
A Novel Immune-Related Gene Prognostic Index (IRGPI) in Pancreatic Adenocarcinoma (PAAD) and Its Implications in the Tumor Microenvironment
 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
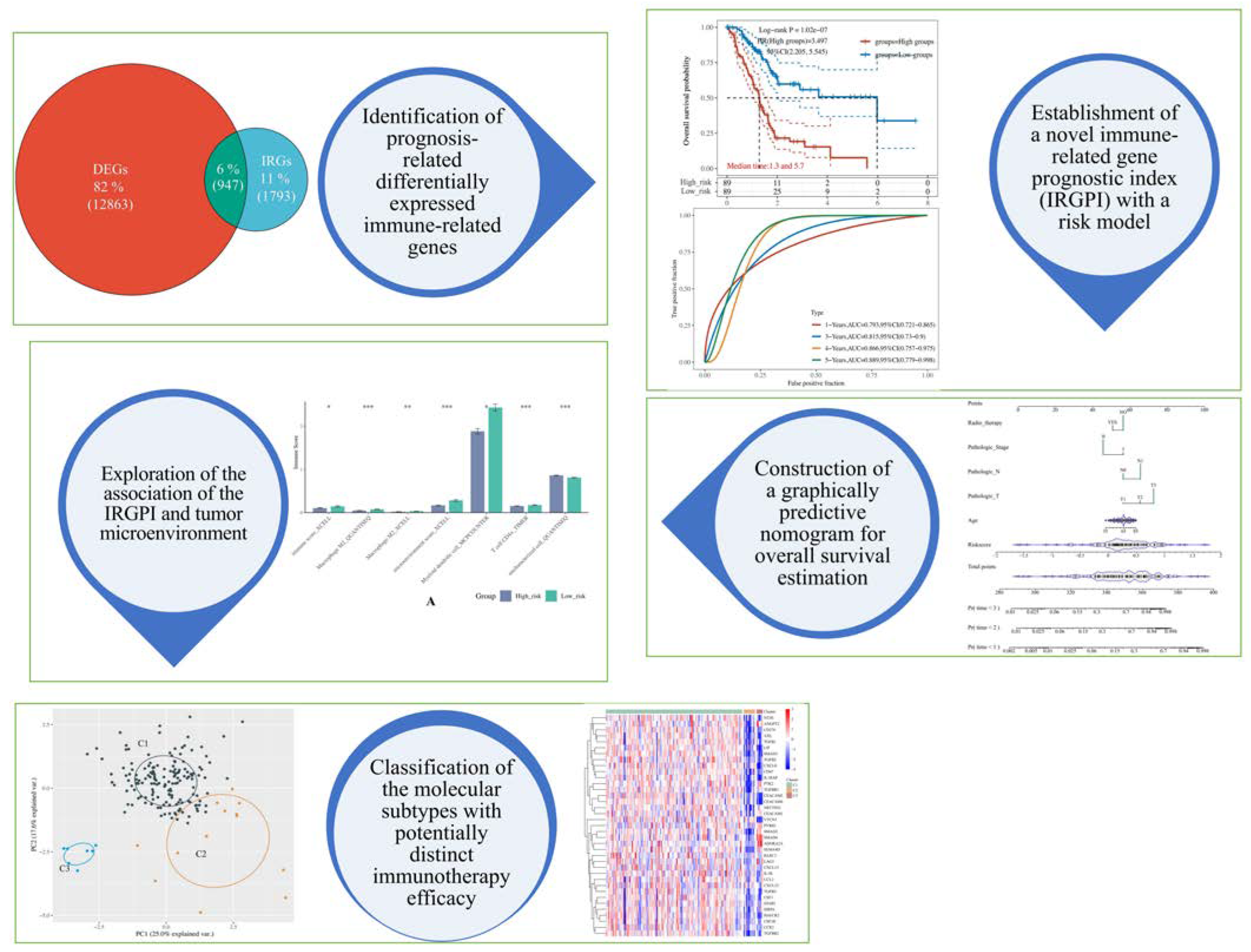
2.2. Identification of Prognosis-Related Differentially Expressed Immune-Related Genes (DEIRGs)
2.3. Establishment of Immune-Related Gene Prognostic Index (IRGPI) and Risk Model
2.4. Comparison with Previously Proposed Predictors
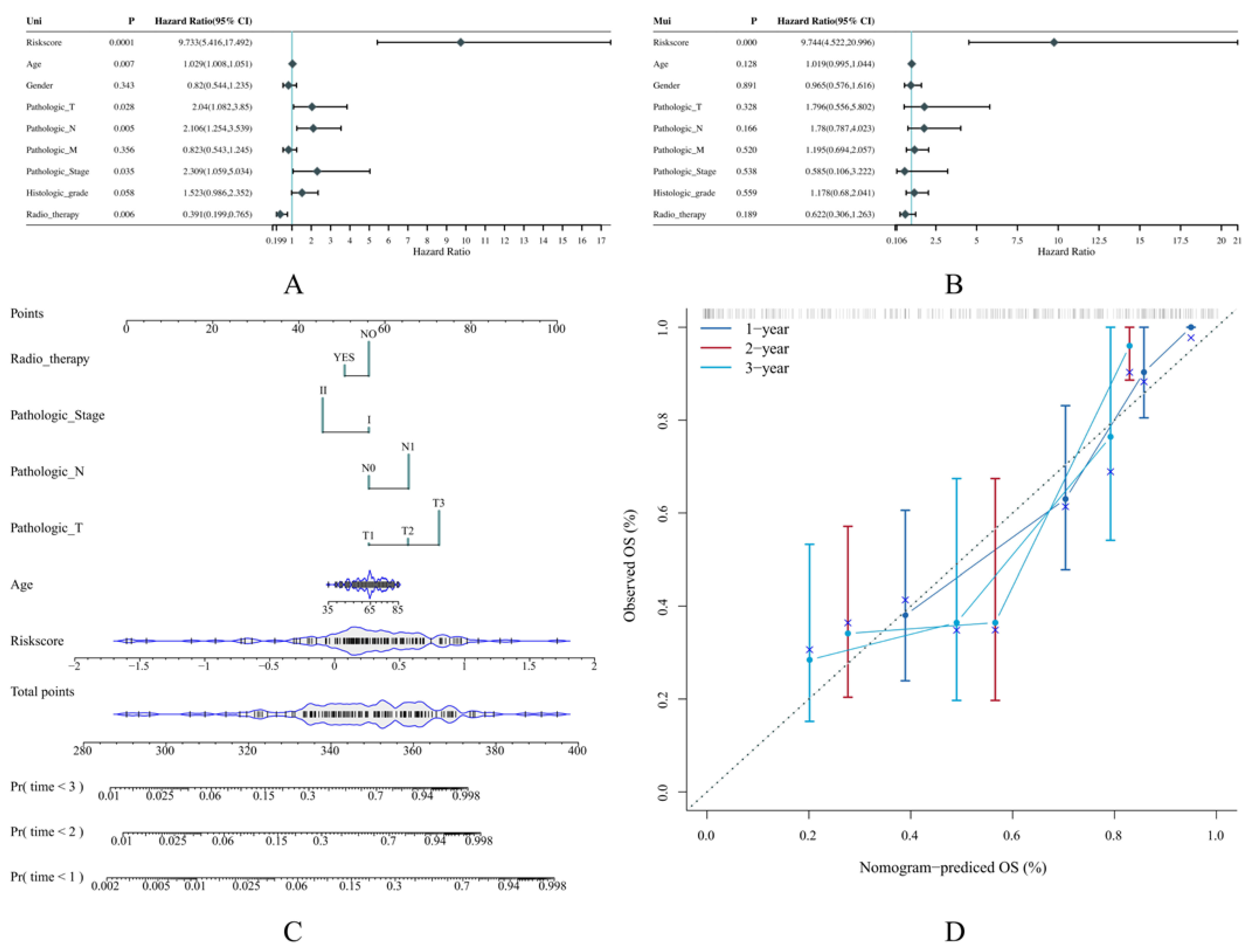
2.5. Construction of Predictive Nomogram According to the Risk Model
2.6. Assessment of the Immunological Tumor Microenvironment
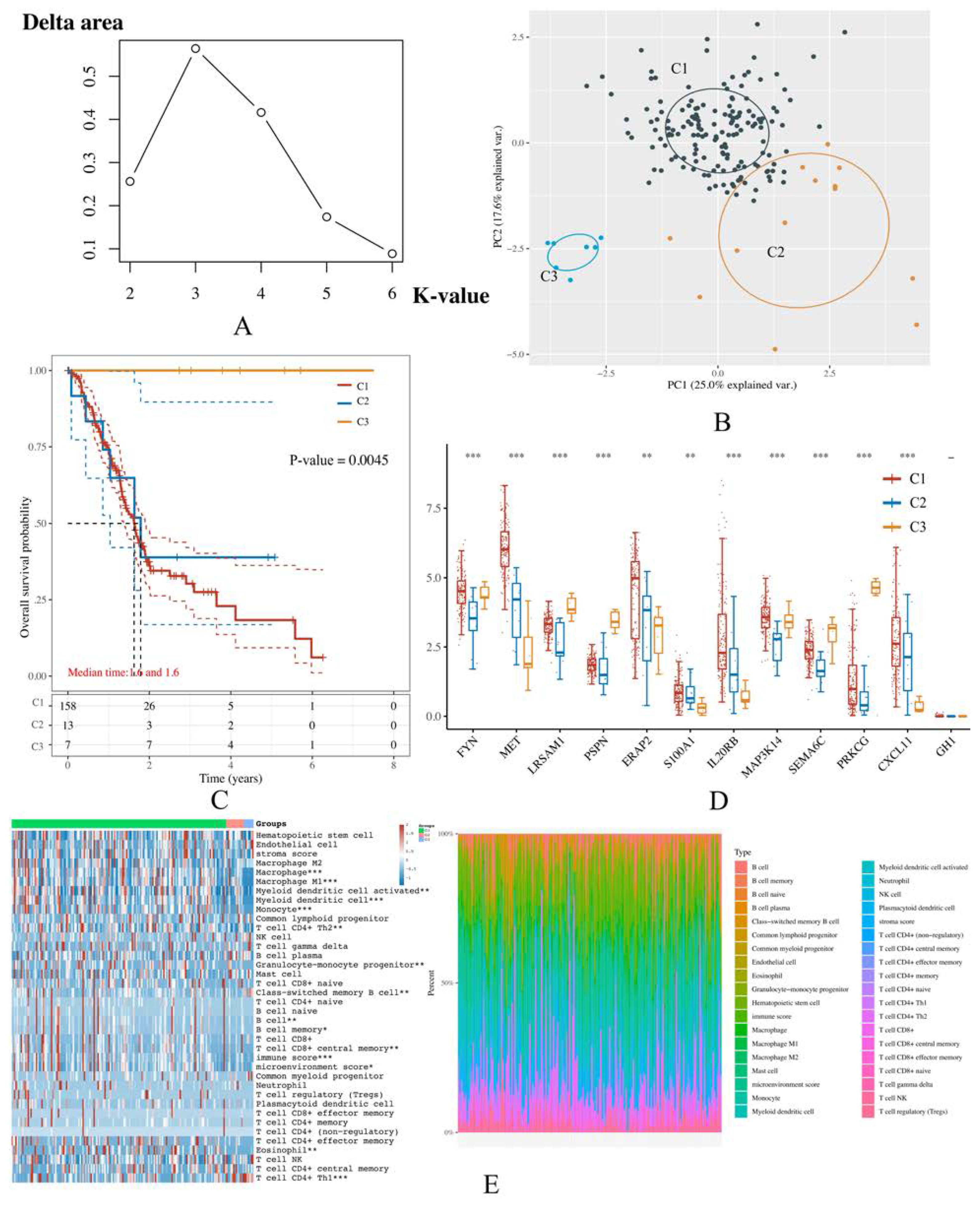
2.7. Unsupervised Consensus Clustering
3. Results
3.1. 12 Feature Genes Were Selected to Construct the Prognosis Predictor through the LASSO Algorithm
3.2. IRGPI-Based Risk Model Demonstrated a Strong Predictive Power
3.3. The Predictive Performance of the IRGPI-Based Risk Model Is Superior to That of the Ferroptosis- and Pyroptosis-Derived Model
3.4. The Risk Score Can Serve as an Independent Prognostic Indicator
