Isolated BAP1 Genomic Alteration in Malignant Pleural Mesothelioma Predicts Distinct Immunogenicity with Implications for Immunotherapeutic Response

 ,
,  ,
, 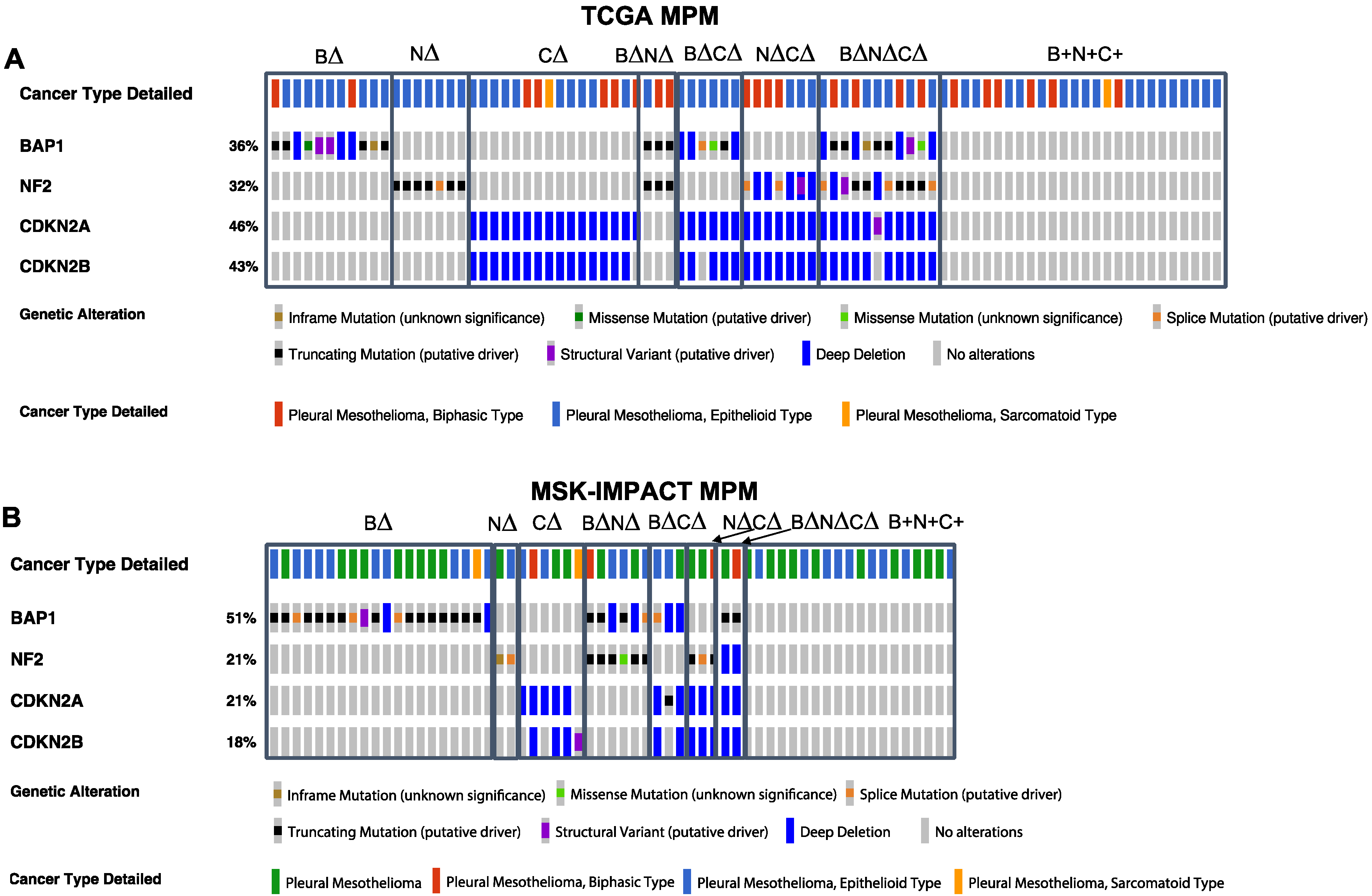{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Association of Patient Survival with TSG Genotypic Groups in MPM
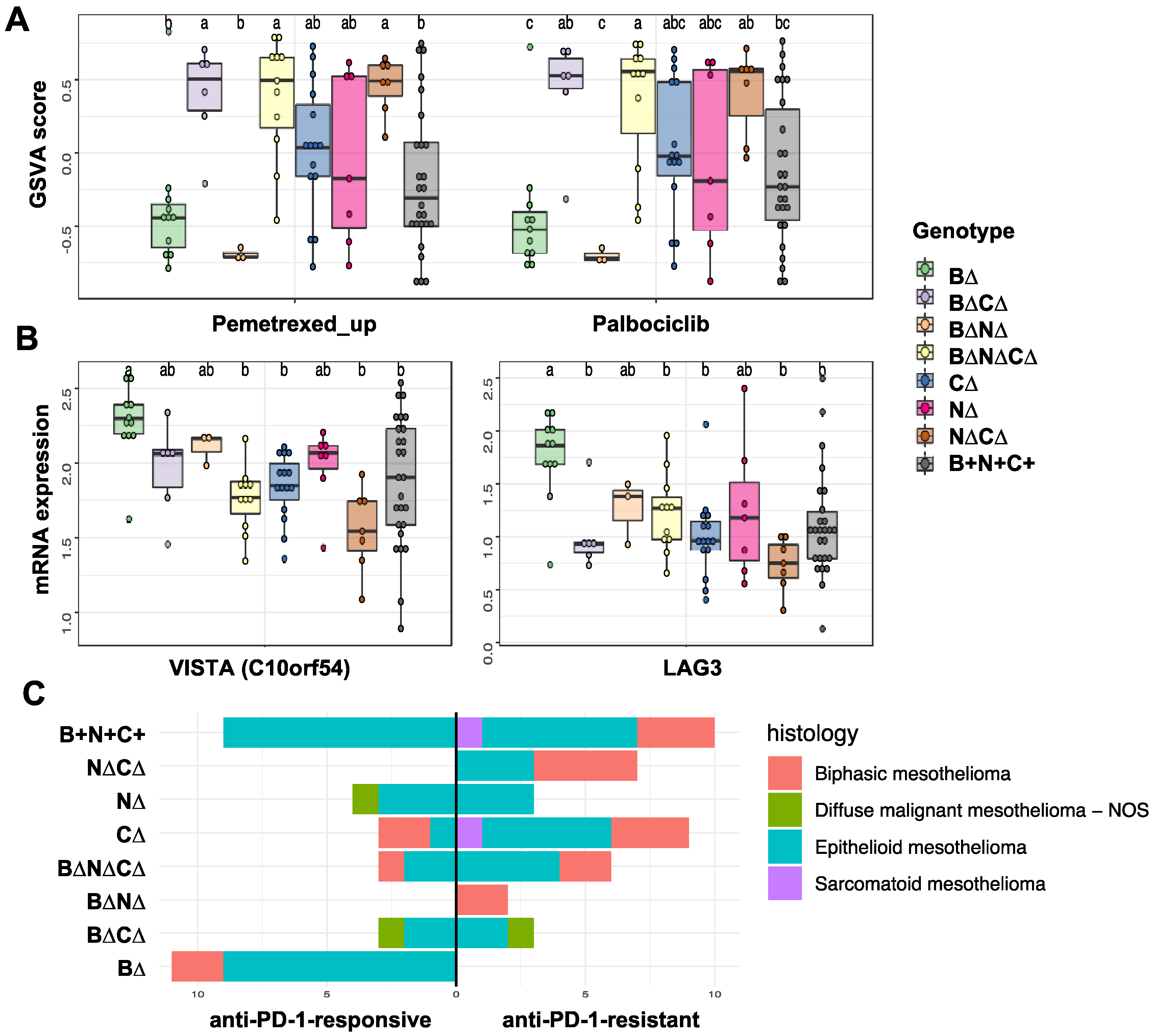
2.2. Association of Gene Signature Predictive of Response to Therapy with TSG Genotypic Groups in MPM
2.3. Transcription Factors and Pathways Associated with TSG Genotypic Groups in MPM
3. Discussion
4. Materials and Methods
4.1. Data and Preprocessing
4.2. Survival Analyses
4.3. Motif Activity Analysis
4.4. Gene Set Enrichment Analysis
4.5. Analysis of the Tumor Immune Microenvironment (TIME)
4.6. Statistical Analysis and Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baumann, F.; Ambrosi, J.P.; Carbone, M. Asbestos is not just asbestos: An unrecognised health hazard. Lancet Oncol. 2013, 14, 576–578. [Google Scholar] [CrossRef]
- Lacourt, A.; Leveque, E.; Guichard, E.; Gilg Soit Ilg, A.; Sylvestre, M.P.; Leffondre, K. Dose-time-response association between occupational asbestos exposure and pleural mesothelioma. Occup. Environ. Med. 2017, 74, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Galateau-Salle, F.; Churg, A.; Roggli, V.; Travis, W.D.; World Health Organization Committee for Tumors of the Pleura. The 2015 World Health Organization Classification of Tumors of the Pleura: Advances since the 2004 Classification. J. Thorac. Oncol. 2016, 11, 142–154. [Google Scholar] [CrossRef]
- Robinson, B.W.; Lake, R.A. Advances in malignant mesothelioma. N. Engl. J. Med. 2005, 353, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Burki, T. Health experts concerned over India’s asbestos industry. Lancet 2010, 375, 626–627. [Google Scholar] [CrossRef]
- Chernova, T.; Murphy, F.A.; Galavotti, S.; Sun, X.M.; Powley, I.R.; Grosso, S.; Schinwald, A.; Zacarias-Cabeza, J.; Dudek, K.M.; Dinsdale, D.; et al. Long-Fiber Carbon Nanotubes Replicate Asbestos-Induced Mesothelioma with Disruption of the Tumor Suppressor Gene Cdkn2a (Ink4a/Arf). Curr. Biol. 2017, 27, 3302–3314.e6. [Google Scholar] [CrossRef]
- Barbarino, M.; Giordano, A. Assessment of the Carcinogenicity of Carbon Nanotubes in the Respiratory System. Cancers 2021, 13, 1318. [Google Scholar] [CrossRef]
