
BMN673 Is a PARP Inhibitor with Unique Radiosensitizing Properties: Mechanisms and Potential in Radiation Therapy

 , ,
, , 

Simple Summary
Abstract
1. Introduction
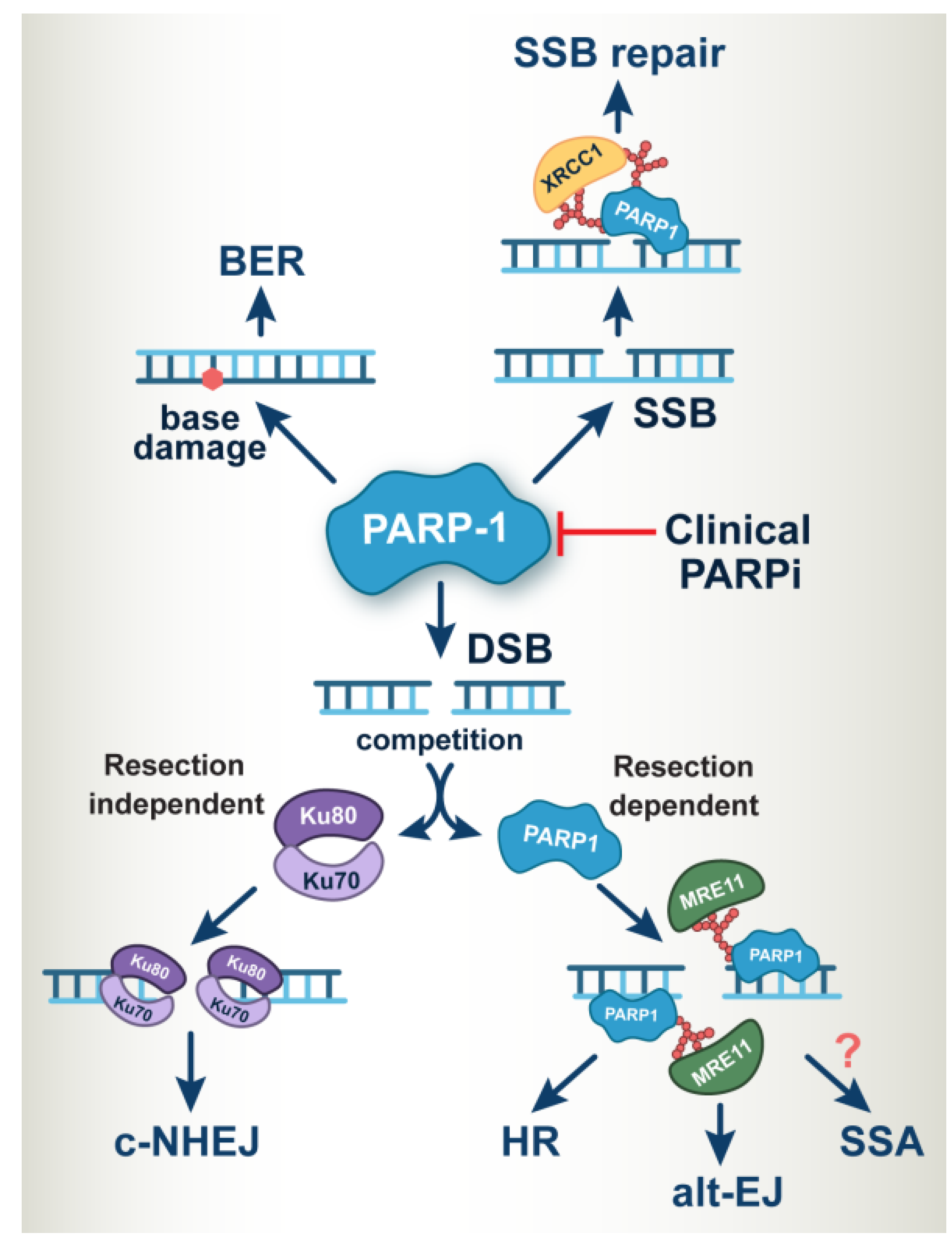
2. PARP-1 Operates at the Heart of DNA Damage Response and DNA Repair
3. Development of Clinically Relevant PARP Inhibitors
4. Rationale for Combining PARP Inhibitors with Radiotherapy
5. BMN673 Is a Superior Radiosensitizer
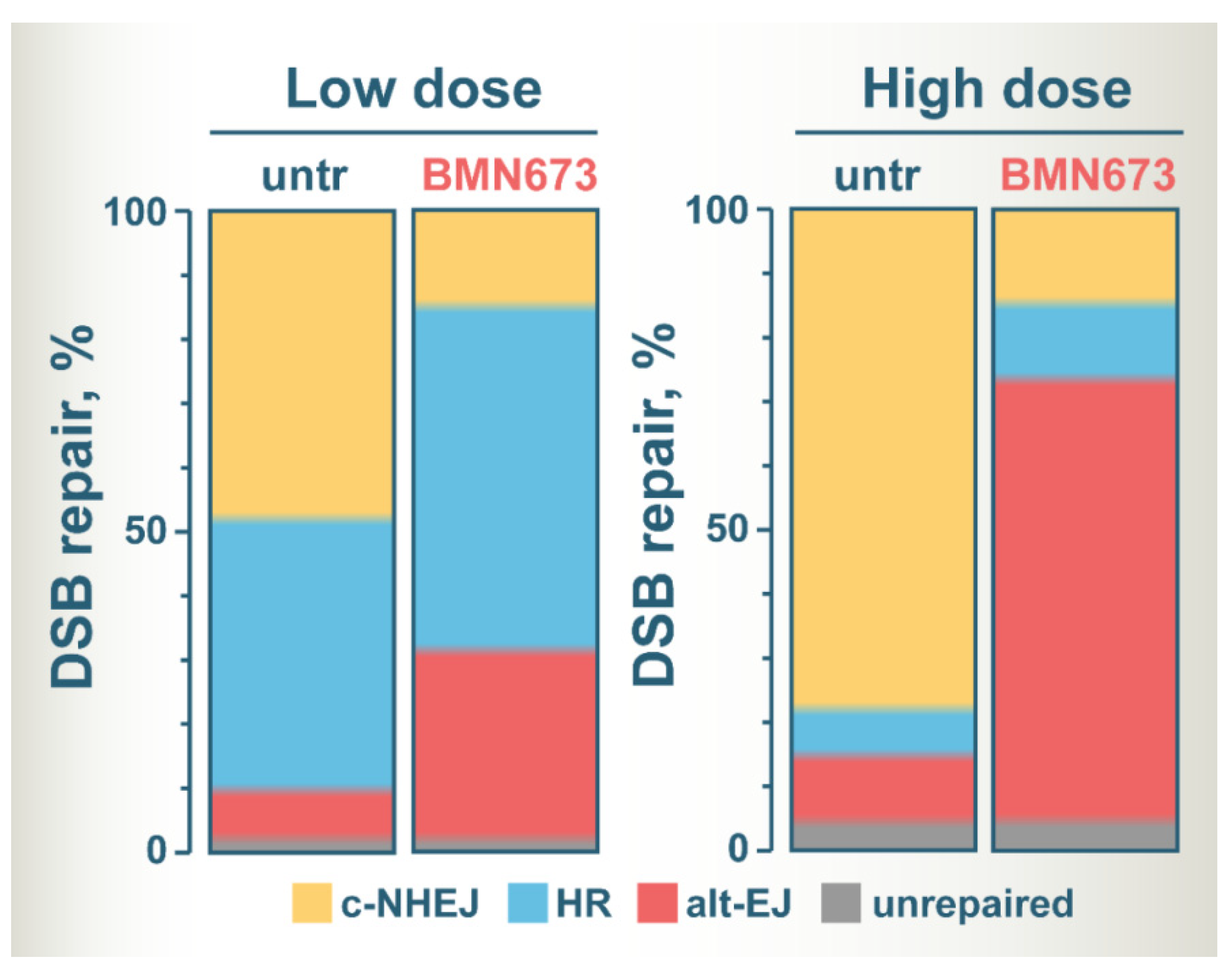
6. Shift of Balance to Error-Prone DSB Repair Pathways Is Likely the Key to BMN673-Mediated Radiosensitization
6.1. BMN673 Increases DNA End-Resection after IR
6.2. Key Questions Remain Unanswered Regarding the Effects of BMN673 on HR
6.3. BMN673 Inhibits c-NHEJ
6.4. BMN673 Shunts DSBs to Error-Prone Alt-EJ
7. Speculations on the Mechanism of BMN673-Induced Radiosensitization
8. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Helleday, T. DNA REPAIR. Drugging DNA repair. Science 2016, 352, 1178–1179. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Jones, D.; Lee, S.-H.; Williamson, E.A.; Hromas, R. Drugging the Cancers Addicted to DNA Repair. JNCI J. Natl. Cancer Inst. 2017, 109, djx059. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one (BMN 673, Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar]
- Shen, Y.; Rehman, F.L.; Feng, Y.; Boshuizen, J.; Bajrami, I.; Elliott, R.; Wang, B.; Lord, C.J.; Post, L.E.; Ashworth, A. BMN 673, a Novel and Highly Potent PARP1/2 Inhibitor for the Treatment of Human Cancers with DNA Repair Deficiency. Clin. Cancer Res. 2013, 19, 5003–5015. [Google Scholar] [CrossRef]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Thomas, H. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef] [PubMed]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Xie, T.; Dickson, K.-A.; Yee, C.; Ma, Y.; Ford, C.E.; Bowden, N.A.; Marsh, D.J. Targeting Homologous Recombination Deficiency in Ovarian Cancer with PARP Inhibitors: Synthetic Lethal Strategies That Impact Overall Survival. Cancers 2022, 14, 4621. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic lethality as an engine for cancer drug target discovery. Nat. Rev. Drug Discov. 2019, 19, 23–38. [Google Scholar] [CrossRef]
- Lord, C.J.; Tutt, A.N.; Ashworth, A. Synthetic Lethality and Cancer Therapy: Lessons Learned from the Development of PARP Inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- Zhu, H.; Tang, Y.-D.; Zhan, G.; Su, C.; Zheng, C. The Critical Role of PARPs in Regulating Innate Immune Responses. Front. Immunol. 2021, 12, 712556. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. Mol. Mech. Mutagen. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Pazzaglia, S.; Pioli, C. Multifaceted Role of PARP-1 in DNA Repair and Inflammation: Pathological and Therapeutic Implications in Cancer and Non-Cancer Diseases. Cells 2019, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Benafif, S. An update on PARP inhibitors for the treatment of cancer. Onco Targets Ther. 2015, 8, 519–528. [Google Scholar] [CrossRef]
- Du, Y.; Yamaguchi, H.; Hsu, J.L.; Hung, M.-C. PARP inhibitors as precision medicine for cancer treatment. Natl. Sci. Rev. 2017, 4, 576–592. [Google Scholar] [CrossRef][Green Version]
- Angel, M.; Zarba, M.; Sade, J.P. PARP inhibitors as a radiosensitizer: A future promising approach in prostate cancer? Ecancermedicalscience 2021, 15, ed118. [Google Scholar] [CrossRef] [PubMed]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. PARP Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef] [PubMed]
- Knelson, E.H.; Patel, S.A.; Sands, J.M. PARP Inhibitors in Small-Cell Lung Cancer: Rational Combinations to Improve Responses. Cancers 2021, 13, 727. [Google Scholar] [CrossRef]
- Lin, L.L.; Lakomy, D.S.; Ning, M.S.; Simpkins, F.; Jhingran, A. Combining novel agents with radiotherapy for gynecologic malignancies: Beyond the era of cisplatin. Int. J. Gynecol. Cancer Off. J. Int. Gynecol. Cancer Soc. 2020, 30, 409–423. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; Austry, J.-B.; Waissi, W.; Burckel, H.; Noël, G.; Habrand, J.-L.; Saintigny, Y.; Joly, F. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: A systematic review of pre-clinical and clinical human studies. Oncotarget 2017, 8, 69105–69124. [Google Scholar] [CrossRef]