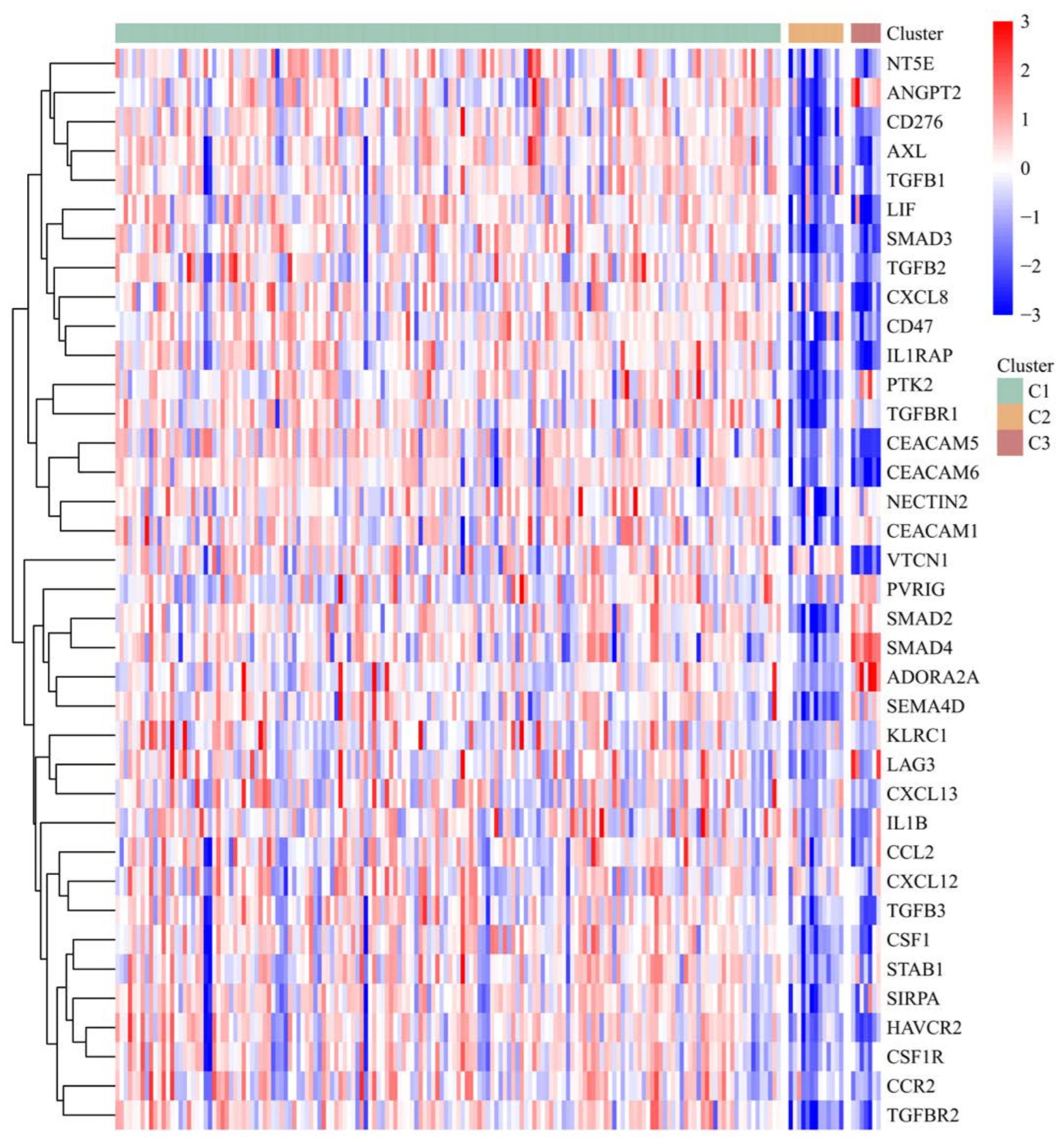
3.5. The Risk Score and IRGPI Genes Are Tightly Associated with the Tumor Microenvironment
3.6. PAAD Could Be More Precisely Divided into 3 Molecular Subtypes According to the Expression of IRGPI Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Karamitopoulou, E. Molecular Pathology of Pancreatic Cancer. Cancers 2022, 14, 1523. [Google Scholar] [CrossRef] [PubMed]
- Nevala-Plagemann, C.; Hidalgo, M.; Garrido-Laguna, I. From state-of-the-art treatments to novel therapies for advanced-stage pancreatic cancer. Nat. Rev. Clin. Oncol. 2020, 17, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Tian, W.; Tang, Y.; Zou, Y.; Zheng, S.; Wu, L.; Zeng, Y.; Wu, S.; Xie, X.; Xie, X. Establishment of a Cell Necroptosis Index to Predict Prognosis and Drug Sensitivity for Patients With Triple-Negative Breast Cancer. Front. Mol. Biosci. 2022, 9. [Google Scholar] [CrossRef]
- Nollmann, F.I.; Ruess, D.A. Targeting Mutant KRAS in Pancreatic Cancer: Futile or Promising? Biomedicines 2020, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Chandan, S.; Deliwala, S.S.; Facciorusso, A.; Mohan, B.P.; Ramai, D.; Kochhar, G.S. Association