

Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling
 , and
, and 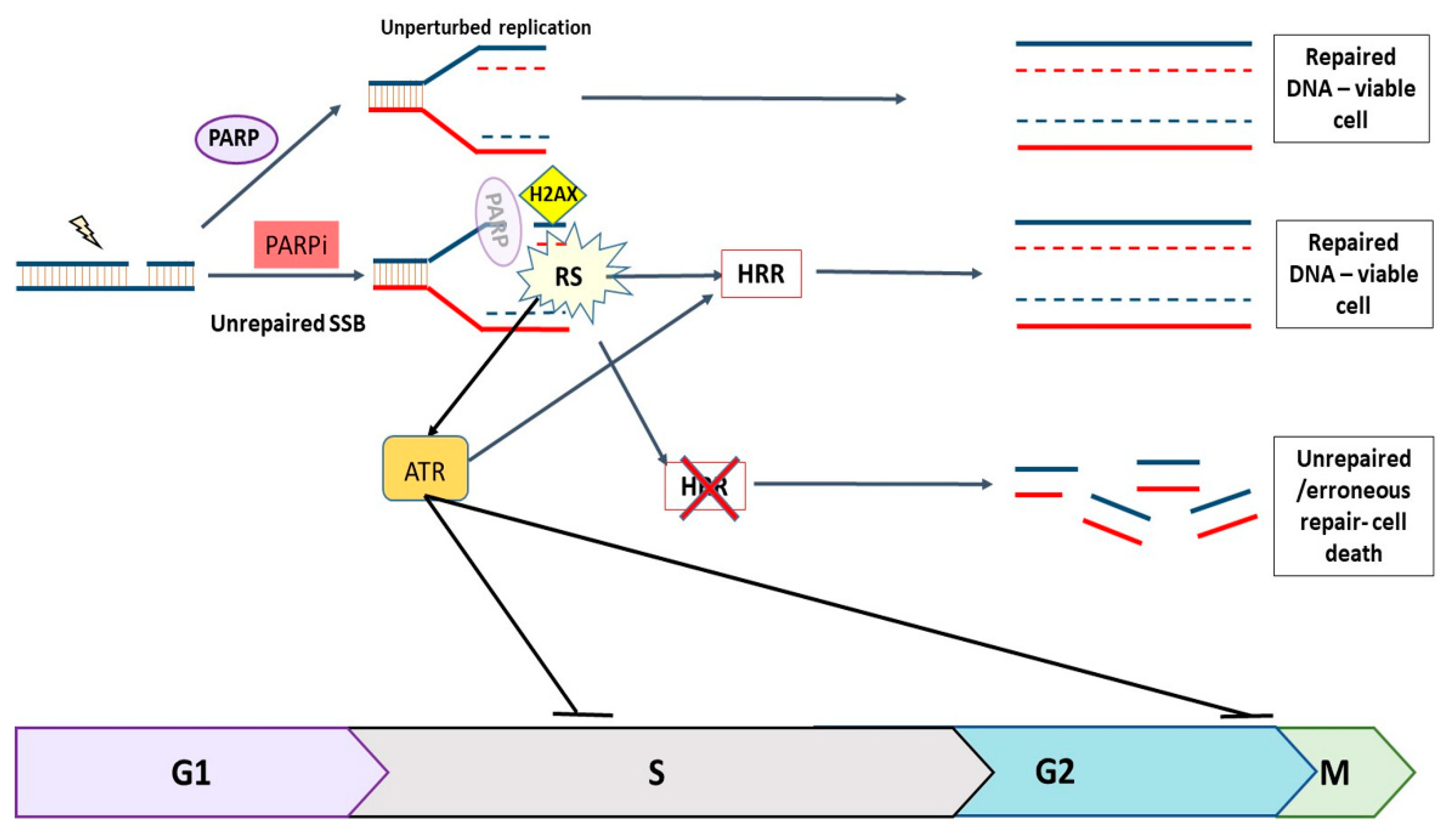{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. PARP Activity Assay
2.4. Western Blot Assay
2.5. Cytotoxicity Scheduling Assays
3. Results
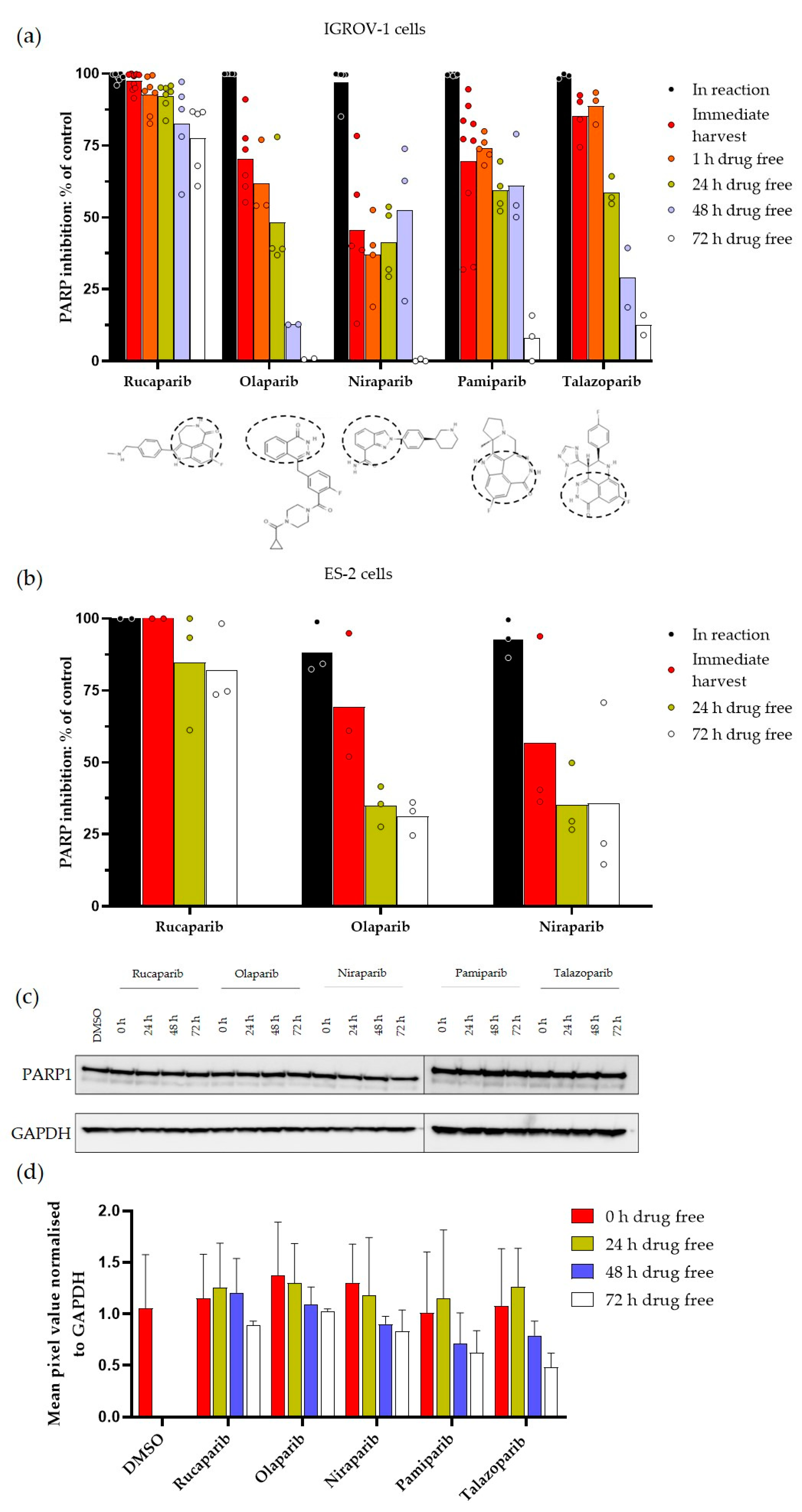
3.1. Durability of PARP Inhibition by Rucaparib, Olaparib, Niraparib, Pamiparib and Talazoparib
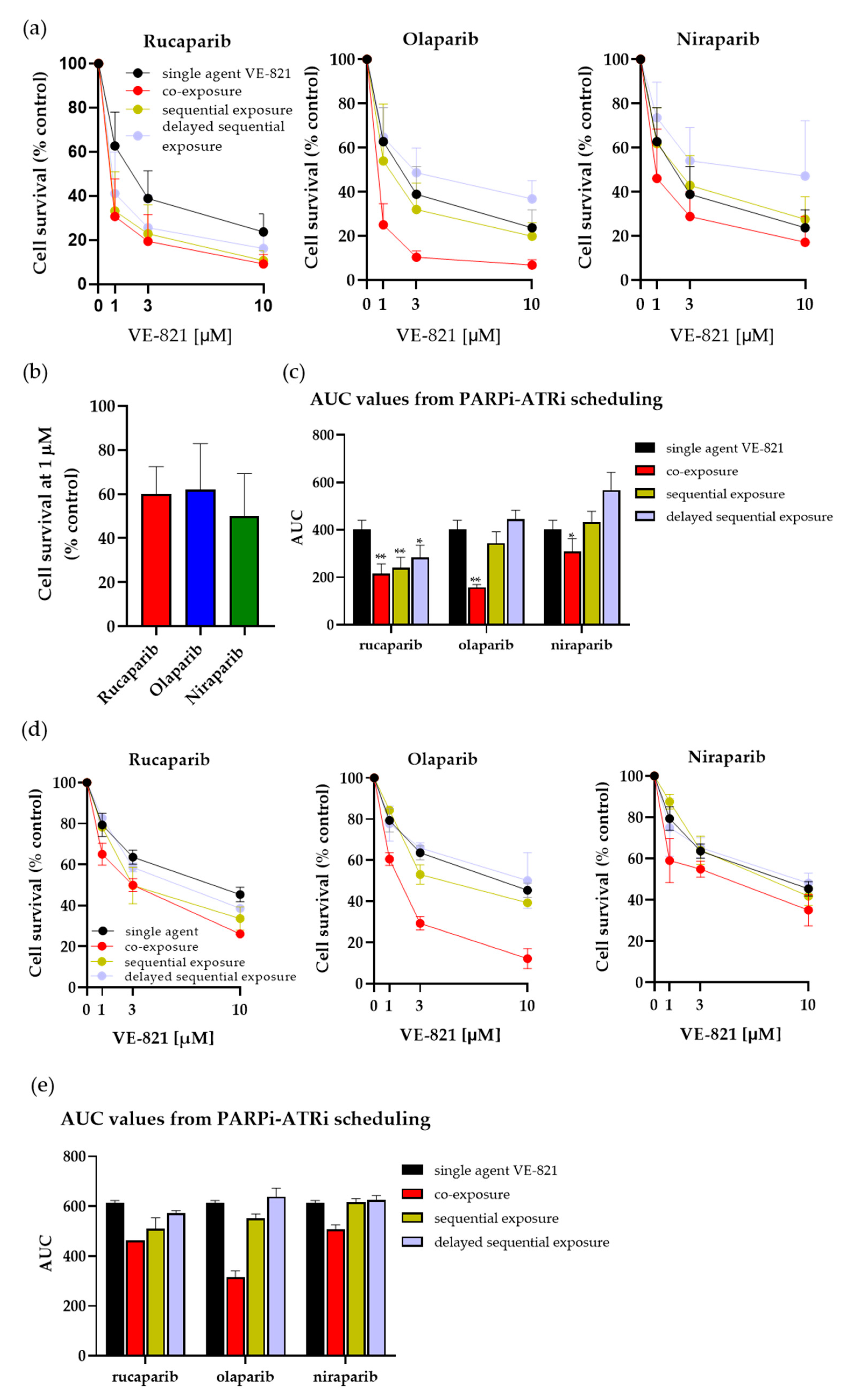
3.2. Investigating Schedules of PARPi and ATRi
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 564–601. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Mikuła, M.; Ostrowski, J.; Śliwińska, A.; Rogalska, A. PARP Inhibition Increases the Reliance on ATR/CHK1 Checkpoint Signaling Leading to Synthetic Lethality-An Alternative Treatment Strategy for Epithelial Ovarian Cancer Cells Independent from HR Effectiveness. Int. J. Mol. Sci. 2020, 21, 9715. [Google Scholar] [CrossRef]
- Smith, H.L.; Southgate, H.; Tweddle, D.A.; Curtin, N.J. DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 2020, 22, e2. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly (ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Setton, J.; Zinda, M.; Riaz, N.; Durocher, D.; Zimmermann, M.; Koehler, M.; Reis-Filho, J.S.; Powell, S.N. Synthetic Lethality in Cancer Therapeutics: The Next Generation. Cancer Discov. 2021, 11, 1626–1635. [Google Scholar] [CrossRef]
- Markham, A. Pamiparib: First approval. Drugs 2021, 81, 1343–1348. [Google Scholar] [CrossRef]
- Lee, A. Fuzuloparib: First Approval. Drugs 2021, 81, 1221–1226. [Google Scholar] [CrossRef]