- Chalmers, A.; Johnston, P.; Woodcock, M.; Joiner, M.; Marples, B. PARP-1, PARP-2, and the cellular response to low doses of ionizing radiation. Int. J. Radiat. Oncol. 2004, 58, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Dungey, F.A.; Löser, D.A.; Chalmers, A.J. Replication-Dependent Radiosensitization of Human Glioma Cells by Inhibition of Poly(ADP-Ribose) Polymerase: Mechanisms and Therapeutic Potential. Int. J. Radiat. Oncol. 2008, 72, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Karnak, D.; Engelke, C.G.; Parsels, L.A.; Kausar, T.; Wei, D.; Robertson, J.R.; Marsh, K.B.; Davis, M.A.; Zhao, L.; Maybaum, J.; et al. Combined Inhibition of Wee1 and PARP1/2 for Radiosensitization in Pancreatic Cancer. Clin. Cancer Res. 2014, 20, 5085–5096. [Google Scholar] [CrossRef]
- Kötter, A.; Cornils, K.; Borgmann, K.; Dahm-Daphi, J.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Inhibition of PARP1-dependent end-joining contributes to Olaparib-mediated radiosensitization in tumor cells. Mol. Oncol. 2014, 8, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.M.; Cao, C.; Kim, K.W.; Willey, C.D.; Geng, L.; Xiao, D.; Wang, H.; Sandler, A.; Johnson, D.H.; Colevas, A.D.; et al. Inhibition of Poly(ADP-Ribose) Polymerase Enhances Cell Death and Improves Tumor Growth Delay in Irradiated Lung Cancer Models. Clin. Cancer Res. 2007, 13, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Gross, N.; Li, Y.; Li, G.; Wang, Z.; Zhong, S.; Li, Y.; Hu, G. PARP inhibitor Olaparib increases the sensitization to radiotherapy in FaDu cells. J. Cell. Mol. Med. 2020, 24, 2444–2450. [Google Scholar] [CrossRef] [PubMed]
- Michmerhuizen, A.R.; Pesch, A.M.; Moubadder, L.; Chandler, B.C.; Wilder-Romans, K.; Cameron, M.; Olsen, E.; Thomas, D.G.; Zhang, A.; Hirsh, N.; et al. PARP1 Inhibition Radiosensitizes Models of Inflammatory Breast Cancer to Ionizing Radiation. Mol. Cancer Ther. 2019, 18, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Nile, D.L.; Rae, C.; Hyndman, I.J.; Gaze, M.N.; Mairs, R.J. An evaluation in vitro of PARP-1 inhibitors, rucaparib and olaparib, as radiosensitisers for the treatment of neuroblastoma. BMC Cancer 2016, 16, 1–13. [Google Scholar] [CrossRef]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O’Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by Olaparib (AZD2281) Increases the Radiosensitivity of a Lung Tumor Xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef]
- Soni, A.; Li, F.; Wang, Y.; Grabos, M.; Krieger, L.M.; Chaudhary, S.; Hasan, M.S.M.; Ahmed, M.; Coleman, C.N.; Teicher, B.A.; et al. Inhibition of Parp1 by BMN673 Effectively Sensitizes Cells to Radiotherapy by Upsetting the Balance of Repair Pathways Processing DNA Double-Strand Breaks. Mol. Cancer Ther. 2018, 17, 2206–2216. [Google Scholar] [CrossRef]
- Laird, J.H.; Lok, B.H.; Ma, J.; Bell, A.; de Stanchina, E.; Poirier, J.T.; Rudin, C.M. Talazoparib Is a Potent Radiosensitizer in Small Cell Lung Cancer Cell Lines and Xenografts. Clin. Cancer Res. 2018, 24, 5143–5152. [Google Scholar] [CrossRef]
- Venneker, S.; Kruisselbrink, A.B.; Bruijn, I.H.B.-D.; de Jong, Y.; van Wijnen, A.J.; Danen, E.H.; Bovée, J.V. Inhibition of PARP Sensitizes Chondrosarcoma Cell Lines to Chemo- and Radiotherapy Irrespective of the IDH1 or IDH2 Mutation Status. Cancers 2019, 11, 1918. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Trovesi, C.; Manfrini, N.; Falcettoni, M.; Longhese, M.P. Regulation of the DNA Damage Response by Cyclin-Dependent Kinases. J. Mol. Biol. 2013, 425, 4756–4766. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, K.; Schell, M.; Hoppe, T.; Kashkar, H. Regulation of the DNA damage response by ubiquitin conjugation. Front. Genet. 2015, 6, 98. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef]
- Gourley, C.; Balmaña, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef]
- Lans, H.; Hoeijmakers, J.H.J.; Vermeulen, W.; Marteijn, J.A. The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol. 2019, 20, 766–784. [Google Scholar] [CrossRef]
- Perina, D.; Mikoč, A.; Ahel, J.; Ćetković, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, F.; Tang, T.; Guo, C. The role of PARP1 in the DNA damage response and its application in tumor therapy. Front. Med. 2012, 6, 156–164. [Google Scholar] [CrossRef] [PubMed]
