
Amino Acids in Cancer and Cachexia: An Integrated View

{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
1.1. Impact of AAs on Cancer Metabolism and Mitochondrial Function
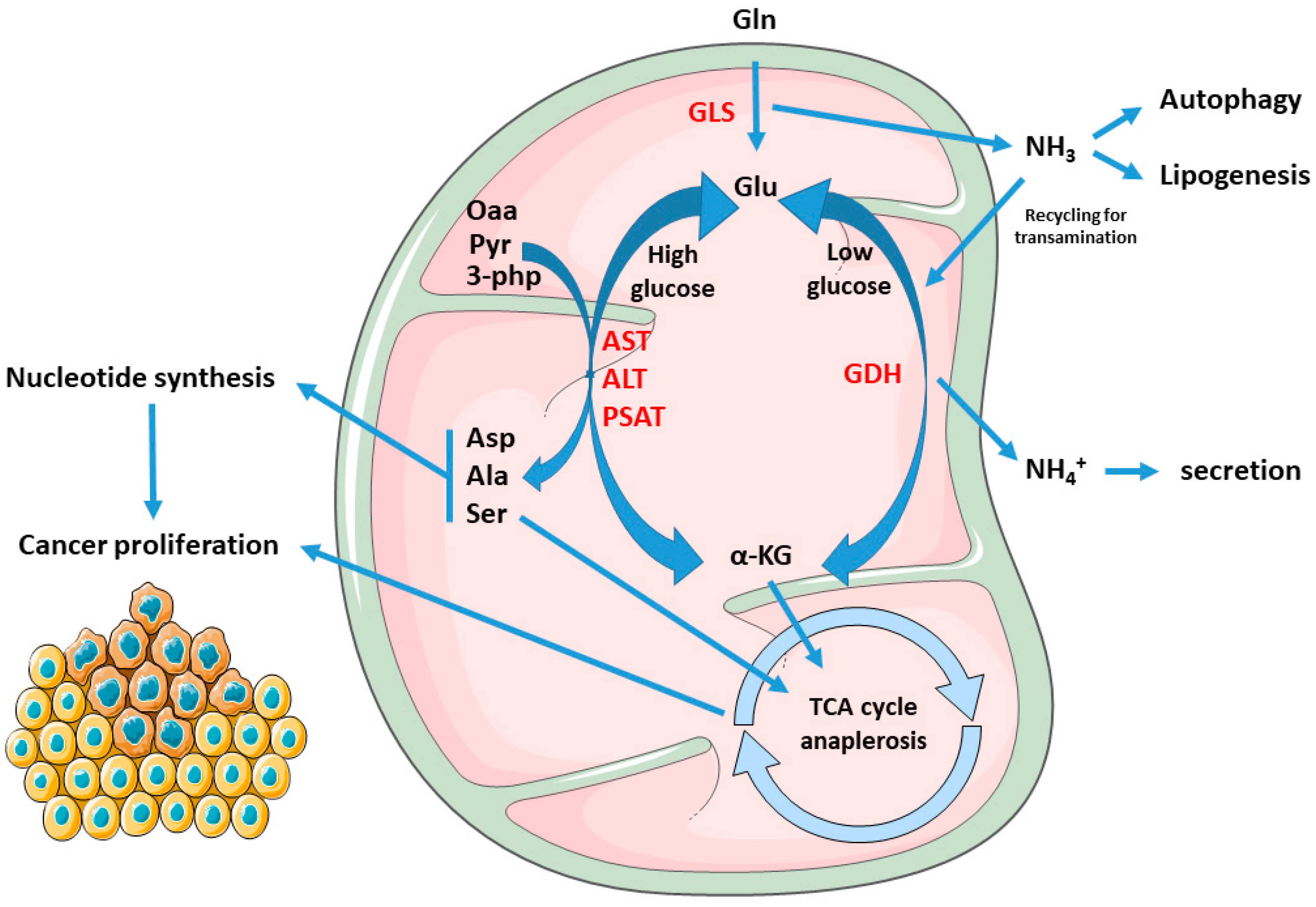
1.2. Cancer Mitochondria Support Biosynthesis of Macromolecules through Glutamine-Dependent Anaplerosis
1.3. Mitochondrial Electron Transport Chain and AA Metabolism
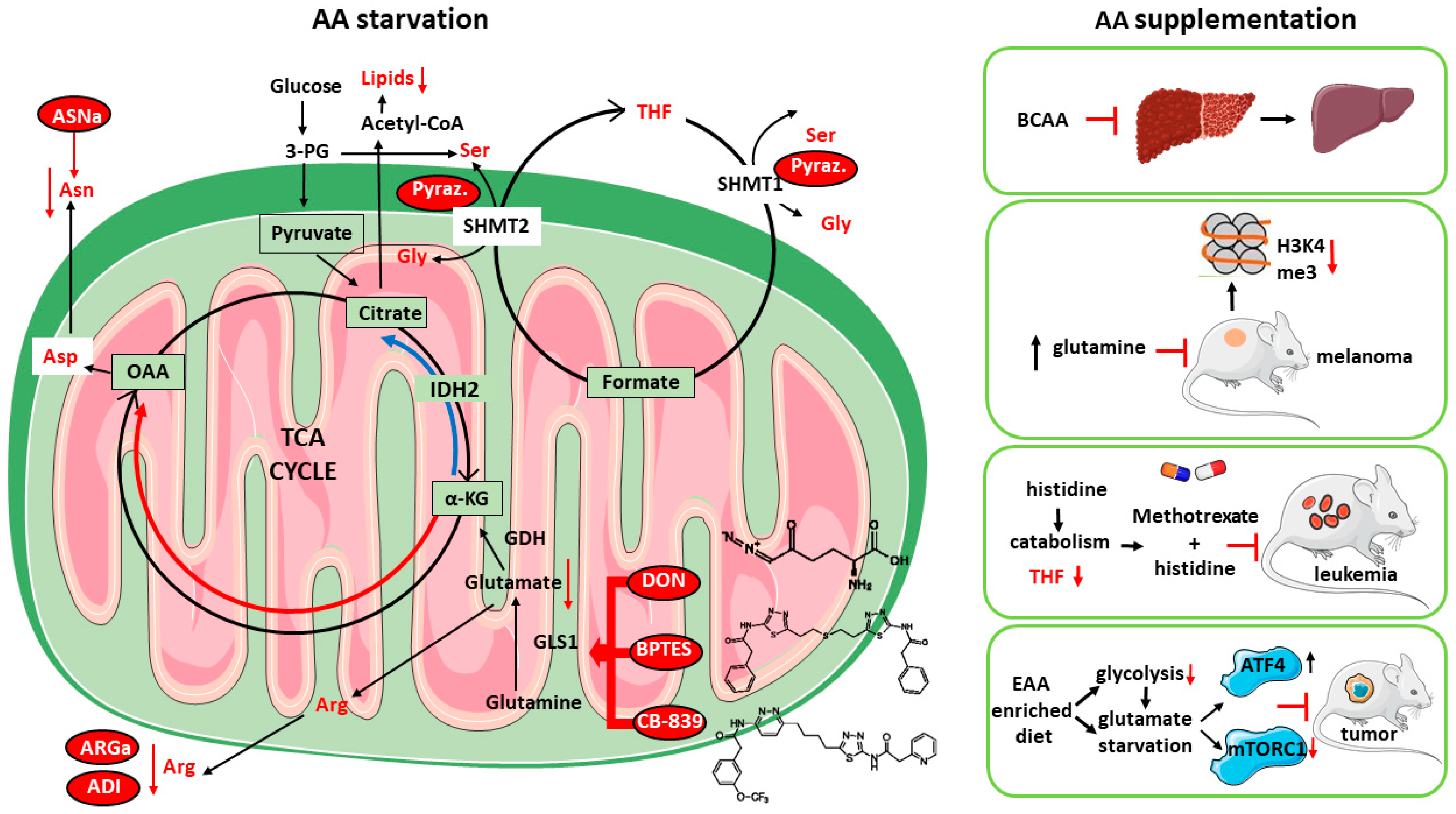
1.4. Targeting AA Metabolism as Anti-Cancer Strategy
2. Amino Acid Supplementation as Anticancer Therapy: Targeting Multiple and Complex Metabolic Networks
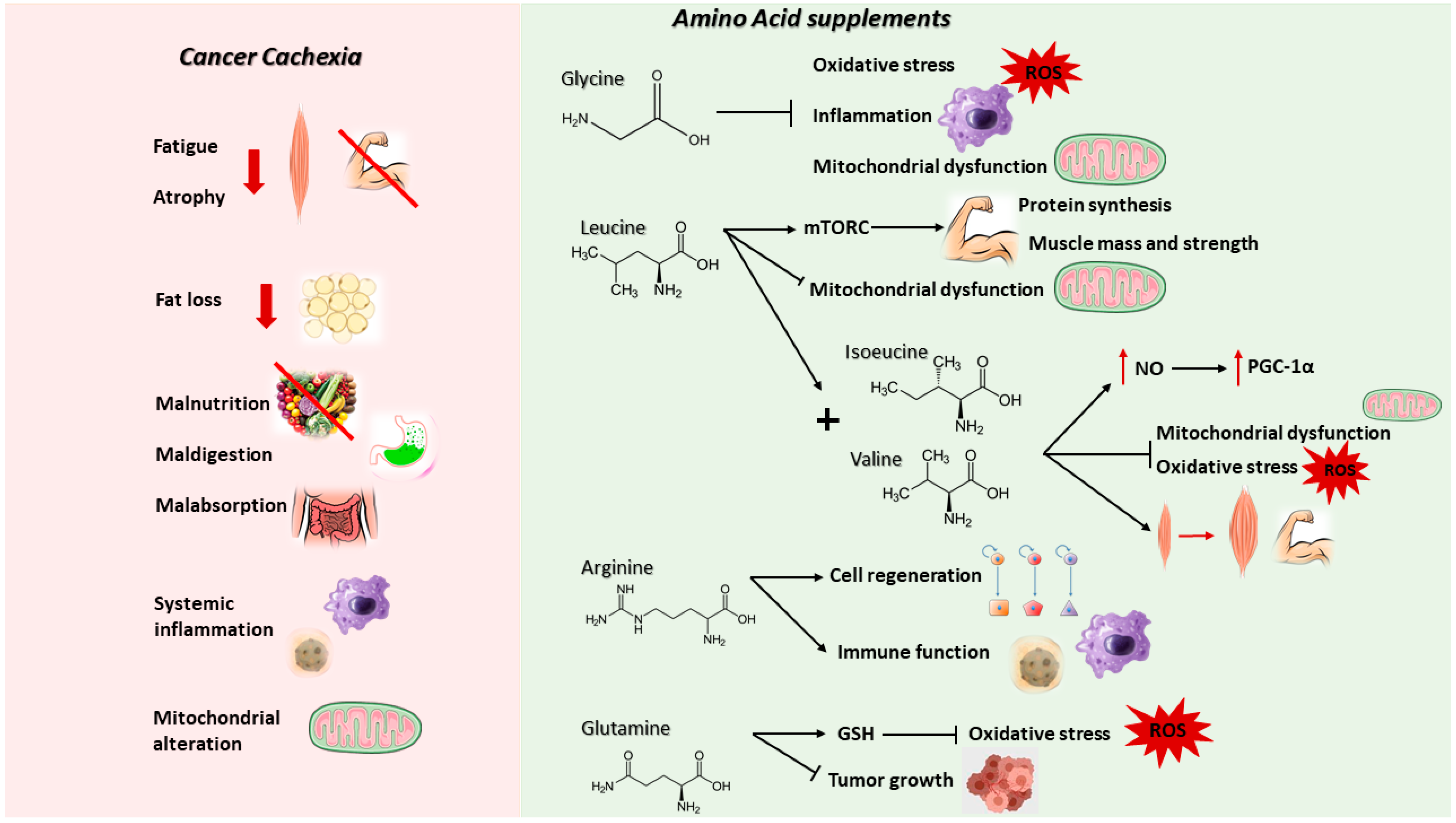
2.1. Cancer-Induced Cachexia
2.2. Nutritional (AA-Based) Anti-Cachexia Supportive Interventions
2.3. HMB
2.4. GLY
2.5. LEU
2.6. ARG
2.7. GLN
2.8. Essential AAs
3. Concluding Remarks and Open Challenges
Author Contributions
Funding
Conflicts of Interest
References
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. The metabolism of carcinoma cells 1. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Hosios, A.M.; Hecht, V.C.; Danai, L.V.; Johnson, M.O.; Rathmell, J.C.; Steinhauser, M.L.; Manalis, S.R.; Vander Heiden, M.G. Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 2016, 36, 540–549. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, cs222570. [Google Scholar] [CrossRef]
- Kim, E. Mechanisms of amino acid sensing in mTOR signaling pathway. Nutr. Res. Pract. 2009, 3, 64. [Google Scholar] [CrossRef]
- Shen, J.Z.; Wu, G.; Guo, S. Amino Acids in Autophagy: Regulation and Function. Adv. Exp. Med. Biol. 2021, 1332, 51–66. [Google Scholar] [CrossRef]
- Eagle, B.H. The Minimum Vitamin Requirements of the L and I~A Cells in Tissue Culture, The Production of Specific Vitamin Deficiencies, and Their Cure (From the Section on Experimental Therapeutics, Laboratory of Infergious Diseases, National Microbiological Institute, National Institutes of Health, * Bethesda). J. Exp. Med. 1955, 102, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Reitzer, L.J.; Wice, B.M.; Kennell, D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979, 254, 2669–2676. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10, 728. [Google Scholar] [CrossRef]
- Cardoso, H.J.; Figueira, M.I.; Vaz, C.V.; Carvalho, T.M.A.; Brás, L.A.; Madureira, P.A.; Oliveira, P.J.; Sardão, V.A.; Socorro, S. Glutaminolysis is a metabolic route essential for survival and growth of prostate cancer cells and a target of 5α-dihydrotestosterone regulation. Cell Oncol. 2021, 44, 385–403. [Google Scholar] [CrossRef]
- Van Geldermalsen, M.; Wang, Q.; Nagarajah, R.; Marshall, A.D.; Thoeng, A.; Gao, D.; Ritchie, W.; Feng, Y.; Bailey, C.G.; Deng, N.; et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 2016, 35, 3201–3208. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.H.M. Vander Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef]
- Chendong, Y.; Sudderth, J.; Tuyen, D.; Bachoo, R.G.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef]
- Choo, A.Y.; Kim, S.G.; Vander Heiden, M.G.; Mahoney, S.J.; Vu, H.; Yoon, S.O.; Cantley, L.C.; Blenis, J. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol. Cell 2010, 38, 487–499. [Google Scholar] [CrossRef]
