholar] [CrossRef]
- Hou, Y.Q.; Yao, Y.; Bao, Y.L.; Song, Z.B.; Yang, C.; Gao, X.L.; Zhang, W.J.; Sun, L.G.; Yu, C.L.; Huang, Y.X.; et al. Juglanthraquinone C Induces Intracellular ROS Increase and Apoptosis by Activating the Akt/Foxo Signal Pathway in HCC Cells. Oxidative Med. Cell Longev. 2016, 2016, 4941623. [Google Scholar] [CrossRef]
- Marquardt, J.U.; Seo, D.; Andersen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stage acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef]
- Wang, M.; Wei, J.; Shang, F.; Zang, K.; Zhang, P. Down-regulation of lncRNA SNHG5 relieves sepsis-induced acute kidney injury by regulating the miR-374a-3p/TLR4/NF-kappaB pathway. J. Biochem. 2021, 169, 575–583. [Google Scholar] [CrossRef]
- Rowther, F.B.; Wei, W.; Dawson, T.P.; Ashton, K.; Singh, A.; Madiesse-Timchou, M.P.; Thomas, D.G.; Darling, J.L.; Warr, T. Cyclic nucleotide phosphodiesterase-1C (PDE1C) drives cell proliferation, migration and invasion in glioblastoma multiforme cells in vitro. Mol. Carcinog. 2016, 55, 268–279. [Google Scholar] [CrossRef]
- Kanda, M.; Nomoto, S.; Okamura, Y.; Nishikawa, Y.; Sugimoto, H.; Kanazumi, N.; Takeda, S.; Nakao, A. Detection of metallothionein 1G as a methylated tumor suppressor gene in human hepatocellular carcinoma using a novel method of double combination array analysis. Int. J. Oncol. 2009, 35, 477–483. [Google Scholar] [CrossRef]
- Neumann, O.; Kesselmeier, M.; Geffers, R.; Pellegrino, R.; Radlwimmer, B.; Hoffmann, K.; Ehemann, V.; Schemmer, P.; Schirmacher, P.; Lorenzo Bermejo, J.; et al. Methylome analysis and integrative profiling of human HCCs identify novel protumorigenic factors. Hepatology 2012, 56, 1817–1827. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Jiang, J.; Zhang, X.; Zhang, M.; Fu, Y. Comprehensive Analysis of IGFBPs as Biomarkers in Gastric Cancer. Front. Oncol. 2021, 11, 723131. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Situ, H.J.; Ma, W.C.; Liu, X.; Wang, L.L. Decreased DHRS1 expression is a novel predictor of poor survival in patients with hepatocellular carcinoma. Biomark. Med. 2021, 15, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Pan, Y.; Zheng, X. Identification of hub genes-based predictive model in hepatocellular carcinoma by robust rank aggregation and regression analysis. J. Cancer 2021, 12, 1884–1893. [Google Scholar] [CrossRef]
- Al-Hanawi, M.K.; Mwale, M.L.; Alshareef, N.; Qattan, A.M.N.; Angawi, K.; Almubark, R.; Alsharqi, O. Psychological Distress Amongst Health Workers and the General Public During the COVID-19 Pandemic in Saudi Arabia. Risk Manag. Healthc. Policy 2020, 13, 733–742. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, S.Q.; Chen, J.; Li, Z.B.; Chen, J.X.; Lu, Q.Q.; Han, Y.S.; Dai, W.; Xie, C.; Li, J.C. Identifying Prognostic Significance of RCL1 and Four-Gene Signature as Novel Potential Biomarkers in HCC Patients. J. Oncol. 2021, 2021, 5574150. [Google Scholar] [CrossRef]
- Xie, S.; Jiang, X.; Zhang, J.; Xie, S.; Hua, Y.; Wang, R.; Yang, Y. Identification of significant gene and pathways involved in HBV-related hepatocellular carcinoma by bioinformatics analysis. PeerJ 2019, 7, e7408. [Google Scholar] [CrossRef]
- Ren, X.; Cui, H.; Wu, J.; Zhou, R.; Wang, N.; Liu, D.; Xie, X.; Zhang, H.; Liu, D.; Ma, X.; et al. Identification of a combined apoptosis and hypoxia gene signature for predicting prognosis and immune infiltration in breast cancer. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Brownstein, C.A.; Smith, R.S.; Rodan, L.H.; Gorman, M.P.; Hojlo, M.A.; Garvey, E.A.; Li, J.; Cabral, K.; Bowen, J.J.; Rao, A.S.; et al. RCL1 copy number variants are associated with a range of neuropsychiatric phenotypes. Mol. Psychiatry 2021, 26, 1706–1718. [Google Scholar] [CrossRef]