- de Murcia, G.; Menissier de Murcia, J. Poly(ADP-ribose) polymerase: A molecular nick-sensor. Trends Biochem. Sci. 1994, 19, 172–173. [Google Scholar] [CrossRef]
- Schreiber, V.; Amé, J.-C.; Dollé, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Ménissier-De Murcia, J.; de Murcia, G. Poly(ADP-ribose) Polymerase-2 (PARP-2) Is Required for Efficient Base Excision DNA Repair in Association with PARP-1 and XRCC1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar] [CrossRef] [PubMed]
- Allinson, S.L.; Dianova, I.I.; Dianov, G.L. Poly(ADP-ribose) polymerase in base excision repair: Always engaged, but not essential for DNA damage processing. Acta Biochim. Pol. 2003, 50, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Le Page, F.; Schreiber, V.; Dherin, C.; de Murcia, G.; Boiteux, S. Poly(ADP-ribose) Polymerase-1 (PARP-1) Is Required in Murine Cell Lines for Base Excision Repair of Oxidative DNA Damage in the Absence of DNA Polymerase ß. J. Biol. Chem. 2003, 278, 18471–18477. [Google Scholar] [CrossRef] [PubMed]
- Sukhanova, M.; Khodyreva, S.; Lavrik, O. Poly(ADP-ribose) polymerase 1 regulates activity of DNA polymerase b in long patch base excision repair. Mutat. Res. /Fundam. Mol. Mech. Mutagen. 2010, 685, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.K.; Stefanick, D.F.; Prasad, R.; Gassman, N.R.; Kedar, P.S.; Wilson, S.H. Base Excision Repair Defects Invoke Hypersensitivity to PARP Inhibition. Mol. Cancer Res. 2014, 12, 1128–1139. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, O.I. PARPs’ impact on base excision DNA repair. DNA Repair 2020, 93, 102911. [Google Scholar] [CrossRef]
- Seeberg, E.; Eide, L.; Bjoras, M. The base excision repair pathway. Trends Biochem. Sci. 1995, 20, 391–397. [Google Scholar] [CrossRef]
- Grasso, S.; Tell, G. Base excision repair in Archaea: Back to the future in DNA repair. DNA Repair 2014, 21, 148–157. [Google Scholar] [CrossRef]
- Kennedy, E.; Caffrey, P.J.; Delaney, S. Initiating base excision repair in chromatin. DNA Repair 2018, 71, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Mullins, E.; Rodriguez, A.; Bradley, N.P.; Eichman, B.F. Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci. 2019, 44, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Ball, S.S.; Bowater, R.P.; Wormstone, I.M. PARP-1 inhibition influences the oxidative stress response of the human lens. Redox Biol. 2016, 8, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Ström, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.-K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2010, 39, 3166–3175. [Google Scholar] [CrossRef]
- Reynolds, P.; Cooper, S.; Lomax, M.; O’Neill, P. Disruption of PARP1 function inhibits base excision repair of a sub-set of DNA lesions. Nucleic Acids Res. 2015, 43, 4028–4038. [Google Scholar] [CrossRef]
- Satoh, M.S.; Lindahl, T. Role of poly(ADP-ribose) formation in DNA repair. Nature 1992, 356, 356–358. [Google Scholar] [CrossRef]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Marintchev, A.; Robertson, A.; Dimitriadis, E.K.; Prasad, R.; Wilson, S.H.; Mullen, G.P. Domain specific interaction in the XRCC1-DNA polymerase beta complex. Nucleic Acids Res. 2000, 28, 2049–2059. [Google Scholar] [CrossRef]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Oei, S.L.; Ziegler, M. ATP for the DNA Ligation Step in Base Excision Repair Is Generated from Poly(ADP-ribose). J. Biol. Chem. 2000, 275, 23234–23239. [Google Scholar] [CrossRef]
- Ruscetti, T.; Lehnert, B.E.; Halbrook, J.; Le Trong, H.; Hoekstra, M.F.; Chen, D.J.; Peterson, S.R. Stimulation of the DNA-dependent Protein Kinase by Poly(ADP-Ribose) Polymerase. J. Biol. Chem. 1998, 273, 14461–14467. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, A.; Tohda, H.; Kanai, M.; Miwa, M.; Sugimura, T. Inhibitors of poly(adenosine diphosphate ribose) polymerase induce sister chromatid exchanges. Biochem. Biophys. Res. Commun. 1980, 97, 1311–1316. [Google Scholar] [CrossRef]
- Zhang, F.; Shi, J.; Bian, C.; Yu, X. Poly(ADP-Ribose) Mediates the BRCA2-Dependent Early DNA Damage Response. Cell Rep. 2015, 13, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Petit, S.A.; Ficarro, S.B.; Toomire, K.J.; Xie, A.; Lim, E.; Cao, S.A.; Park, E.; Eck, M.J.; Scully, R.; et al. PARP1-Driven Poly-ADP-Ribosylation Regulates BRCA1 Function in Homologous Recombination–Mediated DNA Repair. Cancer Discov. 2014, 4, 1430–1447. [Google Scholar] [CrossRef]