- Asciak, R.; George, V.; Rahman, N.M. Update on biology and management of mesothelioma. Eur. Respir. Rev. 2021, 30, 200226. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Vellanki, P.J.; Larkins, E.; Chatterjee, S.; Mishra-Kalyani, P.S.; Bi, Y.; Qosa, H.; Liu, J.; Zhao, H.; Biable, M.; et al. FDA Approval Summary: Nivolumab in Combination with Ipilimumab for the Treatment of Unresectable Malignant Pleural Mesothelioma. Clin. Cancer Res. 2021, 28, 446–451. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Peters, S.; Scherpereel, A.; Cornelissen, R.; Oulkhouir, Y.; Greillier, L.; Kaplan, M.A.; Talbot, T.; Monnet, I.; Hiret, S.; Baas, P.; et al. First-line nivolumab plus ipilimumab versus chemotherapy in patients with unresectable malignant pleural mesothelioma: 3-year outcomes from CheckMate 743. Ann. Oncol. 2022, 33, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [PubMed]
- Jean, D.; Daubriac, J.; Le Pimpec-Barthes, F.; Galateau-Salle, F.; Jaurand, M.C. Molecular changes in mesothelioma with an impact on prognosis and treatment. Arch. Pathol. Lab. Med. 2012, 136, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Luo, J.L.; Sun, Q.; Harber, J.; Dawson, A.G.; Nakas, A.; Busacca, S.; Sharkey, A.J.; Waller, D.; Sheaff, M.T.; et al. Clonal architecture in mesothelioma is prognostic and shapes the tumour microenvironment. Nat. Commun. 2021, 12, 1751. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Martin, A.; Egger, J.; Rizvi, H.; Offin, M.; Rimner, A.; Adusumilli, P.S.; Rusch, V.W.; Kris, M.G.; Sauter, J.L.; et al. The use of a next-generation sequencing-derived machine-learning risk-prediction model (OncoCast-MPM) for malignant pleural mesothelioma: A retrospective study. Lancet Digit. Health 2021, 3, e565–e576. [Google Scholar] [CrossRef]
- Hiltbrunner, S.; Fleischmann, Z.; Sokol, E.S.; Zoche, M.; Felley-Bosco, E.; Curioni-Fontecedro, A. Genomic Landscape of Pleural and Peritoneal Mesothelioma Tumors. Br. J. Cancer 2022. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Hou, J.; Lambers, M.; den Hamer, B.; den Bakker, M.A.; Hoogsteden, H.C.; Grosveld, F.; Hegmans, J.; Aerts, J.; Philipsen, S. Expression profiling-based subtyping identifies novel non-small cell lung cancer subgroups and implicates putative resistance to pemetrexed therapy. J. Thorac. Oncol. 2012, 7, 105–114. [Google Scholar] [CrossRef]