- Eng, C.H.; Yu, K.; Lucas, J.; White, E.; Abraham, R.T. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci. Signal. 2010, 3, ra31. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Geng, F.; Li, Z.; Zhong, Y.; Wang, H.; Cheng, X.; Zhao, Y.; Mo, X.; Horbinski, C.; Duan, W.; et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat. Metab. 2022, 4, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shestov, A.A.; Swain, P.; Yang, C.; Parker, S.J.; Wang, Q.A.; Terada, L.S.; Adams, N.D.; McCabe, M.T.; Pietrak, B.; et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 2016, 73, 389–400. [Google Scholar] [CrossRef]
- Metallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011, 481, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenasedependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef]
- Gameiro, P.A.; Yang, J.; Metelo, A.M.; Pérez-Carro, R.; Baker, R.; Wang, Z.; Arreola, A.; Rathmell, W.K.; Olumi, A.; López-Larrubia, P.; et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013, 17, 372–385. [Google Scholar] [CrossRef]
- Dasgupta, S.; Putluri, N.; Long, W.; Zhang, B.; Wang, J.; Kaushik, A.K.; Arnold, J.M.; Bhowmik, S.K.; Stashi, E.; Brennan, C.A.; et al. Coactivator SRC-2’dependent metabolic reprogramming mediates prostate cancer survival and metastasis. J. Clin. Invest. 2015, 125, 1174–1188. [Google Scholar] [CrossRef]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Vander Heiden, M.G. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Pachnis, P.; Wu, Z.; Faubert, B.; Tasdogan, A.; Gu, W.; Shelton, S.; Solmonson, A.; Rao, A.D.; Kaushik, A.K.; Rogers, T.J.; et al. In vivo isotope tracing reveals a requirement for the electron transport chain in glucose and glutamine metabolism by tumors. Sci. Adv. 2022, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Heiden, M.G.V.; et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef]
- Cardaci, S.; Zheng, L.; Mackay, G.; Van Den Broek, N.J.F.; Mackenzie, E.D.; Nixon, C.; Stevenson, D.; Tumanov, S.; Bulusu, V.; Kamphorst, J.J.; et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 2015, 17, 1317–1326. [Google Scholar] [CrossRef]
- Altea-Manzano, P.; Vandekeere, A.; Edwards-Hicks, J.; Roldan, M.; Abraham, E.; Lleshi, X.; Guerrieri, A.N.; Berardi, D.; Wills, J.; Junior, J.M.; et al. Reversal of mitochondrial malate dehydrogenase 2 enables anaplerosis via redox rescue in respiration-deficient cells. Mol. Cell 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Diehl, F.F.; Lewis, C.A.; Fiske, B.P.; Vander Heiden, M.G. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat. Metab. 2019, 1, 861–867. [Google Scholar] [CrossRef]
- Kilberg, M.S.; Balasubramanian, M.; Fu, L.; Shan, J. The transcription factor network associated with the amino acid response in mammalian cells. Adv. Nutr. 2012, 3, 295–306. [Google Scholar] [CrossRef]
- Bao, X.R.; Ong, S.E.; Goldberger, O.; Peng, J.; Sharma, R.; Thompson, D.A.; Vafai, S.B.; Cox, A.G.; Marutani, E.; Ichinose, F.; et al. Mitochondrial dysfunction remodels one-carbon metabolism in human cells. Elife 2016, 5, e10575. [Google Scholar] [CrossRef]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef]