- Soni, A.; Siemann, M.; Grabos, M.; Murmann, T.; Pantelias, G.E.; Iliakis, G. Requirement for Parp-1 and DNA ligases 1 or 3 but not of Xrcc1 in chromosomal translocation formation by backup end joining. Nucleic Acids Res. 2014, 42, 6380–6392. [Google Scholar] [CrossRef]
- Soni, A.; Siemann, M.; Pantelias, G.E.; Iliakis, G. Marked cell cycle-dependent contribution of alternative end joining to formation of chromosome translocations by stochastically induced DNA double strand breaks in human cells. Mutat. Res. 2015, 793, 2–8. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Yang, G.; Liu, C.; Chen, S.-H.; Kassab, M.A.; Hoff, J.D.; Walter, N.G.; Yu, X. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 2018, 46, 3446–3457. [Google Scholar] [CrossRef]
- Luedeman, M.E.; Stroik, S.; Feng, W.; Luthman, A.J.; Gupta, G.P.; Ramsden, D.A. Poly(ADP) ribose polymerase promotes DNA polymerase theta-mediated end joining by activation of end resection. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2015, 26, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, K.A.; Alabdullah, M.; Griffin, M.; Toss, M.S.; Fatah, T.M.A.; Alblihy, A.; Moseley, P.; Chan, S.; Rakha, E.A.; Madhusudan, S. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol. Oncol. 2019, 153, 416–424. [Google Scholar] [CrossRef] [PubMed]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Vu, T.V.; Das, S.; Nguyen, C.C.; Kim, J.; Kim, J.Y. Single-strand annealing: Molecular mechanisms and potential applications in CRISPR-Cas-based precision genome editing. Biotechnol. J. 2022, 17, e2100413. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef]
- Domchek, S.M.; Aghajanian, C.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; et al. Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol. Oncol. 2016, 140, 199–203. [Google Scholar] [CrossRef]
- Sisay, M.; Edessa, D. PARP inhibitors as potential therapeutic agents for various cancers: Focus on niraparib and its first global approval for maintenance therapy of gynecologic cancers. Gynecol. Oncol. Res. Pract. 2017, 4, 1–13. [Google Scholar] [CrossRef]
- Yoshihama, T.; Kuroda, Y.; Chiyoda, T.; Takahashi, M.; Yoshimura, T.; Saotome, K.; Nanki, Y.; Sakai, K.; Kobayashi, Y.; Yamagami, W.; et al. Efficacy and safety of olaparib maintenance monotherapy for Japanese patients with platinum-sensitive relapsed ovarian, fallopian tube, and primary peritoneal cancer. Int. J. Clin. Oncol. 2022, 27, 1644–1650. [Google Scholar] [CrossRef]
- Dockery, L.; Gunderson, C.; Moore, K. Rucaparib: The past, present, and future of a newly approved PARP inhibitor for ovarian cancer. Onco Targets Ther. 2017, 10, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Deeks, E.D. Olaparib: First global approval. Drugs 2015, 75, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Eskiler, G.G. Talazoparib to treat BRCA-positive breast cancer. Drugs Today 2019, 55, 459–467. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.-Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP Trapping by BMN 673 and Comparison with Olaparib and Rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Kupczyk, P.; Simiczyjew, A.; Marczuk, J.; Dratkiewicz, E.; Beberok, A.; Rok, J.; Pieniazek, M.; Biecek, P.; Nevozhay, D.; Slowikowski, B.; et al. PARP1 as a Marker of an Aggressive Clinical Phenotype in Cutaneous Melanoma—A Clinical and an In Vitro Study. Cells 2021, 10, 286. [Google Scholar] [CrossRef]
- Raleigh, D.; Ahmed, K.M.; Zhang, H.; Ziaee, S.; Park, C.C. PARP-1 modulates β1-integrin/NF-κB-mediated radioresistance in human breast cancer. J. Cancer Ther. Res. 2016, 5, 1. [Google Scholar] [CrossRef]
- Liu, X.; Han, E.K.; Anderson, M.; Shi, Y.; Semizarov, D.; Wang, G.; McGonigal, T.; Roberts, L.; Lasko, L.; Palma, J.; et al. Acquired Resistance to Combination Treatment with Temozolomide and ABT-888 Is Mediated by Both Base Excision Repair and Homologous Recombination DNA Repair Pathways. Mol. Cancer Res. 2009, 7, 1686–1692. [Google Scholar] [CrossRef]