of BRCA Mutations and Pancreatic Cancer: Review of Literature and Meta-analysis. Pancreas 2022, 51, e8–e10. [Google Scholar] [CrossRef]
- Cellini, F.; Arcelli, A.; Simoni, N.; Caravatta, L.; Buwenge, M.; Calabrese, A.; Brunetti, O.; Genovesi, D.; Mazzarotto, R.; Deodato, F.; et al. Basics and Frontiers on Pancreatic Cancer for Radiation Oncology: Target Delineation, SBRT, SIB technique, MRgRT, Particle Therapy, Immunotherapy and Clinical Guidelines. Cancers 2020, 12, 1729. [Google Scholar] [CrossRef]
- Ambe, C.; Fulp, W.; Springett, G.; Hoffe, S.; Mahipal, A. A Meta-analysis of Randomized Clinical Trials of Chemoradiation Therapy in Locally Advanced Pancreatic Cancer. J. Gastrointest. Cancer 2015, 46, 284–290. [Google Scholar] [CrossRef]
- Martín, A.M.; Hidalgo, M.; Alvarez, R.; Arrazubi, V.; Martínez-Galán, J.; Salgado, M.; Macarulla, T.; Carrato, A. From First Line to Sequential Treatment in the Management of Metastatic Pancreatic Cancer. J. Cancer 2018, 9, 1978–1988. [Google Scholar] [CrossRef]