- Javle, M.; Curtin, N.J. The role of PARP in DNA repair and its therapeutic exploitation. Br. J. Cancer 2011, 105, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Michelena, J.; Lezaja, A.; Teloni, F.; Schmid, T.; Imhof, R.; Altmeyer, M. Analysis of PARP inhibitor toxicity by multidimensional fluorescence microscopy reveals mechanisms of sensitivity and resistance. Nat. Commun. 2018, 9, 2678. [Google Scholar] [CrossRef] [PubMed]
- Simoneau, A.; Xiong, R.; Zou, L. The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 2021, 35, 1271–1289. [Google Scholar] [CrossRef] [PubMed]
- Murthy, P.; Muggia, F. Women’s cancers: How the discovery of BRCA genes is driving current concepts of cancer biology and therapeutics. Cancer Med. Sci. 2019, 13, 904. [Google Scholar] [CrossRef]
- Dickson, K.A.; Xie, T.; Evenhuis, C.; Ma, Y.; Marsh, D.J. PARP Inhibitors Display Differential Efficacy in Models of BRCA Mutant High-Grade Serous Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 8506. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Liu, C.; Chang, C.; Shen, C.; Yin, Y.; Yin, X.; Jiang, Z.; Zhao, Z.; Mu, M.; Cao, D.; et al. Comparative safety and tolerability of approved PARP inhibitors in cancer: A systematic review and network meta-analysis. Pharmacol. Res. 2021, 172, 105808. [Google Scholar] [CrossRef] [PubMed]
- Krastev, D.B.; Wicks, A.J.; Lord, C.J. PARP Inhibitors—Trapped in a Toxic Love Affair. Cancer Res. 2021, 81, 5605–5607. [Google Scholar] [CrossRef]
- Plummer, R.; Jones, C.; Middleton, M.; Wilson, R.; Evans, J.; Olsen, A.; Curtin, N.; Boddy, A.; McHugh, P.; Newell, D.; et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin. Cancer Res. 2008, 14, 7917–7923. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.; Thomas, H.; Berry, P.; Kyle, S.; Patterson, M.; Jones, C.; Los, G.; Hostomsky, Z.; Plummer, E.R.; Boddy, A.V.; et al. Tumour cell retention of rucaparib, sustained PARP inhibition and efficacy of weekly as well as daily schedules. Br. J. Cancer 2014, 110, 1977–1984. