
Tau Protein as Therapeutic Target for Cancer? Focus on Glioblastoma

 , ,
, ,  , and
, and
Simple Summary
Abstract
1. Introduction
2. Tau Structure and Regulation
3. Relationship between Tau Expression and Dysfunctions in Cancer
3.1. Tau and Genomic Instability
3.2. Tau in Cell Cycle and Mitosis
3.3. Tau in Cell Migration
3.4. Tau and Angiogenesis
4. Tau in Glioblastoma
4.1. Tau and Altered Pathways in Glioblastoma
4.1.1. The Receptor Tyrosine Kinases Signaling Pathways
Tau and the RTK/Ras/MAPK/ERK Pathway
Tau and the RTK/PI3K/AKT Pathway
Tau and PTEN to Regulate the RTK/PI3K/AKT Pathway
4.1.2. The Src Family Kinases Signaling Pathways
Tau and Src Protein
Tau and Fyn Protein
4.1.3. The p53 Signaling Pathway
4.2. Other Altered Kinase Activity Characterized in Tauopathies
4.2.1. The Glycogen Synthase Kinase 3 Signaling Pathway
4.2.2. The Cyclin-Dependent Kinase 5 Signaling Pathway
5. Innovative Therapeutic Strategies Targeting Tau Protein for Glioblastoma
5.1. Playing on Post-Translational Modifications of Tau
5.1.1. Inhibitors of Tau Hyperphosphorylation
Src Kinase Inhibitor Saractinib
GSK3β Kinase Inhibitors
CDK Inhibitors
5.1.2. Compounds Stimulating Tau Dephosphorylation: Case of PP2A Activation
5.1.3. Compounds Regulating Tau Acetylation/Deacetylation: HAT/HDAC Proteins
5.1.4. Inhibitors of Tau O-GlcNAcetylation
5.2. Reducing Tau Levels in the Cell
5.2.1. Antisense Oligonucleotides Assayed in AD That Could Be Used in GBM
5.2.2. Tau Clearance by the Autophagy-Lysosomal System
5.3. Therapies Primarily Developed for Tauopathies
5.3.1. Modulation of Tau Aggregation
5.3.2. Immunotherapies against Tau Protein
Passive Immunotherapy
Active Immunotherapy
6. Conclusions and Perspective
7. Limitations of This Review
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitchison, T.; Kirschner, M. Dynamic Instability of Microtubule Growth. Nature 1984, 312, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A Protein Factor Essential for Microtubule Assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Drubin, D.G.; Kirschner, M.W. Tau Protein Function in Living Cells. J. Cell Biol. 1986, 103, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Purification of Tau, a Microtubule-Associated Protein That Induces Assembly of Microtubules from Purified Tubulin. J. Mol. Biol. 1977, 116, 207–225. [Google Scholar] [CrossRef]
- Lee, G.; Rook, S.L. Expression of Tau Protein in Non-Neuronal Cells: Microtubule Binding and Stabilization. J. Cell Sci. 1992, 102, 227–237. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.-M.; Mandelkow, E. MARK, a Novel Family of Protein Kinases That Phosphorylate Microtubule-Associated Proteins and Trigger Microtubule Disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Bunker, J.M.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. Modulation of Microtubule Dynamics by Tau in Living Cells: Implications for Development and Neurodegeneration. Mol. Biol. Cell 2004, 15, 2720–2728. [Google Scholar] [CrossRef]
- Feinstein, S.C.; Wilson, L. Inability of Tau to Properly Regulate Neuronal Microtubule Dynamics: A Loss-of-Function Mechanism by Which Tau Might Mediate Neuronal Cell Death. Biochim. Biophys. Acta 2005, 1739, 268–279. [Google Scholar] [CrossRef]
- LeBoeuf, A.C.; Levy, S.F.; Gaylord, M.; Bhattacharya, A.; Singh, A.K.; Jordan, M.A.; Wilson, L.; Feinstein, S.C. FTDP-17 mutations in Tau alter the regulation of microtubule dynamics: An “alternative core” model for normal and pathological Tau action. J. Biol. Chem. 2008, 283, 36406–36415. [Google Scholar] [CrossRef]