- Perales-Patón, J.; Piñeiro-Yañez, E.; Tejero, H.; López-Casas, P.P.; Hidalgo, M.; Gómez-López, G.; Al-Shahrour, F. Pancreas Cancer Precision Treatment Using Avatar Mice from a Bioinformatics Perspective. Public Health Genom. 2017, 20, 81–91. [Google Scholar] [CrossRef]
- Singh, K.; Pruski, M.; Bland, R.; Younes, M.; Guha, S.; Thosani, N.; Maitra, A.; Cash, B.D.; McAllister, F.; Logsdon, C.D.; et al. Kras mutation rate precisely orchestrates ductal derived pancreatic intraepithelial neoplasia and pancreatic cancer. Lab. Invest. 2021, 101, 177–192. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Christenson, E.S.; Jaffee, E.; Azad, N.S. Current and emerging therapies for patients with advanced pancreatic ductal adenocarcinoma: A bright future. Lancet Oncol. 2020, 21, e135–e145. [Google Scholar] [CrossRef]
- Dardare, J.; Witz, A.; Merlin, J.L.; Gilson, P.; Harlé, A. SMAD4 and the TGFβ pathway in patients with pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 3534. [Google Scholar] [CrossRef] [PubMed]
- Connor, A.A.; Denroche, R.E.; Jang, G.H.; Lemire, M.; Zhang, A.; Chan-Seng-Yue, M.; Wilson, G.; Grant, R.C.; Merico, D.; Lungu, I.; et al. Integration of genomic and transcriptional features in pancreatic cancer reveals increased cell cycle progression in metastases. Cancer Cell 2019, 35, 267–282.e267. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A new frontier in cancer. Biomed. Pharmacother. 2019, 121, 109595. [Google Scholar] [CrossRef]
- Yang, F.; Bettadapura, S.N.; Smeltzer, M.S.; Zhu, H.; Wang, S. Pyroptosis and pyroptosis-inducing cancer drugs. Acta Pharmacol. Sin. 2022, 43, 10. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998, Erratum in Nature 2010, 465, 966.. [Google Scholar] [CrossRef]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Dunn, P.; Thomas, C.G.; Smith, B.; Schaefer, H.; Chen, J.; Hu, Z.; Zalocusky, K.A.; Shankar, R.D.; Shen-Orr, S.S.; et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci. Data 2018, 5, 180015. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Vokes, S.; Dang, C.; Ji, H. Turning publicly available gene expression data into discoveries using gene set context analysis. Nucleic Acids Res. 2015, 44, gkv873. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 13 May 2022).