- Pollari, S.; Käkönen, S.M.; Edgren, H.; Wolf, M.; Kohonen, P.; Sara, H.; Guise, T.; Nees, M.; Kallioniemi, O. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 2011, 125, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Snell, K. Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzyme Regul. 1984, 22, 325–400. [Google Scholar] [CrossRef]
- Locasale, J.W.; Grassian, A.R.; Melman, T.; Lyssiotis, C.A.; Mattaini, K.R.; Bass, A.J.; Heffron, G.; Metallo, C.M.; Muranen, T.; Sharfi, H.; et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 2011, 43, 869–874. [Google Scholar] [CrossRef] [PubMed]
- Zahra, K.; Dey, T.; Ashish; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Chaneton, B.; Hillmann, P.; Zheng, L.; Martin, A.C.L.; Maddocks, O.D.K.; Chokkathukalam, A.; Coyle, J.E.; Jankevics, A.; Holding, F.P.; Vousden, K.H.; et al. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 2012, 491, 458–462. [Google Scholar] [CrossRef]
- Lee, G.Y.; Haverty, P.M.; Li, L.; Kljavin, N.M.; Bourgon, R.; Lee, J.; Stern, H.; Modrusan, Z.; Seshagiri, S.; Zhang, Z.; et al. Comparative oncogenomics identifies psmb4 and shmt2 as potential cancer driver genes. Cancer Res. 2014, 74, 3114–3126. [Google Scholar] [CrossRef]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K.; et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621.e5. [Google Scholar] [CrossRef]
- Morscher, R.J.; Ducker, G.S.; Li, S.H.J.; Mayer, J.A.; Gitai, Z.; Sperl, W.; Rabinowitz, J.D. Mitochondrial translation requires folate-dependent tRNA methylation. Nature 2018, 554, 128–132. [Google Scholar] [CrossRef]
- Lucas, S.; Chen, G.; Aras, S.; Wang, J. Serine catabolism is essential to maintain mitochondrial respiration in mammalian cells. Life Sci. Alliance 2018, 1, e201800036. [Google Scholar] [CrossRef] [PubMed]
- Tani, H.; Ohnishi, S.; Shitara, H.; Mito, T.; Yamaguchi, M.; Yonekawa, H.; Hashizume, O.; Ishikawa, K.; Nakada, K.; Hayashi, J.I. Mice deficient in the Shmt2 gene have mitochondrial respiration defects and are embryonic lethal. Sci. Rep. 2018, 8, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, F.; Fan, N.; Zhou, C.; Li, D.; Macvicar, T.; Dong, Q.; Bruns, C.J.; Zhao, Y. Targeting Glutaminolysis: New Perspectives to Understand Cancer Development and Novel Strategies for Potential Target Therapies. Front. Oncol. 2020, 10, 589508. [Google Scholar] [CrossRef] [PubMed]
- Sallan, S.E.; Hitchcock-Bryan, S.; Gelber, R.; Cassady, J.R.; Frei, E.; Nathan, D.G. Influence of Intensive Asparaginase in the Treatment of Childhood Non-T-Cell Acute Lymphoblastic Leukemia. Cancer Res. 1983, 43, 5601–5607. [Google Scholar] [PubMed]
- Nachman, J.B.; Harland, N.S.; Sensel, M.G.; Trigg, M.E.; Cherlow, J.M.; Lunkens, J.N.; Wolfl, L.; Uckun, F.M.; Gaynon, P.S. Augmented Post-Induction Therapy for Children With High-Risk To Initial Therapy. N. Engl. J. Med. 1998, 338, 1663–1671. [Google Scholar] [CrossRef]
- Cools, J. Improvements in the survival of children and adolescents with acute lymphoblastic leukemia. Haematologica 2012, 97, 635. [Google Scholar] [CrossRef]
- Curran, E.; Stock, W. How I treat acute lymphoblastic leukemia in older adolescents and young adults. Blood 2015, 125, 3702–3710. [Google Scholar] [CrossRef]