- Huang, L.C.; Ye, J.C.; Hsieh, C.H.; Chen, L.M.; Lin, T.Y.; Hung, Y.C.; Chang, W.C. PTEN, Tau-AP-3, Thymidylate Synthase Immunohistochemistry Scoring Expression in Patients with Uterine Leiomyomas, Uterine Smooth Muscle Tumors of Uncertain Malignancy Potential and Uterine Leiomyosarcomas. Eur. J. Gynaecol. Oncol. 2011, 32, 496–499. [Google Scholar]
- Zaatiti, H.; Abdallah, J.; Nasr, Z.; Khazen, G.; Sandler, A.; Abou-Antoun, T.J. Tumorigenic Proteins Upregulated in the MYCN-Amplified IMR-32 Human Neuroblastoma Cells Promote Proliferation and Migration. Int. J. Oncol. 2018, 52, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.; Wang, B.; Clark, E.; Lee, H.; Rouzier, R.; Pusztai, L. Microtubule Associated Protein (MAP)-Tau: A Novel Mediator of Paclitaxel Sensitivity In Vitro and In Vivo. Cell Cycle 2005, 4, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhang, Z.; Sun, L.; Fang, Y.; Xu, X.; Zhou, G. MiR-186 Regulates Chemo-Sensitivity to Paclitaxel via Targeting MAPT in Non-Small Cell Lung Cancer (NSCLC). Mol. Biosyst. 2016, 12, 3417–3424. [Google Scholar] [CrossRef]
- Souter, S.; Lee, G. Microtubule-Associated Protein Tau In Human Prostate Cancer Cells: Isoforms, Phosphorylation, And Interactions. J. Cell. Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef]
- Miyazono, M.; Iwaki, T.; Kitamoto, T.; Shin, R.-W.; Fukui, M.; Tateishi, J. Widespread Distribution of Tau in the Astrocytic Elements of Glial Tumors. Acta Neuropathol. 1993, 86, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Rouzier, R.; Rajan, R.; Wagner, P.; Hess, K.R.; Gold, D.L.; Stec, J.; Ayers, M.; Ross, J.S.; Zhang, P.; Buchholz, T.A.; et al. Microtubule-Associated Protein Tau: A Marker of Paclitaxel Sensitivity in Breast Cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 8315–8320. [Google Scholar] [CrossRef]
- Pusztai, L.; Jeong, J.-H.; Gong, Y.; Ross, J.S.; Kim, C.; Paik, S.; Rouzier, R.; Andre, F.; Hortobagyi, G.N.; Wolmark, N.; et al. Evaluation of Microtubule-Associated Protein-Tau Expression As a Prognostic and Predictive Marker in the NSABP-B 28 Randomized Clinical Trial. J. Clin. Oncol. 2009, 27, 4287–4292. [Google Scholar] [CrossRef] [PubMed]
- Lara-Velazquez, M.; Al-Kharboosh, R.; Jeanneret, S.; Vazquez-Ramos, C.; Mahato, D.; Tavanaiepour, D.; Rahmathulla, G.; Quinones-Hinojosa, A. Advances in Brain Tumor Surgery for Glioblastoma in Adults. Brain Sci. 2017, 7, 166. [Google Scholar] [CrossRef]
- De Vleeschouwer, S.; Bergers, G. Glioblastoma: To Target the Tumor Cell or the Microenvironment? In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 315–340. ISBN 978-0-9944381-2-6. [Google Scholar]
- Figarella-Branger, D.; Chappe, C.; Padovani, L.; Mercurio, S.; Colin, C.; Forest, F.; Bouvier, C. Glial and glioneuronal tumors in adults and children: Main genetic alterations and towards a histomolecular classification. Bull. Cancer 2013, 100, 715–726. [Google Scholar] [CrossRef]
- Eramo, A.; Ricci-Vitiani, L.; Zeuner, A.; Pallini, R.; Lotti, F.; Sette, G.; Pilozzi, E.; Larocca, L.M.; Peschle, C.; De Maria, R. Chemotherapy Resistance of Glioblastoma Stem Cells. Cell Death Differ. 2006, 13, 1238–1241. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma Multiforme: Pathogenesis and Treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liang, R.F.; Wang, X.; Mao, Q.; Liu, Y.H. BKM120 Sensitizes C6 Glioma Cells to Temozolomide via Suppression of the PI3K/Akt/NF-κB/MGMT Signaling Pathway. Oncol. Lett. 2017, 14, 6597–6603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gao, Y.-T.; Chen, X.-B.; Liu, H.-L. Up-Regulation of MiR-370-3p Restores Glioblastoma Multiforme Sensitivity to Temozolomide by Influencing MGMT Expression. Sci. Rep. 2016, 6, 32972. [Google Scholar] [CrossRef] [PubMed]