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 5, 25–29. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Xu, F.; Feng, X.; Wu, Y.; Lv, J.; Shi, Y.; Pei, H. Pyroptosis regulators exert crucial functions in prognosis, progression and immune microenvironment of pancreatic adenocarcinoma: A bioinformatic and in vitro research. Bioengineered 2022, 13, 1717–1735. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Mode; Springer: New York, NY, USA, 2000; ISBN 0-387-98784-3. [Google Scholar]
- Blanche, P.; Dartigues, J.; Jacqmin-Gadda, H. Estimating and Comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. Available online: http://onlinelibrary.wiley.com/doi/10.1002/sim.5958/full (accessed on 13 May 2022). [CrossRef] [PubMed]
- Yang, J.; Wei, X.; Hu, F.; Dong, W.; Sun, L. Development and validation of a novel 3-gene prognostic model for pancreatic adenocarcinoma based on ferroptosis-related genes. Cancer Cell Int. 2022, 22, 21. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res 2020, 48, W509–W514. [Google Scholar] [CrossRef] [PubMed]
- Sturm, G.; Finotello, F.; List, M. Immunedeconv: An R Package for Unified Access to Computational Methods for Estimating Immune Cell Fractions from Bulk RNA-Sequencing Data. Methods Mol. Biol. 2020, 2120, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Miao, Y.-R.; Xia, M.; Luo, M.; Luo, T.; Yang, M.; Guo, A.-Y. ImmuCellAI-mouse: A tool for comprehensive prediction of mouse immune cell abundance and immune microenvironment depiction. Bioinformatics 2021, 38, btab711. [Google Scholar] [CrossRef]
- Wilkerson, D.M.; Hayes Neil, D. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. Available online: http://bioinformatics.oxfordjournals.org/content/26/12/1572.abstract (accessed on 13 May 2022). [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Fitzgerald, M.; Saville, B.R.; Lewis, R.J. Decision Curve Analysis. In JAMA Guide to Statistics and Methods; Livingston, E.H., Lewis, R.J., Eds.; McGraw Hill: New York, NY, USA, 2019; Available online: https://jamaevidence.mhmedical.com/content.aspx?bookid=2742§ionid=233567832 (accessed on 5 May 2022).
- Vickers, A.J.; van Calster, B.; Steyerberg, E.W. A simple, step-by-step guide to interpreting decision curve analysis. Diagn. Progn. Res. 2019, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rousson, V.; Lee, W.C.; Ferdynus, C.; Chen, M.; Qian, X.; Guo, Y. Decision curve analysis: A technical note. Ann. Transl. Med. 2018, 6, 308. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles Posed by the Tumor Microenvironment to T cell Activity: A Case for Synergistic Therapies. Cancer Cell 2017, 3, 311–325. [Google Scholar] [CrossRef] [PubMed]
- Hosein, A.N.; Brekken, R.A.; Maitra, A. Pancreatic cancer stroma: An update on therapeutic targeting strategies. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Liot, S.; Balas, J.; Aubert, A.; Prigent, L.; Mercier-Gouy, P.; Verrier, B.; Bertolino, P.; Hennino, A.; Valcourt, U.; Lambert, E. Stroma Involvement in Pancreatic Ductal Adenocarcinoma: An Overview Focusing on Extracellular Matrix Proteins. Front. Immunol. 2021, 12, 612271. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Fukunaga, A.; Miyamoto, M.; Cho, Y.; Murakami, S.; Kawarada, Y.; Oshikiri, T.; Kato, K.; Kurokawa, T.; Suzuoki, M.; Nakakubo, Y.; et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas 2004, 28, e26–e31. [Google Scholar] [CrossRef]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 18, 5057–5069. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Bear, A.S. Tumor-Derived Myeloid Cell Chemoattractants and T Cell Exclusion in Pancreatic Cancer. Front. Immunol. 2020, 11, 605619. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 3, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 3, 579–596. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 50, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, S.; Yanagimoto, H.; Satoi, S.; Yamamoto, T.; Toyokawa, H.; Yamaki, S.; Yui, R.; Inoue, K.; Michiura, T.; Kwon, A.-H. The role of circulating dendritic cells in patients with unresectable pancreatic cancer. Anticancer Res. 2011, 11, 3827–3834. [Google Scholar]
