1. Introduction
Prostate cancer is the most common male cancer. Despite tremendous advancements made on improving early diagnosis and treatment, resistance to current chemotherapy drugs still occurs [
1]. Currently, this is an incurable disease and the only strategy available is the use of chemotherapy and androgen deprivation therapy [
2,
3,
4]. Targeted therapies such as tyrosine kinase inhibitors hold promise but are only under clinical trials and not standard of care for prostate cancer. There is need for better therapeutic strategies to treat prostate cancer.
Lysosomotropic agents are molecules able to penetrate lysosome membranes and induce lysosome membrane permeabilization (LMP). Many of these molecules were designed for clinical use as antihistamines or antidepressants that have LMP properties [
5]. Many of these lysosomotropic agents were capable of penetrating and accumulating within lysosomes and inducing cell death through the accumulation of reactive oxygen species in many cancer cell lines [
6]. In contrast, lysosomotropic agents are less effective in non-malignant cell lines [
7,
8]. In addition, these lysosomotropic agents were effective at inducing cell death in in-vitro models of cancer such as breast, lung, chronic lymphocytic leukemia (CLL) and glioblastoma at relatively low doses [
7,
9,
10]. Of these lysosomotropic agents, siramesine is one of the most potent inducers of lipid reactive oxygen species (ROS) and cell death in many different cancer cells [
11]. In CLL, acute myeloid leukemia (AML) and breast cancer cells treatment with low doses of siramesine was shown to induce massive lipid peroxidation and mitochondria dysfunction since lipid peroxidation can decrease the mitochondrial membrane potential, leading to cell death [
8,
10,
12]. It is unknown whether prostate cancer cells are as sensitive to lysosomotropic agents alone or in combination with other anti-cancer drugs.
Lapatinib is a small molecule targeting these two receptors simultaneously and is currently approved by the Food and Drug Administration (FDA) for the treatment of breast cancer. Given its effectiveness in clinical trials for the treatment of breast cancer, and its mechanism of action targeting HER2 receptors altered in prostate cancer, lapatinib was considered as a new strategy for the treatment of advanced prostate cancer [
13,
14,
15]. However, the single therapy therapeutic agent lapatinib was not effective in clinical trials. In one phase II clinical trial, treatment with lapatinib in patients with hormone-sensitive prostate cancer showed no significant antitumor activity although the drug was well tolerated by patients. In a separate phase II trial, a small population of patients with castration-resistant prostate cancer showed some reduction in bone metastasis (7 out of 29) and PSA levels (1 out of 21). Since lapatinib showed small positive results in some patients, combination treatments are therefore being investigated to test its effectiveness [
13]. Lysosome-disrupting agents in combination with tyrosine kinase inhibitors were shown to cause synergistic cell death in cancer types such as breast, lung, CLL and glioblastoma cell lines [
7,
8,
9,
12,
16]. It is still unknown whether a combination with lysosomotropic agents may lead to triggering events of cell death in advanced prostate cancer cells.
Herein, we demonstrate that the lysosomotropic agent, sirasemine, was effective at inducing cell death in a variety of prostate cancer cell lines mediated by ROS. Moreover, the combination of siramesine and Lapatinib induced an increase in ROS, mitochondrial dysfunction and apoptotic cell death in prostate cancers.
2. Materials and Methods
2.1. Cell Culture
The three cell lines used were PC3, DU145 and LNCaP which were purchased from ATCC. Cell lines were maintained in a humidified 5% CO2 environment (normoxia = 21% O2) at 37 °C. PC3 cells were cultured in DMEM/F-12 (Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12; Gibco, Life Technologies, Indianapolis, IN, USA), DU145 and LNCaP cells were cultured in RPMI 1640 Medium (Gibco, Life Technologies) and all cell lines were supplemented with 5% (v/v) fetal bovine serum (FBS; Life Technologies) and 1% Penicillin/Streptomycin (Pen/Strep) (Gibco, Life Technologies). Cells were grown in 100 × 20 mm2 tissue culture plates (Sarstedt, Newton, MA, USA, Version Five). Generally, cells were passaged upon reaching ~80% confluency (see below). RWPE prostate endothelial cells were purchased from ATCC and grown in Keratinocyte Serum Free Medium (K-SFM) with bovine pituitary extract (BPE) and human recombinant epidermal growth factor (EGF) added per instructions.