- Bach, S.J.; Simon-Reuss, I. Arginase, an antimitotic agent in tissue culture. Biochim. Biophys. Acta 1953, 11, 396–402. [Google Scholar] [CrossRef]
- Feun, L.G.; Marini, A.; Walker, G.; Elgart, G.; Moffat, F.; Rodgers, S.E.; Wu, C.J.; You, M.; Wangpaichitr, M.; Kuo, M.T.; et al. Negative argininosuccinate synthetase expression in melanoma tumours may predict clinical benefit from arginine-depleting therapy with pegylated arginine deiminase. Br. J. Cancer 2012, 106, 1481–1485. [Google Scholar] [CrossRef]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and Distribution of Argininosuccinate Synthetase Deficiency in Human Cancers: A Method for Identifying Cancers Sensitive to Arginine Deprivation. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Klabatsa, A.; Pallaska, A.; Sheaff, M.; Smith, P.; Crook, T.; Grimshaw, M.J.; Steele, J.P.; Rudd, R.M.; Balkwill, F.R.; et al. In vivo loss of expression of argininosuccinate synthetase in malignant pleural mesothelioma is a biomarker for susceptibility to arginine depletion. Clin. Cancer Res. 2006, 12, 7126–7131. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.Y.; Shim, Y.J.; Kim, E.H.; Lee, J.H.; Won, N.H.; Kim, J.H.; Park, I.S.; Yoon, D.K.; Min, B.H. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int. J. Cancer 2007, 120, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.; Lamb, J.; Smith, S.; Wheatley, D.N. Single amino acid (arginine) deprivation: Rapid and selective death of cultured transformed and malignant cells. Br. J. Cancer 2000, 83, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Kawatra, A.; Dhankhar, R.; Gulati, P. Microbial arginine deiminase: A multifaceted green catalyst in biomedical sciences. Int. J. Biol. Macromol. 2022, 196, 151–162. [Google Scholar] [CrossRef]
- Farber, S.; Diamond, L.K.; Mercer, R.D.; Sylvester, R.F.; Wolff, J.A. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-aminopteroyl-glutamic acid. N. Engl. J. Med. 1948, 238, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Howard, S.C.; McCormick, J.; Pui, C.-H.; Buddington, R.K.; Harvey, R.D. Preventing and Managing Toxicities of High-Dose Methotrexate. Oncologist 2016, 21, 1471–1482. [Google Scholar] [CrossRef]
- Dekhne, A.S.; Hou, Z.; Gangjee, A.; Matherly, L.H. Therapeutic targeting of mitochondrial one-carbon metabolism in cancer. Mol. Cancer Ther. 2020, 19, 2245–2255. [Google Scholar] [CrossRef]
- Marani, M.; Paone, A.; Fiascarelli, A.; Macone, A.; Gargano, M.; Rinaldo, S.; Giardina, G.; Pontecorvi, V.; Koes, D.; McDermott, L.; et al. A pyrazolopyran derivative preferentially inhibits the activity of human cytosolic serine hydroxymethyltransferase and induces cell death in lung cancer cells. Oncotarget 2016, 7, 4570–4583. [Google Scholar] [CrossRef]
- Tajan, M.; Hennequart, M.; Cheung, E.C.; Zani, F.; Hock, A.K.; Legrave, N.; Maddocks, O.D.K.; Ridgway, R.A.; Athineos, D.; Suárez-Bonnet, A.; et al. Serine synthesis pathway inhibition cooperates with dietary serine and glycine limitation for cancer therapy. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Vilstrup, H.; Amodio, P.; Bajaj, J.; Cordoba, J.; Ferenci, P.; Mullen, K.D.; Weissenborn, K.; Wong, P. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study Of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014, 60, 715–735. [Google Scholar] [CrossRef]