- Volinia, S.; Galasso, M.; Sana, M.E.; Wise, T.F.; Palatini, J.; Huebner, K.; Croce, C.M. Breast cancer signatures for invasiveness and prognosis defined by deep sequencing of microRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 3024–3029. [Google Scholar] [CrossRef]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis-Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2012, 229, 422–429. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, A.; Sharip, A.; Sharip, A.; Jiang, J.; Yang, Q.; Xie, Y. Reverse the Resistance to PARP Inhibitors. Int. J. Biol. Sci. 2017, 13, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Montoni, A.; Robu, M.; Pouliot, E.; Shah, G.M. Resistance to PARP-Inhibitors in Cancer Therapy. Front. Pharmacol. 2013, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Plummer, R.; Azad, N.S.; Helleday, T. The DNA Damaging Revolution: PARP Inhibitors and Beyond. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 185–195. [Google Scholar] [CrossRef]
- Jannetti, S.A.; Zeglis, B.M.; Zalutsky, M.R.; Reiner, T. Poly(ADP-Ribose)Polymerase (PARP) Inhibitors and Radiation Therapy. Front. Pharmacol. 2020, 11, 170. [Google Scholar] [CrossRef]
- Zhang, N.; Gao, Y.; Zeng, Z.; Luo, Y.; Jiang, X.; Zhang, J.; Li, J.; Zhang, J.; Gong, Y.; Xie, C. PARP inhibitor niraparib as a radiosensitizer promotes antitumor immunity of radiotherapy in EGFR-mutated non-small cell lung cancer. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2021, 23, 1827–1837. [Google Scholar] [CrossRef]
- Lesueur, P.; Chevalier, F.; El-Habr, E.A.; Junier, M.-P.; Chneiweiss, H.; Castera, L.; Müller, E.; Stefan, D.; Saintigny, Y. Radiosensitization Effect of Talazoparib, a Parp Inhibitor, on Glioblastoma Stem Cells Exposed to Low and High Linear Energy Transfer Radiation. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Abbotts, R.; Topper, M.J.; Biondi, C.; Fontaine, D.; Goswami, R.; Stojanovic, L.; Choi, E.Y.; McLaughlin, L.; Kogan, A.A.; Xia, L.; et al. DNA methyltransferase inhibitors induce a BRCAness phenotype that sensitizes NSCLC to PARP inhibitor and ionizing radiation. Proc. Natl. Acad. Sci. USA 2019, 116, 22609–22618. [Google Scholar] [CrossRef]
- Carter, R.; Cheraghchi-Bashi, A.; Westhorpe, A.; Yu, S.; Shanneik, Y.; Seraia, E.; Ouaret, D.; Inoue, Y.; Koch, C.; Wilding, J.; et al. Identification of anticancer drugs to radiosensitise BRAF-wild-type and mutant colorectal cancer. Cancer Biol. Med. 2019, 16, 234–246. [Google Scholar]
- Gerossier, L.; Dubois, A.; Paturel, A.; Fares, N.; Cohen, D.; Merle, P.; Lachuer, J.; Wierinckx, A.; Saintigny, P.; Bancel, B.; et al. PARP inhibitors and radiation potentiate liver cell death in vitro. Do hepatocellular carcinomas have an achilles’ heel? Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101553. [Google Scholar] [CrossRef]
- Jonuscheit, S.; Jost, T.; Gajdošová, F.; Wrobel, M.; Hecht, M.; Fietkau, R.; Distel, L. PARP Inhibitors Talazoparib and Niraparib Sensitize Melanoma Cells to Ionizing Radiation. Genes 2021, 12, 849. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Mina, L.A.; Gonzalez, M.; Curtin, N.J.; Wang, E.; Henshaw, J.W.; Chadha, M.; Sachdev, J.C.; Matei, D.; Jameson, G.S.; et al. First-in-human trial of novel oral PARP inhibtior BMN673 in patients with solid tumors. J. Clin. Oncol. 2013, 31, 2580. [Google Scholar] [CrossRef]
- Carney, B.; Kossatz, S.; Lok, B.H.; Schneeberger, V.; Gangangari, K.K.; Pillarsetty, N.V.K.; Weber, W.A.; Rudin, C.M.; Poirier, J.T.; Reiner, T. Target engagement imaging of PARP inhibitors in small-cell lung cancer. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Loap, P.; Loirat, D.; Berger, F.; Ricci, F.; Vincent-Salomon, A.; Ezzili, C.; Mosseri, V.; Fourquet, A.; Ezzalfani, M.; Kirova, Y. Combination of Olaparib and Radiation Therapy for Triple Negative Breast Cancer: Preliminary Results of the RADIOPARP Phase 1 Trial. Int. J. Radiat. Oncol. 2020, 109, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Abson, C.; Moschetta, M.; Rassy, E.; Karathanasi, A.; Bhat, T.; Ghumman, F.; Sheriff, M.; Pavlidis, N. Poly (ADP-Ribose) Polymerase Inhibitors: Talazoparib in Ovarian Cancer and Beyond. Drugs RD 2020, 20, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Ettl, J.; Quek, R.G.W.; Lee, K.H.; Rugo, H.S.; Hurvitz, S.; Gonçalves, A.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: Patient-reported outcomes from the EMBRACA phase III trial. Ann. Oncol. 2018, 29, 1939–1947. [Google Scholar] [CrossRef]