- Malorni, L.; Piazza, S.; Ciani, Y.; Guarducci, C.; Bonechi, M.; Biagioni, C.; Hart, C.D.; Verardo, R.; Di Leo, A.; Migliaccio, I. A gene expression signature of retinoblastoma loss-of-function is a predictive biomarker of resistance to palbociclib in breast cancer cell lines and is prognostic in patients with ER positive early breast cancer. Oncotarget 2016, 7, 68012–68022. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Truong, C.Y.; Lo, E.M.; Holmes, H.M.; Ramos, D.; Ramineni, M.; Lee, J.S.; Wang, D.Y.; Pietropaolo, M.; Ripley, R.T.; et al. Inhibition of CDK4/6 Overcomes Primary Resistance to PD-1 Blockade in Malignant Mesothelioma. Ann. Thorac. Surg. 2021, 114, 1842–1852. [Google Scholar] [CrossRef] [PubMed]
- Balwierz, P.J.; Pachkov, M.; Arnold, P.; Gruber, A.J.; Zavolan, M.; van Nimwegen, E. ISMARA: Automated modeling of genomic signals as a democracy of regulatory motifs. Genome. Res. 2014, 24, 869–884. [Google Scholar] [CrossRef]
- Gu, Y.; Li, A.; Sun, H.; Li, X.; Zha, H.; Zhao, J.; Xie, J.; Zeng, Z.; Zhou, L. BCL6B suppresses proliferation and migration of colorectal carcinoma cells through inhibition of the PI3K/AKT signaling pathway. Int. J. Mol. Med. 2018, 41, 2660–2668. [Google Scholar] [CrossRef]
- Li, X.; Yu, J.; Brock, M.V.; Tao, Q.; Herman, J.G.; Liang, P.; Guo, M. Epigenetic silencing of BCL6B inactivates p53 signaling and causes human hepatocellular carcinoma cell resist to 5-FU. Oncotarget 2015, 6, 11547–11560. [Google Scholar] [CrossRef] [PubMed]
- Hartatik, T.; Okada, S.; Okabe, S.; Arima, M.; Hatano, M.; Tokuhisa, T. Binding of BAZF and Bc16 to STAT6-binding DNA sequences. Biochem. Biophys. Res. Commun. 2001, 284, 26–32. [Google Scholar] [CrossRef]
- Budhwani, M.; Mazzieri, R.; Dolcetti, R. Plasticity of Type I Interferon-Mediated Responses in Cancer Therapy: From Anti-tumor Immunity to Resistance. Front. Oncol. 2018, 8, 322. [Google Scholar] [CrossRef]
- Talens, F.; Van Vugt, M. Inflammatory signaling in genomically instable cancers. Cell Cycle 2019, 18, 1830–1848. [Google Scholar] [CrossRef]
- Shrestha, R.; Nabavi, N.; Lin, Y.Y.; Mo, F.; Anderson, S.; Volik, S.; Adomat, H.H.; Lin, D.; Xue, H.; Dong, X.; et al. BAP1 haploinsufficiency predicts a distinct immunogenic class of malignant peritoneal mesothelioma. Genome. Med. 2019, 11, 8. [Google Scholar] [CrossRef]