2.2. Passaging of Cells
All three cell lines were passaged at a 1:4 ratio after reaching ~80% confluency. To detach strongly adherent PC3 cells from culture plates, 3 mL of Trypsin-Ethylenediaminetetraacetic acid (EDTA) (0.05%) (Gibco) was added and incubated at 37 °C for 5–6 min. For DU145 cells, 2 mL of trypsin-EDTA was added and incubated for 5 min. Trypsin-sensitive LNCaP cells were incubated with 2 mL of trypsin for a maximum of 2 min. To stop the proteolytic reaction, cells were neutralized with media up to 10 mL in total. Cells were centrifuged at 1200 rpm for 5 min and resuspended in 10 mL of fresh growth media and dispensed into new 100 × 20 mm tissue culture plates. Fresh growth media was added every 2–3 days.
2.3. Drug Treatments
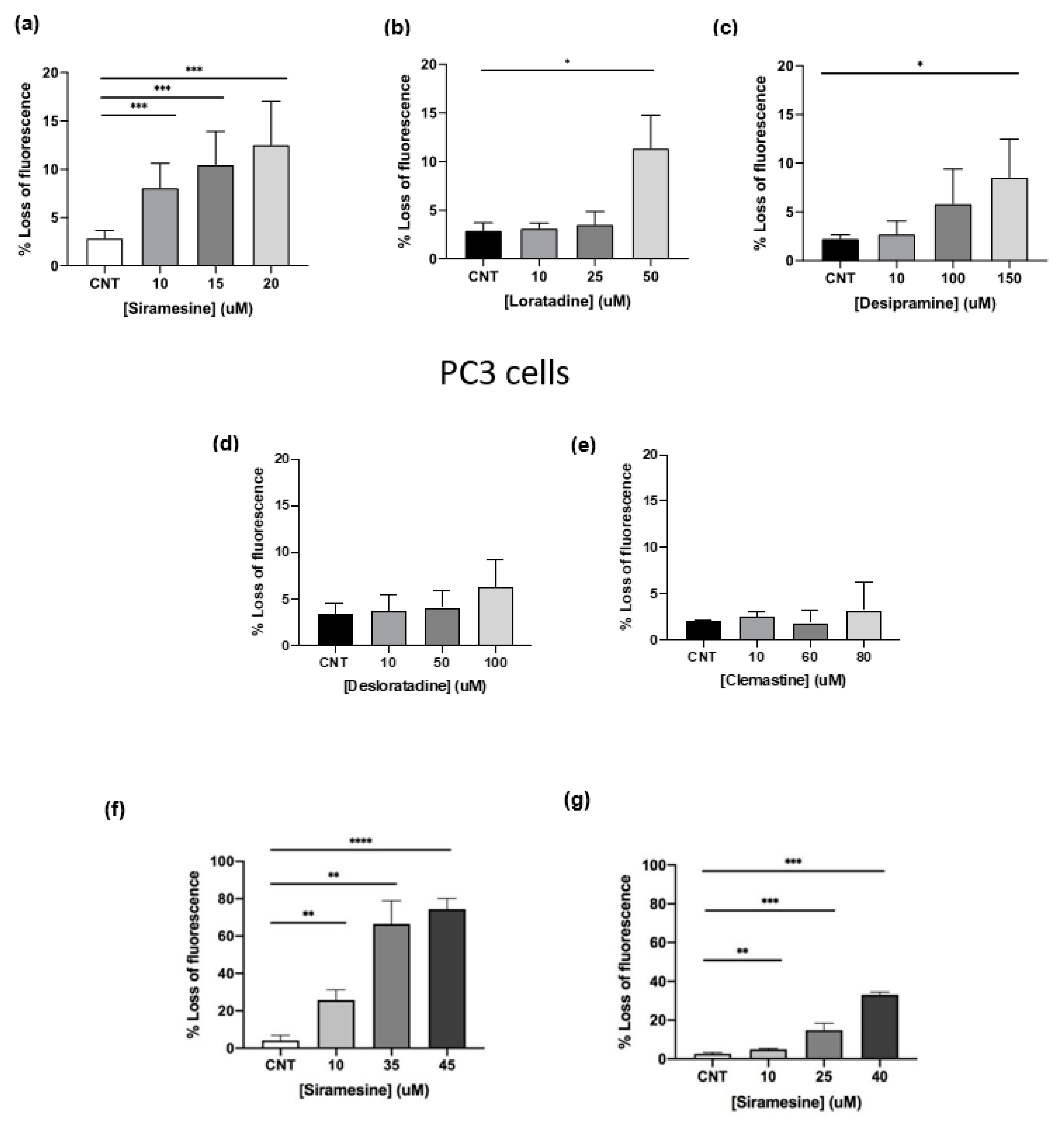
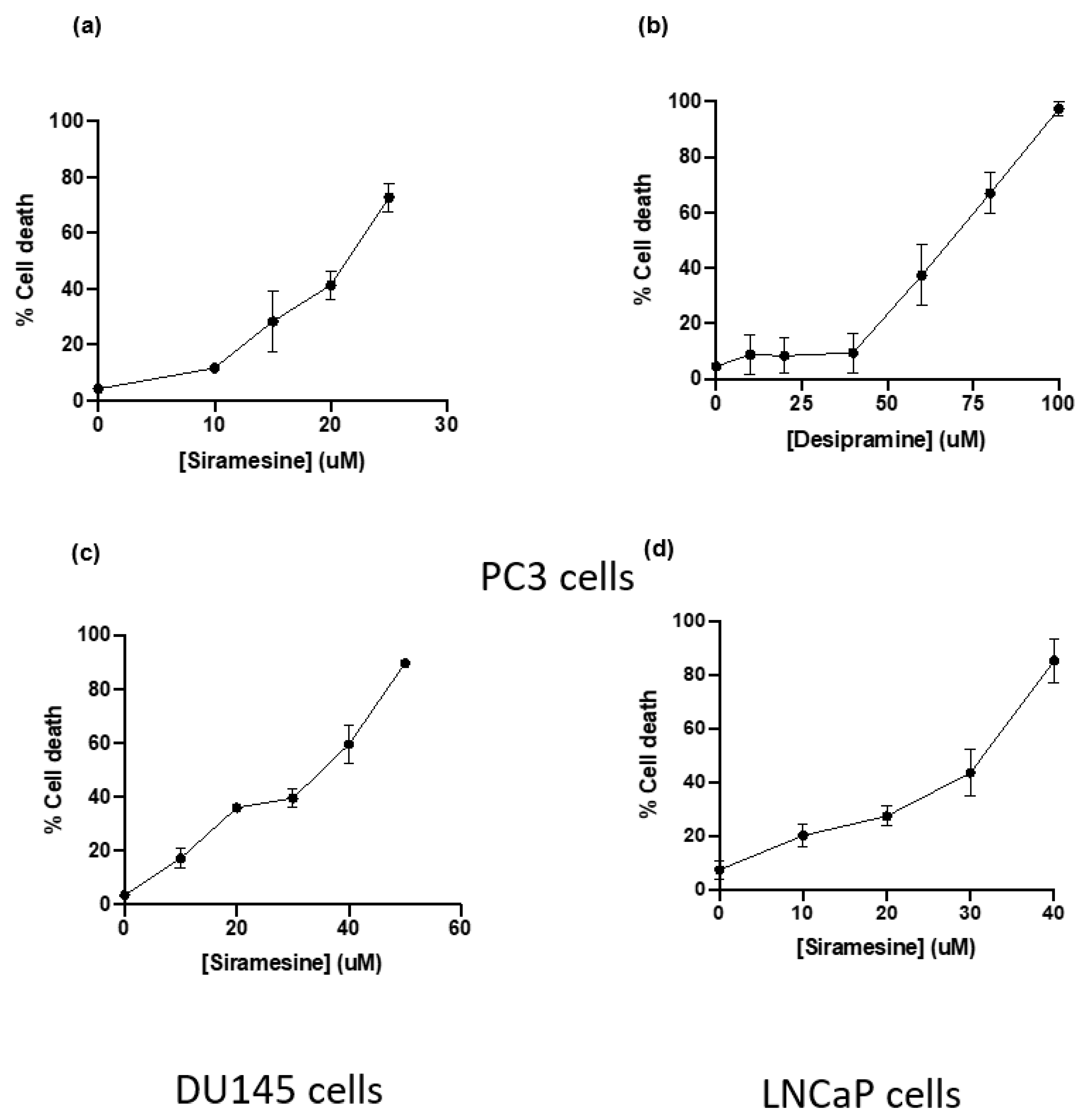
Drugs were stored in single use aliquots and were used fresh for each experiment. A 24 h dose–response curve was generated for PC3 cells using lysosomotropic agents (siramesine, desipramine, clemastine, loratadine and desloratadine), tyrosine kinase inhibitors (lapatinib, gefitinib and sorafenib), and chemotherapy drugs (paclitaxel and etoposide). A dose–response curve for siramesine was tested in all three cell lines. In addition, the LC50 for siramesine, desipramine and clemastine was determined for assays to study their effects on cell lines. For drug combination experiments, the concentrations used were based on the lowest concentration of each drug that induced between 10–15% cell death at 24 h.