- Jagsi, R.; Griffith, K.A.; Bellon, J.R.; Woodward, W.; Horton, J.K.; Ho, A.; Feng, F.Y.; Speers, C.; Overmoyer, B.; Sabel, M.; et al. Concurrent Veliparib With Chest Wall and Nodal Radiotherapy in Patients With Inflammatory or Locoregionally Recurrent Breast Cancer: The TBCRC 024 Phase I Multicenter Study. J. Clin. Oncol. 2018, 36, 1317–1322. [Google Scholar] [CrossRef]
- Czito, B.G.; Deming, D.A.; Jameson, G.S.; Mulcahy, M.F.; Vaghefi, H.; Dudley, M.W.; Holen, K.D.; DeLuca, A.; Mittapalli, R.K.; Munasinghe, W.; et al. Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: A phase 1b study. Lancet Gastroenterol. Hepatol. 2017, 2, 418–426. [Google Scholar] [CrossRef]
- Chabot, P.; Hsia, T.C.; Ryu, J.S.; Gorbunova, V.; Belda-Iniesta, C.; Ball, D.; Kio, E.; Mehta, M.; Papp, K.; Qin, Q.; et al. Veliparib in combination with whole-brain radiation therapy for patients with brain metastases from non-small cell lung cancer: Results of a randomized, global, placebo-controlled study. J. Neurooncol. 2017, 131, 105–115. [Google Scholar] [CrossRef]
- Baxter, P.A.; Su, J.M.; Onar-Thomas, A.; Billups, C.A.; Li, X.-N.; Poussaint, T.Y.; Smith, E.R.; Thompson, P.; Adesina, A.; Ansell, P.; et al. A phase I/II study of veliparib (ABT-888) with radiation and temozolomide in newly diagnosed diffuse pontine glioma: A Pediatric Brain Tumor Consortium study. Neuro-Oncology 2020, 22, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.W.; McDonald, K.L.; Lwin, Z.; Barnes, E.H.; Rosenthal, M.; Foote, M.C.; Koh, E.S.; Back, M.; Wheeler, H.; Sulman, E.P.; et al. A randomized phase II trial of veliparib, radiotherapy, and temozolomide in patients with unmethylated MGMT glioblastoma: The VERTU study. Neuro-Oncology 2021, 23, 1736–1749. [Google Scholar] [CrossRef] [PubMed]
- Karam, S.D.; Reddy, K.; Blatchford, P.J.; Waxweiler, T.; DeLouize, A.M.; Oweida, A.; Somerset, H.; Marshall, C.; Young, C.; Davies, K.D.; et al. Final Report of a Phase I Trial of Olaparib with Cetuximab and Radiation for Heavy Smoker Patients with Locally Advanced Head and Neck Cancer. Clin. Cancer Res. 2018, 24, 4949–4959. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Mladenova, V.; Mladenov, E.; Stuschke, M.; Iliakis, G. DNA Damage Clustering after Ionizing Radiation and Consequences in the Processing of Chromatin Breaks. Molecules 2022, 27, 1540. [Google Scholar] [CrossRef]
- Lourenco, L.H.M.; Jiang, Y.; Drobnitzky, N.; Green, M.; Cahill, F.; Patel, A.; Shanneik, Y.; Moore, J.; Ryan, A.J. PARP Inhibition Combined With Thoracic Irradiation Exacerbates Esophageal and Skin Toxicity in C57BL6 Mice. Int. J. Radiat. Oncol. 2018, 100, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Kim, W.; Kloeber, J.A.; Lou, Z. DNA end resection and its role in DNA replication and DSB repair choice in mammalian cells. Exp. Mol. Med. 2020, 52, 1705–1714. [Google Scholar] [CrossRef]
- Caron, M.-C.; Sharma, A.K.; O’Sullivan, J.; Myler, L.R.; Ferreira, M.T.; Rodrigue, A.; Coulombe, Y.; Ethier, C.; Gagné, J.-P.; Langelier, M.-F.; et al. Poly(ADP-ribose) polymerase-1 antagonizes DNA resection at double-strand breaks. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Kong, E.A.Y.; Xu, C.; Sun, X.; Sun, H.; Zhao, X.; He, N.; Ji, K.; Wang, Q.; Du, L.; Wang, J.; et al. BLM helicase inhibition synergizes with PARP inhibition to improve the radiosensitivity of olaparib resistant non-small cell lung cancer cells by inhibiting homologous recombination repair. Cancer Biol. Med. 2021, 18, 1–32. [Google Scholar] [CrossRef]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2019, 48, 1905–1924. [Google Scholar] [CrossRef] [PubMed]
- Albala, J.S.; Thelen, M.P.; Prange, C.; Fan, W.; Christensen, M.; Thompson, L.H.; Lennon, G.G. Identification of a novel human RAD51 homolog, RAD51B. Genomics 1997, 48, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Dardillac, E.; Muhammad, A.; Chailleux, C.; Sesma-Sanz, L.; Ragu, S.; Le Cam, E.; Canitrot, Y.; Masson, J.Y.; Dupaigne, P.; et al. RAD51 protects against nonconservative DNA double-strand break repair through a nonenzymatic function. Nucleic Acids Res. 2022, 50, 2651–2666. [Google Scholar] [CrossRef]
- Kijas, A.W.; Lim, Y.C.; Bolderson, E.; Cerosaletti, K.; Gatei, M.; Jakob, B.; Tobias, F.; Taucher-Scholz, G.; Gueven, N.; Oakley, G.; et al. ATM-dependent phosphorylation of MRE11 controls extent of resection during homology directed repair by signalling through Exonuclease 1. Nucleic Acids Res. 2015, 43, 8352–8367. [Google Scholar] [CrossRef]