- Xu, P.; Sun, D.; Gao, Y.; Jiang, Y.; Zhong, M.; Zhao, G.; Chen, J.; Wang, Z.; Liu, Q.; Hong, J.; et al. Germline mutations in a DNA repair pathway are associated with familial colorectal cancer. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Girard, E.; Eon-Marchais, S.; Olaso, R.; Renault, A.L.; Damiola, F.; Dondon, M.G.; Barjhoux, L.; Goidin, D.; Meyer, V.; Le Gal, D.; et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int. J. Cancer 2019, 144, 1962–1974. [Google Scholar] [CrossRef] [PubMed]
- Bonache, S.; Esteban, I.; Moles-Fernandez, A.; Tenes, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; Lopez-Fernandez, A.; et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef] [PubMed]
- Worns, M.A.; Galle, P.R. HCC therapies--lessons learned. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 447–452. [Google Scholar] [CrossRef]
- Shimada, S.; Mogushi, K.; Akiyama, Y.; Furuyama, T.; Watanabe, S.; Ogura, T.; Ogawa, K.; Ono, H.; Mitsunori, Y.; Ban, D.; et al. Comprehensive molecular and immunological characterization of hepatocellular carcinoma. EBioMedicine 2019, 40, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. [Google Scholar] [CrossRef]
- Foerster, F.; Hess, M.; Gerhold-Ay, A.; Marquardt, J.U.; Becker, D.; Galle, P.R.; Schuppan, D.; Binder, H.; Bockamp, E. The immune contexture of hepatocellular carcinoma predicts clinical outcome. Sci. Rep. 2018, 8, 5351. [Google Scholar] [CrossRef]
- Buonaguro, L.; Mauriello, A.; Cavalluzzo, B.; Petrizzo, A.; Tagliamonte, M. Immunotherapy in hepatocellular carcinoma. Ann. Hepatol. 2019, 18, 291–297. [Google Scholar] [CrossRef]
- Motta, J.M.; Rumjanek, V.M. Sensitivity of Dendritic Cells to Microenvironment Signals. J. Immunol. Res. 2016, 2016, 4753607. [Google Scholar] [CrossRef]
- Liu, P.; Chen, L.; Zhang, H. Natural Killer Cells in Liver Disease and Hepatocellular Carcinoma and the NK Cell-Based Immunotherapy. J. Immunol. Res. 2018, 2018, 1206737. [Google Scholar] [CrossRef]
- Liao, R.; Sun, J.; Wu, H.; Yi, Y.; Wang, J.X.; He, H.W.; Cai, X.Y.; Zhou, J.; Cheng, Y.F.; Fan, J.; et al. High expression of IL-17 and IL-17RE associate with poor prognosis of hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2013, 32, 3. [Google Scholar] [CrossRef]
- Hong, M.; Jiang, Z.; Zhou, Y.F. Effects of thermotherapy on Th1/Th2 cells in esophageal cancer patients treated with radiotherapy. Asian Pac. J. Cancer Prev. 2014, 15, 2359–2362. [Google Scholar] [CrossRef] [PubMed]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Lin, Y.; Gao, X.; Lu, L.; Huang, X.; Huang, S.; Bai, T.; Wu, G.; Luo, X.; Li, Y.; et al. Prognosis-Related Molecular Subtypes and Immune Features Associated with Hepatocellular Carcinoma. Cancers 2022, 14, 5721. https://doi.org/10.3390/cancers14225721
Ye J, Lin Y, Gao X, Lu L, Huang X, Huang S, Bai T, Wu G, Luo X, Li Y, et al. Prognosis-Related Molecular Subtypes and Immune Features Associated with Hepatocellular Carcinoma. Cancers. 2022; 14(22):5721. https://doi.org/10.3390/cancers14225721
Chicago/Turabian StyleYe, Jiazhou, Yan Lin, Xing Gao, Lu Lu, Xi Huang, Shilin Huang, Tao Bai, Guobin Wu, Xiaoling Luo, Yongqiang Li, and et al. 2022. "Prognosis-Related Molecular Subtypes and Immune Features Associated with Hepatocellular Carcinoma" Cancers 14, no. 22: 5721. https://doi.org/10.3390/cancers14225721
APA StyleYe, J., Lin, Y., Gao, X., Lu, L., Huang, X., Huang, S., Bai, T., Wu, G., Luo, X., Li, Y., & Liang, R. (2022). Prognosis-Related Molecular Subtypes and Immune Features Associated with Hepatocellular Carcinoma. Cancers, 14(22), 5721. https://doi.org/10.3390/cancers14225721