- Maddocks, O.D.K.; Athineos, D.; Cheung, E.C.; Lee, P.; Zhang, T.; Van Den Broek, N.J.F.; Mackay, G.M.; Labuschagne, C.F.; Gay, D.; Kruiswijk, F.; et al. Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 2017, 544, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.T.; Chen, H.H.W.; Feun, L.G.; Savaraj, N. Targeting the proline–glutamine–asparagine–arginine metabolic axis in amino acid starvation cancer therapy. Pharmaceuticals 2021, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Hayaishi, S.; Chung, H.; Kudo, M.; Ishikawa, E.; Takita, M.; Ueda, T.; Kitai, S.; Inoue, T.; Yada, N.; Hagiwara, S.; et al. Oral branched-chain amino acid granules reduce the incidence of hepatocellular carcinoma and improve event-free survival in patients with liver cirrhosis. Dig. Dis. 2011, 29, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Poon, R.T.P.; Yu, W.C.; Fan, S.T.; Wong, J. Long-term oral branched chain amino acids in patients undergoing chemoembolization for hepatocellular carcinoma: A randomized trial. Aliment. Pharmacol. Ther. 2004, 19, 779–788. [Google Scholar] [CrossRef]
- Park, J.G.; Tak, W.Y.; Park, S.Y.; Kweon, Y.O.; Chung, W.J.; Jang, B.K.; Bae, S.H.; Lee, H.J.; Jang, J.Y.; Suk, K.T.; et al. Effects of Branched-Chain Amino Acid (BCAA) Supplementation on the Progression of Advanced Liver Disease: A Korean Nationwide, Multicenter, Prospective, Observational, Cohort Study. Nutrients 2020, 12, 1429. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Bianchi, G.; Merli, M.; Amodio, P.; Panella, C.; Loguercio, C.; Fanelli, F.R.; Abbiati, R. Nutritional supplementation with branched-chain amino acids in advanced cirrhosis: A double-blind, randomized trial. Gastroenterology 2003, 124, 1792–1801. [Google Scholar] [CrossRef]
- Ishak Gabra, M.B.; Yang, Y.; Li, H.; Senapati, P.; Hanse, E.A.; Lowman, X.H.; Tran, T.Q.; Zhang, L.; Doan, L.T.; Xu, X.; et al. Dietary glutamine supplementation suppresses epigenetically-activated oncogenic pathways to inhibit melanoma tumour growth. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Kanarek, N.; Keys, H.R.; Cantor, J.R.; Lewis, C.A.; Chan, S.H.; Kunchok, T.; Abu-Remaileh, M.; Freinkman, E.; Schweitzer, L.D.; Sabatini, D.M.; et al. Histidine catabolism is a major determinant of methotrexate sensitivity. Nature 2018, 559, 632–636. [Google Scholar] [CrossRef]
- Ragni, M.; Ruocco, C.; Tedesco, L.; Carruba, M.O.; Valerio, A.; Nisoli, E. An amino acid-defined diet impairs tumour growth in mice by promoting endoplasmic reticulum stress and mTOR inhibition. Mol. Metab. 2022, 60, 101478. [Google Scholar] [CrossRef]
- Hamill, M.J.; Afeyan, R.; Chakravarthy, M.V.; Tramontin, T. Endogenous Metabolic Modulators: Emerging Therapeutic Potential of Amino Acids. iScience 2020, 23, 101628. [Google Scholar] [CrossRef]