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstrat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian cancer cell line panel (OCCP): Clinical importance of in vitro morphological subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Stordal, B.; Timms, K.; Farrelly, A.; Gallagher, D.; Busschots, S.; Renaud, M.; Thery, J.; Williams, D.; Potter, J.; Tran, T.; et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol. Oncol. 2013, 7, 567–569. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.S.; Gentles, L.; Bradbury, A.; Brecht, D.; Robinson, R.; O’Donnell, R.; Curtin, N.J.; Drew, Y. Genomic, Transcriptomic and Functional Alterations in DNA Damage Response Pathways as Putative Biomarkers of Chemotherapy Response in Ovarian Cancer. Cancers 2021, 13, 1420. [Google Scholar] [CrossRef] [PubMed]
- Canan, S.; Maegley, K.; Curtin, N.J. Strategies Employed for the Development of PARP Inhibitors. Methods Mol. Biol. 2017, 1608, 271–297. [Google Scholar] [PubMed]
- Schwanhäusser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Long, M.J.; Gollapalli, D.R.; Hedstrom, L. Inhibitor mediated protein degradation. Chem. Biol. 2012, 19, 629–637. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.Ü.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Wang, S.; Han, L.; Han, J.; Li, P.; Ding, Q.; Zhang, Q.J.; Liu, Z.P.; Chen, C.; Yu, Y. Uncoupling of PARP1 trapping and inhibition using selective PARP1 degradation. Nat. Chem. Biol. 2019, 15, 1223–1231. [Google Scholar] [CrossRef]