2.4. Cell Death Assays
PC3, DU145 and LNCaP cells were added to 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. On the day of treatment, fresh media was added before the addition of drugs and incubated at 37 °C for 24 h, unless otherwise indicated. For experiments where inhibitors were used, cells were incubated with inhibitors for 1 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After treatment, cells were collected, together with the media, and resuspended in phosphate saline buffer (PBS) and stained with 10 μL of 0.04% Trypan blue (Sigma, Oakville, ON, Canada). Trypan blue can enter cells through membrane pores of dying cells while being excluded from live ones. Cells were analyzed within 5 min of trypan blue addition using the Novocyte flow cytometer (Acea Biosciences, San Diego, CA, USA). A total of 20,000 events were collected from each sample and gated using the PER-CP channel and data was analyzed using CellQuest software. To detect early and late apoptotic events, cells were resuspended in 1X Binding Buffer with AnnexinV (BD) and 7AAD (BD) dyes diluted in 1X Binding Buffer. Cells were incubated at 37 °C for 15 min and immediately analyzed by flow cytometry. A total of 20,000 events were collected and gated using FITC and PER-CP channels. Events positive for either FITC or PER-CP were considered apoptotic cells.
2.5. Combination Index Method
PC3 cells (1.5 × 10
4) were seeded per well of a 96-well flat bottom plate and grown overnight with 100 μL DMEM F12 (5% FBS, 1% penicillin-streptomycin). Cells were treated with DMSO as a negative control, and varying concentrations of lapatinib (3–10 μM), siramesine (5–30 μM) or a combination of 10 μM siramesine and 0.5 μM lapatinib in 100 μL media for 24 h. An amount of 10 μL MTS assay reagent (CellTiter 96
® AQueous Non-Radioactive Cell Proliferation Assay (MTS), Promega, Madison, WI, USA) was added to each well after treatment, and the plate was incubated for 4 h at 37 °C in the dark before obtaining readings. Wells containing only media were used to normalize background. Percentage cell death was normalized to negative control and plotted against treatment concentrations, and lines of best fit were drawn to obtain the IC
50 for siramesine or lapatinib. Combination index was calculated using the formula
where (
D)
1 and (
D)
2 are the doses of siramesine and lapatinib in combination required to reach the IC
50, respectively, and (
Dx)
1 and (
Dx)
2 are the doses of siramesine and lapatinib alone that are required to reach the IC
50 as single agent treatments, respectively. A combination index of <1 indicates synergy.