- Chernova, T.; Sun, X.M.; Powley, I.R.; Galavotti, S.; Grosso, S.; Murphy, F.A.; Miles, G.J.; Cresswell, L.; Antonov, A.V.; Bennett, J.; et al. Molecular profiling reveals primary mesothelioma cell lines recapitulate human disease. Cell Death Differ. 2016, 23, 1152–1164. [Google Scholar] [CrossRef]
- Sun, S.; Frontini, F.; Qi, W.; Hariharan, A.; Ronner, M.; Wipplinger, M.; Blanquart, C.; Rehrauer, H.; Fonteneau, J.F.; Felley-Bosco, E. Endogenous retrovirus expression activates type-I interferon signaling in an experimental mouse model of mesothelioma development. Cancer Lett. 2021, 507, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xu, D.; Gao, Y.; Schmid, R.A.; Peng, R.W. Oncolytic Viral Therapy for Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2020, 15, e111–e113. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Wu, L.; Murakami, J.; Zhao, Y.; Yun, H.; de Perrot, M. IRF3 Knockout Results in Partial or Complete Rejection of Murine Mesothelioma. J. Clin. Med. 2021, 10, 5196. [Google Scholar] [CrossRef] [PubMed]
- Cantini, L.; Laniado, I.; Murthy, V.; Sterman, D.; Aerts, J. Immunotherapy for mesothelioma: Moving beyond single immune check point inhibition. Lung Cancer 2022, 165, 91–101. [Google Scholar] [CrossRef]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Nowak, E.C.; Lines, J.L.; Varn, F.S.; Deng, J.; Sarde, A.; Mabaera, R.; Kuta, A.; Le Mercier, I.; Cheng, C.; Noelle, R.J. Immunoregulatory functions of VISTA. Immunol. Rev. 2017, 276, 66–79. [Google Scholar] [CrossRef]
- Muller, S.; Victoria Lai, W.; Adusumilli, P.S.; Desmeules, P.; Frosina, D.; Jungbluth, A.; Ni, A.; Eguchi, T.; Travis, W.D.; Ladanyi, M.; et al. V-domain Ig-containing suppressor of T-cell activation (VISTA), a potentially targetable immune checkpoint molecule, is highly expressed in epithelioid malignant pleural mesothelioma. Mod. Pathol. 2020, 33, 303–311. [Google Scholar] [CrossRef]
- Andrews, L.P.; Cillo, A.R.; Karapetyan, L.; Kirkwood, J.M.; Workman, C.J.; Vignali, D.A.A. Molecular Pathways and Mechanisms of LAG-3 in Cancer Therapy. Clin. Cancer Res. 2022. [Google Scholar] [CrossRef]