- Todisco, E.; Gigli, F.; Ronchini, C.; Amato, V.; Sammassimo, S.; Pastano, R.; Parma, G.; Lapresa, M.T.; Bertolini, F.; Corsini, C.; et al. Hematological disorders after salvage PARPi treatment for ovarian cancer: Cytogenetic and molecular defects and clinical outcomes. Int. J. Cancer 2022, 151, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Peasland, A.; Wang, L.Z.; Rowling, E.; Kyle, S.; Chen, T.; Hopkins, A.; Cliby, W.A.; Sarkaria, J.; Beale, G.; Edmondson, R.J.; et al. Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 2011, 105, 372–381. [Google Scholar] [CrossRef]
- Middleton, F.K.; Patterson, M.J.; Elstob, C.J.; Fordham, S.; Herriott, A.; Wade, M.A.; McCormick, A.; Edmondson, R.; May, F.E.; Allan, J.M.; et al. Common cancer-associated imbalances in the DNA damage response confer sensitivity to single agent ATR inhibition. Oncotarget 2015, 6, 32396–32409. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, H.L.; Willmore, E.; Mukhopadhyay, A.; Drew, Y.; Curtin, N.J. Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling. Cancers 2022, 14, 5559. https://doi.org/10.3390/cancers14225559
Smith HL, Willmore E, Mukhopadhyay A, Drew Y, Curtin NJ. Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling. Cancers. 2022; 14(22):5559. https://doi.org/10.3390/cancers14225559
Chicago/Turabian StyleSmith, Hannah L., Elaine Willmore, Asima Mukhopadhyay, Yvette Drew, and Nicola J. Curtin. 2022. "Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling" Cancers 14, no. 22: 5559. https://doi.org/10.3390/cancers14225559
APA StyleSmith, H. L., Willmore, E., Mukhopadhyay, A., Drew, Y., & Curtin, N. J. (2022). Differences in Durability of PARP Inhibition by Clinically Approved PARP Inhibitors: Implications for Combinations and Scheduling. Cancers, 14(22), 5559. https://doi.org/10.3390/cancers14225559