2.6. Cell Viability Assay
PC3 cells (1.5 × 104) were seeded per well of a 96-well flat bottom plate and grown overnight with 100 μL DMEM F12 (5% FBS, 1% penicillin-streptomycin). Cells were treated with DMSO as a negative control, 0.5 μM lapatinib, 10 μM siramesine, or a combination of siramesine and lapatinib in 100 μL media for 24 h. An amount of 10 μL MTS assay reagent (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS), Promega) was added to each well after treatment, and the plate was incubated for 4 h at 37 °C in the dark before obtaining readings. Wells containing only media were used to normalize background. Percentage cell death was normalized to negative control.
2.7. Membrane Permeability Assay
PC3, DU145 and LNCaP cells were seeded in 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After 4 h treatment the media was removed from cells and Lysotracker Deep Red dye (50 nM, Invitrogen, Carlsbad, CA, USA) was diluted in fresh media and added to cells for 15 min at 37 °C in the dark. Cells were collected, together with the media, and resuspended in PBS and analyzed using the Novocyte flow cytometer (Acea Biosciences). An amount of 20,000 events were collected from each sample and gated using the PER-CP channel. Data was analyzed using CellQuest software.
2.8. Lysotracker Assay
PC3 cells (3 × 105) were seeded per 60 mm plate on glass coverslips and treated for 4 h with DMSO as a negative control, 0.5 μM lapatinib, 10 μM siramesine or a combination of siramesine and lapatinib in 2 mL DMEM F12 (5% FBS, 1% penicillin-streptomycin) media. Cells were stained for 30 min at 37 °C in the dark with 100 nM LysoTrackerTM Red DND-99 (Invitrogen) in 2 mL media, washed with PBS, and fixed for 20 min with 1 mL paraformaldehyde at room temperature. Cells were mounted with 10 μL mounting media containing DAPI. Images were obtained by confocal microscopy.
2.9. ROS Detection Assay
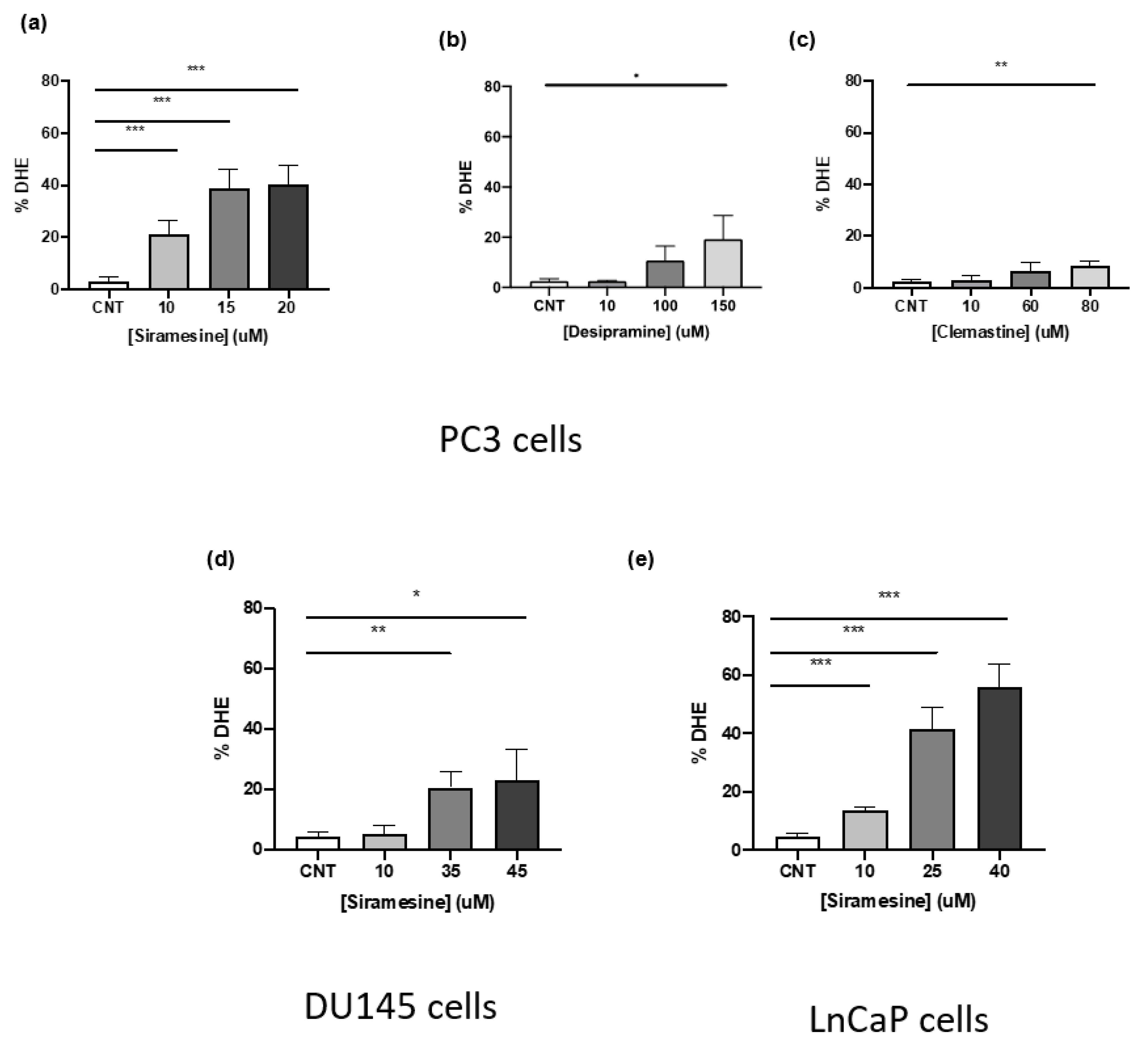