- Marcq, E.; Van Audenaerde, J.R.M.; De Waele, J.; Merlin, C.; Pauwels, P.; van Meerbeeck, J.P.; Fisher, S.A.; Smits, E.L.J. The Search for an Interesting Partner to Combine with PD-L1 Blockade in Mesothelioma: Focus on TIM-3 and LAG-3. Cancers 2021, 13, 282. [Google Scholar] [CrossRef]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Sato, A.; Tsujimura, T.; Emi, M.; Morinaga, T.; Fukuoka, K.; Yamada, S.; Murakami, A.; Kondo, N.; Matsumoto, S.; et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012, 103, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Nasu, M.; Emi, M.; Pastorino, S.; Tanji, M.; Powers, A.; Luk, H.; Baumann, F.; Zhang, Y.A.; Gazdar, A.; Kanodia, S.; et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J. Thorac. Oncol. 2015, 10, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 0-387-98784-3. [Google Scholar]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdottir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Ma, X.; Somasundaram, A.; Qi, Z.; Hartman, D.J.; Singh, H.; Osmanbeyoglu, H.U. SPaRTAN, a computational framework for linking cell-surface receptors to transcriptional regulators. Nucleic. Acids Res. 2021, 49, 9633–9647. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osmanbeyoglu, H.U.; Palmer, D.; Sagan, A.; Sementino, E.; Becich, M.J.; Testa, J.R. Isolated BAP1 Genomic Alteration in Malignant Pleural Mesothelioma Predicts Distinct Immunogenicity with Implications for Immunotherapeutic Response. Cancers 2022, 14, 5626. https://doi.org/10.3390/cancers14225626
Osmanbeyoglu HU, Palmer D, Sagan A, Sementino E, Becich MJ, Testa JR. Isolated BAP1 Genomic Alteration in Malignant Pleural Mesothelioma Predicts Distinct Immunogenicity with Implications for Immunotherapeutic Response. Cancers. 2022; 14(22):5626. https://doi.org/10.3390/cancers14225626
Chicago/Turabian StyleOsmanbeyoglu, Hatice Ulku, Drake Palmer, April Sagan, Eleonora Sementino, Michael J. Becich, and Joseph R. Testa. 2022. "Isolated BAP1 Genomic Alteration in Malignant Pleural Mesothelioma Predicts Distinct Immunogenicity with Implications for Immunotherapeutic Response" Cancers 14, no. 22: 5626. https://doi.org/10.3390/cancers14225626
APA StyleOsmanbeyoglu, H. U., Palmer, D., Sagan, A., Sementino, E., Becich, M. J., & Testa, J. R. (2022). Isolated BAP1 Genomic Alteration in Malignant Pleural Mesothelioma Predicts Distinct Immunogenicity with Implications for Immunotherapeutic Response. Cancers, 14(22), 5626. https://doi.org/10.3390/cancers14225626