- Saha, S.K.; Riazul Islam, S.M.; Abdullah-Al-Wadud, M.; Islam, S.; Ali, F.; Park, K.S. Multiomics analysis reveals that GLS and GLS2 differentially modulate the clinical outcomes of cancer. J. Clin. Med. 2019, 8, 355. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Nishiyama, M.; Ishizaki, S. Branched-chain amino acids prevent insulin-induced hepatic tumor cell proliferation by inducing apoptosis through mTORC1 and mTORC2-dependent mechanisms. J. Cell Physiol. 2012, 227, 2097–2105. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Baum, S.J.; Gunn, N.T.; Younes, Z.H.; Kohli, A.; Patil, R.; Koziel, M.J.; Chera, H.; Zhao, J.; Chakravarthy, M.V. Safety, Tolerability, and Biologic Activity of AXA1125 and AXA1957 in Subjects with Nonalcoholic Fatty Liver Disease. Am. J. Gastroenterol. 2021, 116, 2399–2409. [Google Scholar] [CrossRef] [PubMed]
- Argilés, J.M.; Busquets, S.; Stemmler, B.; López-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Penna, F.; Ballarò, R.; Beltrà, M.; De Lucia, S.; Castillo, L.G.; Costelli, P. The Skeletal Muscle as an Active Player Against Cancer Cachexia. Front. Physiol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Robinder, J.S.; Dhillon, M.D.M.; Sarfaraz, H.M. Pathogenesis and management of sarcopenia. Clin. Geriatr. Med. 2017, 33, 17–36. [Google Scholar] [CrossRef]
- Bruggeman, A.R.; Kamal, A.H.; LeBlanc, T.W.; Ma, J.D.; Baracos, V.E.; Roeland, E.J. Cancer cachexia: Beyond weight loss. J. Oncol. Pract. 2016, 12, 1163–1171. [Google Scholar] [CrossRef]
- Fearon, K.; Arends, J.; Baracos, V. Understanding the mechanisms and treatment options in cancer cachexia. Nat. Rev. Clin. Oncol. 2013, 10, 90–99. [Google Scholar] [CrossRef]
- van der Meij, B.S.; Teleni, L.; Engelen, M.P.K.J.; Deutz, N.E.P. Amino acid kinetics and the response to nutrition in patients with cancer. Int. J. Radiat. Biol. 2019, 95, 480–492. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 601. [Google Scholar] [CrossRef]
- Zanetti, M.; Cappellari, G.G.; Barazzoni, R.; Sanson, G. The impact of protein supplementation targeted at improving muscle mass on strength in cancer patients: A scoping review. Nutrients 2020, 12, 2099. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef]
- Dao, T.; Green, A.E.; Kim, Y.A.; Bae, S.J.; Ha, K.T.; Gariani, K.; Lee, M.R.; Menzies, K.J.; Ryu, D. Sarcopenia and muscle aging: A brief overview. Endocrinol. Metab. 2020, 35, 716–732. [Google Scholar] [CrossRef]
- Beltrà, M.; Pin, F.; Ballarò, R.; Costelli, P.; Penna, F. Mitochondrial Dysfunction in Cancer Cachexia: Impact on Muscle Health and Regeneration. Cells 2021, 10, 3150. [Google Scholar] [CrossRef] [PubMed]
- Sandri, M. Protein breakdown in cancer cachexia. Semin. Cell Dev. Biol. 2016, 54, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Baccino, F.M.; Costelli, P. Coming back: Autophagy in cachexia. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 241–246. [Google Scholar] [CrossRef]
- Argilés, J.M.; López-Soriano, F.J.; Busquets, S. Muscle wasting in cancer: The role of mitochondria. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Costamagna, D.; Pin, F.; Camperi, A.; Fanzani, A.; Chiarpotto, E.M.E.M.; Cavallini, G.; Bonelli, G.; Baccino, F.M.F.M.; Costelli, P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am. J. Pathol. 2013, 182, 1367–1378. [Google Scholar] [CrossRef]
- Ballarò, R.; Beltrà, M.; De Lucia, S.; Pin, F.; Ranjbar, K.; Hulmi, J.J.; Costelli, P.; Penna, F. Moderate exercise in mice improves cancer plus chemotherapy-induced muscle wasting and mitochondrial alterations. FASEB J. 2019, 33, 5482–5494. [Google Scholar] [CrossRef]