PC3, DU145, and LNCaP cells were seeded in 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After 4 h or 24 h treatment, cells were collected, together with the media, and resuspended in PBS and stained with 3.2 μM dihydroethidium (DHE, Invitrogen) for 30 min at 37 °C in the dark. Cells were analyzed using the Novocyte flow cytometer (Acea Biosciences) and Cellquest software. An amount of 20,000 events were collected from each sample and gated using the PER-CP channel.
2.10. Mitochondria Membrane Potential Assay
PC3 cells were seeded in 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After 24 h treatment, cells were collected together with the media and resuspended in PBS and stained with 25 nM tetramethylrhodamine (TMRM, Invitrogen) for 30 min at 37 °C. Cells were analyzed using the Novocyte flow cytometer (Acea Biosciences) and Cellquest software. An amount of 20,000 events were collected from each sample and gated using the PE channel.
2.11. Mitochondria Superoxide Detection Assay
PC3 cells were seeded in 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After 24 h treatment, cells were collected together with the media and resuspended in PBS and stained with 5 μM Mitosox Red (Invitrogen) for 10 min at 37 °C in the dark. Cells were analyzed using the Novocyte flow cytometer (Acea Biosciences) and Cellquest software (Version five). An amount of 20,000 events were collected from each sample and gated using the PE channel.
2.12. Lipid Peroxidation Assay
PC3 cells were seeded in 12-well plates at a concentration of 1.0 × 105 cells/mL and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After 24 h treatment, cells were collected together with the media and resuspended in PBS and stained with 1 μM C11-BODIPY (Invitrogen) for 30 min at 37 °C in the dark. Cells were analyzed using the Novocyte flow cytometer (Acea Biosciences) and Cellquest software. An amount of 20,000 events were collected from each sample and gated using the PE channel.