- Yanagimoto, H.; Takai, S.; Satoi, S.; Toyokawa, H.; Takahashi, K.; Terakawa, N.; Kwon, A.-H.; Kamiyama, Y. Impaired function of circulating dendritic cells in patients with pancreatic cancer. Clin. Immunol. 2005, 1, 52–60. [Google Scholar] [CrossRef]
- Pratt, H.G.; Steinberger, K.J.; Mihalik, N.E.; Ott, S.; Whalley, T.; Szomolay, B.; Boone, B.A.; Eubank, T.D. Macrophage and Neutrophil Interactions in the Pancreatic Tumor Microenvironment Drive the Pathogenesis of Pancreatic Cancer. Cancers 2021, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Martin-Orozco, N.; Zheng, P.; Li, J.; Zhang, P.; Tan, H.; Park, H.J.; Jeong, M.; Chang, S.H.; Kim, B.-S.; et al. Inhibition of the B7-H3 immune checkpoint limits tumor growth by enhancing cytotoxic lymphocyte function. Cell Res. 2017, 27, 1034–1045. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. B7H3 As a Promoter of Metastasis and Promising Therapeutic Target. Front. Oncol. 2018, 8, 264. [Google Scholar] [CrossRef]
- Podojil, J.R.; Miller, S.D. Potential targeting of B7-H4 for the treatment of cancer. Immunol. Rev. 2017, 276, 40–51. [Google Scholar] [CrossRef]
- MacGregor, H.L.; Ohashi, P.S. Molecular Pathways: Evaluating the Potential for B7-H4 as an Immunoregulatory Target. Clin. Cancer Res. 2017, 23, 2934–2941. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Abdul-Ghafar, J.; Ud Din, N.; Saadaat, R.; Ahmad, Z. Metastatic renal cell carcinoma to pancreas and gastrointestinal tract: A clinicopathological study of 3 cases and review of literature. BMC Urol. 2021, 21, 84. [Google Scholar] [CrossRef]
- Ballarin, R.; Spaggiari, M.; Cautero, N.; De Ruvo, N.; Montalti, R.; Longo, C.; Pecchi, A.; Giacobazzi, P.; De Marco, G.; D’Amico, G.; et al. Pancreatic metastases from renal cell carcinoma: The state of the art. World J. Gastroenterol. 2011, 17, 4747–4756. [Google Scholar] [CrossRef] [PubMed]
- Thadani, A.; Pais, S.; Savino, J. Metastasis of renal cell carcinoma to the pancreas 13 years postnenhrectomv. Gastroenterol. Hepatol. 2011, 7, 697–699. [Google Scholar]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Szöllősi, A.G.; Huang, X.; Chang-Chien, Y.-C.; Hajdu, A. A Novel Immune-Related Gene Prognostic Index (IRGPI) in Pancreatic Adenocarcinoma (PAAD) and Its Implications in the Tumor Microenvironment. Cancers 2022, 14, 5652. https://doi.org/10.3390/cancers14225652
Zhou S, Szöllősi AG, Huang X, Chang-Chien Y-C, Hajdu A. A Novel Immune-Related Gene Prognostic Index (IRGPI) in Pancreatic Adenocarcinoma (PAAD) and Its Implications in the Tumor Microenvironment. Cancers. 2022; 14(22):5652. https://doi.org/10.3390/cancers14225652
Chicago/Turabian StyleZhou, Shujing, Attila Gábor Szöllősi, Xufeng Huang, Yi-Che Chang-Chien, and András Hajdu. 2022. "A Novel Immune-Related Gene Prognostic Index (IRGPI) in Pancreatic Adenocarcinoma (PAAD) and Its Implications in the Tumor Microenvironment" Cancers 14, no. 22: 5652. https://doi.org/10.3390/cancers14225652
APA StyleZhou, S., Szöllősi, A. G., Huang, X., Chang-Chien, Y.-C., & Hajdu, A. (2022). A Novel Immune-Related Gene Prognostic Index (IRGPI) in Pancreatic Adenocarcinoma (PAAD) and Its Implications in the Tumor Microenvironment. Cancers, 14(22), 5652. https://doi.org/10.3390/cancers14225652