- Gunn, A.; Stark, J.M. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Methods Mol. Biol. (Clifton N.J.) 2012, 920, 379–391. [Google Scholar]
- Jelinic, P.; Levine, D.A. New Insights into PARP Inhibitors’ Effect on Cell Cycle and Homology-Directed DNA Damage Repair. Mol. Cancer Ther. 2014, 13, 1645–1654. [Google Scholar] [CrossRef]
- Groth, P.; Orta, M.L.; Elvers, I.; Majumder, M.M.; Lagerqvist, A.; Helleday, T. Homologous recombination repairs secondary replication induced DNA double-strand breaks after ionizing radiation. Nucleic Acids Res. 2012, 40, 6585–6594. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA Double-Strand Break Repair during the Mammalian Cell Cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Cho, J.-E.; Carvajal-Garcia, J.; Smith, C.M.; Headley, K.M.; Hathaway, N.; Ramsden, D.A.; Gupta, G.P. Marker-free quantification of repair pathway utilization at Cas9-induced double-strand breaks. Nucleic Acids Res. 2021, 49, 5095–5105. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e806. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Guo, Y.; Liu, X.; Czauderna, F.; Carr, M.I.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. Therapeutic Implications of p53 Status on Cancer Cell Fate Following Exposure to Ionizing Radiation and the DNA-PK Inhibitor M3814. Mol. Cancer Res. 2019, 17, 2457–2468. [Google Scholar] [CrossRef]
- Dickreuter, E.; Eke, I.; Krause, M.; Borgmann, K.; van Vugt, M.A.; Cordes, N. Targeting of beta1 integrins impairs DNA repair for radiosensitization of head and neck cancer cells. Oncogene 2016, 35, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, A.A.; Pannhausen, J.; Dinger, P.; Wirtz, J.; Knüchel, R.; Gaisa, N.T.; Eble, M.J.; Rose, M. Effective Radiosensitization of Bladder Cancer Cells by Pharmacological Inhibition of DNA-PK and ATR. Biomedicines 2022, 10, 1277. [Google Scholar] [CrossRef] [PubMed]
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome breaks generated by low doses of ionizing radiation in G2-phase are processed exclusively by gene conversion. DNA Repair 2020, 89, 102828. [Google Scholar] [CrossRef]
- Murmann-Konda, T.; Soni, A.; Stuschke, M.; Iliakis, G. Analysis of chromatid-break-repair detects a homologous recombination to non-homologous end-joining switch with increasing load of DNA double-strand breaks. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2021, 867, 503372. [Google Scholar] [CrossRef]
- Waters, C.A.; Strande, N.T.; Wyatt, D.W.; Pryor, J.M.; Ramsden, D.A. Nonhomologous end joining: A good solution for bad ends. DNA Repair 2014, 17, 39–51. [Google Scholar] [CrossRef]
- Wu, S.; Gao, F.; Zheng, S.; Zhang, C.; Martinez-Ledesma, E.; Ezhilarasan, R.; Ding, J.; Li, X.; Feng, N.; Multani, A.; et al. EGFR Amplification Induces Increased DNA Damage Response and Renders Selective Sensitivity to Talazoparib (PARP Inhibitor) in Glioblastoma. Clin. Cancer Res. 2020, 26, 1395–1407. [Google Scholar] [CrossRef]
- Bellare, G.P.; Saha, B.; Patro, B.S. Targeting autophagy reverses de novo resistance in homologous recombination repair proficient breast cancers to PARP inhibition. Br. J. Cancer 2021, 124, 1260–1274. [Google Scholar] [CrossRef]
- Pai Bellare, G.; Sankar Patro, B. Resveratrol sensitizes breast cancer to PARP inhibitor, talazoparib through dual inhibition of AKT and autophagy flux. Biochem. Pharmacol. 2022, 199, 115024. [Google Scholar] [CrossRef]
- Oh, S.; Harvey, A.; Zimbric, J.; Wang, Y.; Nguyen, T.; Jackson, P.J.; Hendrickson, E.A. DNA ligase III and DNA ligase IV carry out genetically distinct forms of end joining in human somatic cells. DNA Repair 2014, 21, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Masani, S.; Han, L.; Meek, K.; Yu, K. Redundant function of DNA ligase 1 and 3 in alternative end-joining during immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. USA 2016, 113, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Pol[thgr]-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res./Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef]
- Wray, J.; Williamson, E.A.; Singh, S.B.; Wu, Y.; Cogle, C.; Weinstock, D.M.; Zhang, Y.; Lee, S.-H.; Zhou, D.; Shao, L.; et al. PARP1 is required for chromosomal translocations. Blood 2013, 121, 4359–4365. [Google Scholar] [CrossRef]