2.13. Western Blot
PC3 cells were cultured as described above and seeded in 6-well plates at a concentration of 3.0 × 105 cells/mL, and allowed to grow for 42 h before treatment. Depending on the solvent used to dilute drugs, DMSO or water was added as vehicle control. After treatment, cells were collected and lysed in 1% NP40 Lysis buffer. Protein was quantified using the Denovix DS-11 UV-Vis spectrophotometer. Samples were loaded into pre-made 4–20% acrylamide Mini-PROTEAN TGX Stain-Free Protein gels (Bio-rad, Montreal, QC, Canada). PageRuler Plus Prestained Protein ladder (Thermo Fisher, Waltham, WA, USA) was loaded in one well to determine the molecular weight of proteins. Antibodies against PARP was purchased from Thermofisher (cat. #MA3-950) and caspase 3 (cat. #06-735) and tubulin (cat. #T5168) were purchased from Cell Signaling Inc., Boston, MA, USA. The antibodies were incubated on membranes (1:1000 ratio) per manufacture instructions. Membranes were developed by enhanced chemiluminescence using the Pierce ECL Western Blotting substrate (Thermo Fisher) according to manufacturer’s instructions. The membranes were imaged using the ImageQuant LAS 500 gel imager (GE Healthcare Life Sciences, Chicago, IL, USA). For all western blot membranes, the reference gene, Actin, was used as a loading control. Protein images obtained from the gel imager were quantified using ImageJ software (v. 2.0).
2.14. Statistical Analysis
All graphs were generated using GraphPad Prism 7. Statistical analysis for flow cytometry experiments were conducted using GraphPad Prism 7. Statistical significance was determined using a two-tailed unpaired t-test for all treatments and control samples. A p-value < 0.05 was considered statistically significant (represented by *) in addition to a p-value < 0.01 (represented by **), and a p-value < 0.001 (represented by ***). Error bars represent standard error of the mean for each treatment, and a minimum of three independent replicates were included for each experiment. For normalization of protein expression levels, Microsoft Excel 2017 was used.
4. Discussion
Drug resistance is the main obstacle for effectively treating advanced prostate cancer. New therapeutic strategies need to be developed. Targeting lysosomes in combination with tyrosine kinase inhibitors to treat aggressive cancer were effective in several in-vitro models of breast, lung, glioblastoma and CLL [
7,
8,
9,
10,
21]. However, it has not been investigated in prostate cancer cells. We found the lysosomotropic agent, siramesine, was the most effective at inducing cell death in prostate cancer cells through increased lipid peroxidation. When combined with tyrosine kinase inhibitor lapatinib, the amount of cell death significantly increased. This suggests that lysosomotropic agents in combination with tyrosine kinase inhibitors could be an effective treatment strategy in prostate cancer. The limitation of the study is the use of cell lines and whether these treatment doses are clinically achievable. In the future, we will investigate the clinically achievable doses using animal and organoid models.
The idea of repurposing lysosomotropic agents originally designed to treat health conditions such as depression, or the use of antihistamines with lysosomotropic properties had successful therapeutic results in a wide range of cancer types [
22]. Since then, studies have found several more compounds that accumulate within lysosomes that induce cell death in a relatively short time [
23]. In CLL cells, treatment with antihistamines (loratadine, desloratadine, clemastine) and antidepressants (siramesine and desipramine) significantly increased cell death compared to non-malignant cells [
12,
21]. We found clemastine, desipramine and siramesine to induce to cell death in PC3 cells and siramesine was the most effective drug at low concentrations. Antihistamines desloratadine and loratadine did not induce cell death at any of the concentrations tested despite its effectiveness in other cancer types. These differences in results could be due to prostate cancer being more dependent on lysosome function supported by having higher lysosome numbers, size, and the type and level of toxic content inside lysosomes such as labile iron used to provide cancer cells with additional building blocks.