- Sadeghi, M.; Keshavarz-Fathi, M.; Baracos, V.; Arends, J.; Mahmoudi, M.; Rezaei, N. Cancer cachexia: Diagnosis, assessment, and treatment. Crit. Rev. Oncol. Hematol. 2018, 127, 91–104. [Google Scholar] [CrossRef]
- Madeddu, C.; Maccio, A.; Mantovani, G. Multitargeted treatment of cancer cachexia. Crit. Rev. Oncog. 2012, 17, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Penna, F.; Ballarò, R.; Beltrá, M.; De Lucia, S.; Costelli, P. Modulating Metabolism to Improve Cancer-Induced Muscle Wasting. Oxid. Med. Cell Longev. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Muscaritoli, M.; Arends, J.; Bachmann, P.; Baracos, V.; Barthelemy, N.; Bertz, H.; Bozzetti, F.; Hütterer, E.; Isenring, E.; Kaasa, S.; et al. ESPEN practical guideline: Clinical Nutrition in cancer. Clin. Nutr. 2021, 40, 2898–2913. [Google Scholar] [CrossRef] [PubMed]
- McCurdy, B.; Nejatinamini, S.; Debenham, B.J.; Álvarez-Camacho, M.; Kubrak, C.; Wismer, W.V.; Mazurak, V.C. Meeting Minimum ESPEN Energy Recommendations Is Not Enough to Maintain Muscle Mass in Head and Neck Cancer Patients. Nutrients 2019, 11, 2743. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, C.; Segala, A.; Valerio, A.; Nisoli, E. Essential amino acid formulations to prevent mitochondrial dysfunction and oxidative stress. Curr. Opin. Clin. Nutr. Metab. Care 2021, 24, 88–95. [Google Scholar] [CrossRef]
- Soares, J.D.P.; Howell, S.L.; Teixeira, F.J.; Pimentel, G.D. Dietary Amino Acids and Immunonutrition Supplementation in Cancer-Induced Skeletal Muscle Mass Depletion: A Mini-Review. Curr. Pharm. Des. 2020, 26, 970–978. [Google Scholar] [CrossRef]
- Prado, C.M.; Orsso, C.E.; Pereira, S.L.; Atherton, P.J.; Deutz, N.E.P. Effects of β-hydroxy β-methylbutyrate (HMB) supplementation on muscle mass, function, and other outcomes in patients with cancer: A systematic review. J. Cachexia. Sarcopenia Muscle 2022, 13, 1623–1641. [Google Scholar] [CrossRef]
- Molfino, A.; Gioia, G.; Rossi Fanelli, F.; Muscaritoli, M. Beta-hydroxy-beta-methylbutyrate supplementation in health and disease: A systematic review of randomized trials. Amino Acids 2013, 45, 1273–1292. [Google Scholar] [CrossRef]
- Koopman, R.; Caldow, M.K.; Ham, D.J.; Lynch, G.S. Glycine metabolism in skeletal muscle: Implications for metabolic homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2017, 20, 237–242. [Google Scholar] [CrossRef]
- de Bandt, J.P. Leucine and Mammalian Target of Rapamycin-Dependent Activation of Muscle Protein Synthesis in Aging. J. Nutr. 2016, 146, 2616S–2624S. [Google Scholar] [CrossRef]
- Beaudry, A.G.; Law, M.L. Leucine Supplementation in Cancer Cachexia: Mechanisms and a Review of the Pre-Clinical Literature. Nutrients 2022, 14, 2824. [Google Scholar] [CrossRef] [PubMed]
- Martins, H.A.; Bazotte, R.B.; Vicentini, G.E.; Lima, M.M.; Guarnier, F.A.; Hermes-Uliana, C.; Frez, F.C.V.; Bossolani, G.D.P.; Fracaro, L.; Fávaro, L.D.S.; et al. l-Glutamine supplementation promotes an improved energetic balance in Walker-256 tumor–bearing rats. Tumor Biol. 2017, 39, 1010428317695960. [Google Scholar] [CrossRef]