- Bai, W.; Zhu, G.; Xu, J.; Chen, P.; Meng, F.; Xue, H.; Chen, C.; Dong, J. The 3′-flap endonuclease XPF-ERCC1 promotes alternative end joining and chromosomal translocation during B cell class switching. Cell Rep. 2021, 36, 109756. [Google Scholar] [CrossRef]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nature Reviews. Cancer 2013, 13, 443–454. [Google Scholar] [PubMed]
- Soutoglou, E.; Misteli, T. Mobility and immobility of chromatin in transcription and genome stability. Curr. Opin. Genet. Dev. 2007, 17, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class polymerase theta inhibitor selectively targets homologous-recombination-deficient tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Krastev, D.B.; Li, S.; Sun, Y.; Wicks, A.J.; Hoslett, G.; Weekes, D.; Badder, L.M.; Knight, E.G.; Marlow, R.; Pardo, M.C.; et al. The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin. Nature 2022, 24, 62–73. [Google Scholar] [CrossRef]
- Juhász, S.; Smith, R.; Schauer, T.; Spekhardt, D.; Mamar, H.; Zentout, S.; Chapuis, C.; Huet, S.; Timinszky, G. The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci. Adv. 2020, 6, eabb8626. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial Phase | Disease Setting | HR Status | Treatments | Eligible Enrolled Patients | Efficacy | Ref. |
|---|---|---|---|---|---|---|
| I | Inflammatory or loco-regionally recurrent breast cancer | N/A | RT + veliparib | 30 | 3-year OS-56.6%, PFS -50% | [118] |
| I | Locally advanced rectal cancer | N/A | RT + capecitabine + veliparib | 32 | Tumor downstaging after surgery-71%; pCR-29% | [119] |
| II | Brain metastases from NSCLC | N/A | WBRT +/− veliparib | Randomization (1:1:1); | WBRT alone | [120] |
| 103 in 50mg veliparib arm, 102 in 200 mg veliparib arm, 102 in control arm | Median OS: 185 days | |||||
| WBRT+50 mg veliparib | ||||||
| Median OS: 209 days (p = 0.933) | ||||||
| WBRT+200 mg veliparib | ||||||
| Median OS: 209 days (p = 0.909) | ||||||
| I/II | Diffuse pontine gliomas | N/A | RT + veliparib | 65 | Phase I | [121] |
| PR(0%) SD-91.7% PD(8.3%) | ||||||
| Phase II | ||||||
| PR(13.2%) SD-71.7% PD-9.4% | ||||||
| II | Unmethyated MGMT glioblastoma | N/A | RT+ temozolomide +/− veliparib | Randomization (2:1) | Without veliparib | [122] |
| 84 in veliparib arm and 41 in control arm | PFS at 6 months-31%. Median OS: 12.8months | |||||
| With veliparib | ||||||
| PFS at 6 months (46%). Median OS: 12.7 months | ||||||
| I | Head and neck squamous cell carcinoma | N/A | olaparib + RT + cetuximab | 15 | Median OS: 37 months | [123] |
| 2-year OS (72%), PFS (63%) |
| Trial Phase | Disease Setting | HR Status | Treatments | Trial Status | Identifier |
|---|---|---|---|---|---|
| I | Extensive-stage small cell lung cancer | N/A | BMN673 + low dose consolidative thoracic RT | Recruiting | NCT04170946 |
| I | Locally recurrent gynecologic cancers | N/A | BMN673 + RT | Recruiting | NCT03968406 |
| II | Metastatic triple negative breast cancer | gBRCA 1/2 negative | BMN673 + Atezolizumab + SBRT | Recruiting | NCT04690855 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soni, A.; Lin, X.; Mladenov, E.; Mladenova, V.; Stuschke, M.; Iliakis, G. BMN673 Is a PARP Inhibitor with Unique Radiosensitizing Properties: Mechanisms and Potential in Radiation Therapy. Cancers 2022, 14, 5619. https://doi.org/10.3390/cancers14225619
Soni A, Lin X, Mladenov E, Mladenova V, Stuschke M, Iliakis G. BMN673 Is a PARP Inhibitor with Unique Radiosensitizing Properties: Mechanisms and Potential in Radiation Therapy. Cancers. 2022; 14(22):5619. https://doi.org/10.3390/cancers14225619
Chicago/Turabian StyleSoni, Aashish, Xixi Lin, Emil Mladenov, Veronika Mladenova, Martin Stuschke, and George Iliakis. 2022. "BMN673 Is a PARP Inhibitor with Unique Radiosensitizing Properties: Mechanisms and Potential in Radiation Therapy" Cancers 14, no. 22: 5619. https://doi.org/10.3390/cancers14225619
APA StyleSoni, A., Lin, X., Mladenov, E., Mladenova, V., Stuschke, M., & Iliakis, G. (2022). BMN673 Is a PARP Inhibitor with Unique Radiosensitizing Properties: Mechanisms and Potential in Radiation Therapy. Cancers, 14(22), 5619. https://doi.org/10.3390/cancers14225619