Prostate cancers can be androgen sensitive or resistant. We found that PC3 and DU145 cells that lack the androgen receptor were more sensitive to siramesine, where LNCaP cells that have an androgen receptor were the least sensitive. Androgen receptor has been shown to be a target for TFEB, a master regulator of lysosome biogenesis [
24] and might contribute to lysosomotropic agent resistance. This will need to be the focus of future investigation
Some cancer cell lines possess increased lysosomal genetic alterations in key enzymes such as Hsp70 which provides lysosome membranes with extra protection from lysosome-induced cell death [
25]. The lower number of lysosomotropic agents reported to kill prostate cancer cells compared to other cell lines could suggest that either prostate cancer is less susceptible to lysosome damage or antihistamines loratadine and desloratadine were much more effective at killing CLL cells [
26]. Perhaps these drugs do not readily diffuse through lysosome membranes and are more readily tolerated and processed efficiently by lysosomes. Siramesine was effective as a lysosomotropic agent in prostate cancer cell lines compared to lysosomotropic agent [
7,
9]. Siramesine is not FDA-approved for cancer therapy since it was developed as an anti-depressant drug and did not achieve significant clinical results; however, several researchers and this group have found their potential as a lysosomotropic agent and an inducer of cell death in many cancer cells [
11,
16,
27,
28]. Breast, CLL, lung and glioblastoma cell lines showed significant cell death after 24 h when treated with the lysosomotropic agent siramesine. In these cases, cell death was associated by an increased in lysosome membrane disruption, lipid ROS and mitochondrial damage [
7,
8,
9,
12]. Despite the number of reports conducted on the effects of siramesine on cancer cell lines, its mechanism of action is not fully understood yet as it displays heterogeneity on its effects on cellular processes, which seems to work in a cell- and context-dependent manner [
17,
23] This will be the focus of future investigations.
Lysosomotropic agents can also induce other cellular events leading to cell death. One of the most widely characterized consequences of lysosome membrane permeabilization is the release of lysosomal proteases into the cytosol, as the membrane becomes porous for lysosome contents to diffuse into the cytosol and among them cathepsin proteases are one of the most toxic enzymes [
29]. Their function is to degrade molecules destined to recycling within the lysosome but when they are released into the cytosol, they are capable of activating proteins associated with cell death signals, such as Bid and Bak [
30]. Treatment with siramesine triggers the release of cathepsins into the cytosol followed by a decrease in mitochondrial membrane potential and ultimately, cell death [
11]. In a study conducted using immortalized keratinocyte HaCaT cells and glioblastoma U-87MG cells, cathepsin release into the cytosol was not observed after treatment with higher concentrations of siramesine than the one used in this study [
7,
9,
12,
16].
As the accumulation of lysosomotropic agents within lysosomes occurs, this often leads to an increase in ROS as these drugs destabilize lysosome membranes by intercalating within membranes, inhibiting or activating key enzymes, and in some cases working as a detergent [
5,
31]. Production of ROS is not only tied to lysosomes as it can interfere with cytosolic components or lipid in other organelles, such as mitochondria destabilizing the mitochondria membrane potential [
30,
32]. We found this to be the case when we treated prostate cancer cell lines with siramesine with significant increases in lipid ROS accompanied by decreased mitochondrial membrane potential and increased levels of mitochondria superoxide. We also observed a decrease in Mcl-1 protein levels that might play a role in mitochondrial dysfunction and will be the focus of future investigation. In the same study conducted with HaCaT and U87 cells, siramesine induced mitochondrial damage and induction of lipid peroxidation. Treatment with the lipophilic antioxidant alpha-tocopherol before addition of siramesine was able to decrease cell death and restore mitochondrial function [
16]. In this study, we investigated the role of lipid ROS as a trigger for siramesine-induced cell death by treating with alpha-tocopherol and measuring cell death. Our results agree with the previous study as the same concentration of alpha-tocopherol decreased cell death after 24 h treatment with siramesine. This indicates the main mechanism lysosomotropic agents induce cell death is through increased ROS. The precise mechanism(s) for lysosomotropic agent-induced cell death in prostate cancer cells will be the focus of future studies.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