- Martins, H.A.; Sehaber, C.C.; Hermes-Uliana, C.; Mariani, F.A.; Guarnier, F.A.; Vicentini, G.E.; Bossolani, G.D.P.; Jussani, L.A.; Lima, M.M.; Bazotte, R.B.; et al. Supplementation with l-glutamine prevents tumor growth and cancer-induced cachexia as well as restores cell proliferation of intestinal mucosa of Walker-256 tumor-bearing rats. Amino Acids 2016, 48, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- D’Antona, G.; Ragni, M.; Cardile, A.; Tedesco, L.; Dossena, M.; Bruttini, F.; Caliaro, F.; Corsetti, G.; Bottinelli, R.; Carruba, M.O.; et al. Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 2010, 12, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.R.; Anthony, S.; Johanssen, V.A.; Yeo, T.; Sealey, M.; Yates, A.G.; Smith, C.F.; Claridge, T.D.W.; Nicholson, B.D.; Moreland, J.A.; et al. Metabolomic Biomarkers in Blood Samples Identify Cancers in a Mixed Population of Patients with Nonspecific Symptoms. Clin. Cancer Res. 2022, 28, 1651–1661. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, E.; Ricciardi, W.; Cadoni, G.; Arzani, D.; Petrelli, L.; Paludetti, G.; Brennan, P.; Luce, D.; Stucker, I.; Matsuo, K.; et al. Recommended European Society of Parenteral and Enteral Nutrition protein and energy intakes and weight loss in patients with head and neck cancer. Head Neck 2014, 36, 1391. [Google Scholar] [CrossRef]
- Ala, M. Tryptophan metabolites modulate inflammatory bowel disease and colorectal cancer by affecting immune system. Int. Rev. Immunol. 2022, 41, 326–345. [Google Scholar] [CrossRef]
- Ni, Y.; Lohinai, Z.; Heshiki, Y.; Dome, B.; Moldvay, J.; Dulka, E.; Galffy, G.; Berta, J.; Weiss, G.J.; Sommer, M.O.A.; et al. Distinct composition and metabolic functions of human gut microbiota are associated with cachexia in lung cancer patients. ISME J. 2021, 15, 3207–3220. [Google Scholar] [CrossRef]
- Genton, L.; Mareschal, J.; Charretier, Y.; Lazarevic, V.; Bindels, L.B.; Schrenzel, J. Targeting the Gut Microbiota to Treat Cachexia. Front. Cell Infect. Microbiol. 2019, 9, 305. [Google Scholar] [CrossRef]
- Vernieri, C.; Fucà, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frigè, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [Google Scholar] [CrossRef]
- Seki, T.; Yang, Y.; Sun, X.; Lim, S.; Xie, S.; Guo, Z.; Xiong, W.; Kuroda, M.; Sakaue, H.; Hosaka, K.; et al. Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature 2022, 608, 421–428. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ragni, M.; Fornelli, C.; Nisoli, E.; Penna, F. Amino Acids in Cancer and Cachexia: An Integrated View. Cancers 2022, 14, 5691. https://doi.org/10.3390/cancers14225691
Ragni M, Fornelli C, Nisoli E, Penna F. Amino Acids in Cancer and Cachexia: An Integrated View. Cancers. 2022; 14(22):5691. https://doi.org/10.3390/cancers14225691
Chicago/Turabian StyleRagni, Maurizio, Claudia Fornelli, Enzo Nisoli, and Fabio Penna. 2022. "Amino Acids in Cancer and Cachexia: An Integrated View" Cancers 14, no. 22: 5691. https://doi.org/10.3390/cancers14225691
APA StyleRagni, M., Fornelli, C., Nisoli, E., & Penna, F. (2022). Amino Acids in Cancer and Cachexia: An Integrated View. Cancers, 14(22), 5691. https://doi.org/10.3390/cancers14225691