- Couchie, D.; Fages, C.; Bridoux, A.M.; Rolland, B.; Tardy, M.; Nunez, J. Microtubule-Associated Proteins and in Vitro Astrocyte Differentiation. J. Cell Biol. 1985, 101, 2095–2103. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple Isoforms of Human Microtubule-Associated Protein Tau: Sequences and Localization in Neurofibrillary Tangles of Alzheimer’s Disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Goode, B.L.; Feinstein, S.C. Identification of a Novel Microtubule Binding and Assembly Domain in the Developmentally Regulated Inter-Repeat Region of Tau. J. Cell Biol. 1994, 124, 769–782. [Google Scholar] [CrossRef]
- Gustke, N.; Trinczek, B.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. Domains of Tau Protein and Interactions with Microtubules. Biochemistry 1994, 33, 9511–9522. [Google Scholar] [CrossRef]
- Pedersen, J.T.; Sigurdsson, E.M. Tau Immunotherapy for Alzheimer’s Disease. Trends Mol. Med. 2015, 21, 394–402. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and Sequencing of the CDNA Encoding an Isoform of Microtubule-Associated Protein Tau Containing Four Tandem Repeats: Differential Expression of Tau Protein MRNAs in Human Brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [CrossRef]
- Ittner, L.M.; Götz, J. Amyloid-β and Tau—A Toxic Pas de Deux in Alzheimer’s Disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimers Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-Translational Modifications of Tau Protein: Implications for Alzheimer’s Disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Lu, S.X.; Ouyang, X.; Melchor, J.; Lee, J.; Terracina, G.; Wang, X.; Hyde, L.; Hess, J.F.; Parker, E.M.; et al. Analysis of Tau Post-Translational Modifications in RTg4510 Mice, a Model of Tau Pathology. Mol. Neurodegener. 2015, 10, 14. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of Separate Isoforms of Human Tau Protein: Correlation with the Tau Pattern in Brain and Effects on Tubulin Polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally Regulated Expression of Specific Tau Sequences. Neuron 1989, 2, 1389–1397. [Google Scholar] [CrossRef]
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Hwo, S.Y.; Kirschner, M.W. Physical and Chemical Properties of Purified Tau Factor and the Role of Tau in Microtubule Assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Woody, R.W.; Clark, D.C.; Roberts, G.C.K.; Martin, S.R.; Bayley, P.M. Molecular Flexibility in Microtubule Proteins: Proton Nuclear Magnetic Resonance Characterization. Biochemistry 1983, 22, 2186–2192. [Google Scholar] [CrossRef]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural Studies of Tau Protein and Alzheimer Paired Helical Filaments Show No Evidence for Beta-Structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar] [CrossRef]
- Jeganathan, S.; Chinnathambi, S.; Mandelkow, E.-M.; Mandelkow, E. Conformations of Microtubule-Associated Protein Tau Mapped by Fluorescence Resonance Energy Transfer. In Amyloid Proteins: Methods and Protocols; Sigurdsson, E.M., Calero, M., Gasset, M., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 85–99. ISBN 978-1-61779-551-0. [Google Scholar]
- De Bessa, T.; Breuzard, G.; Allegro, D.; Devred, F.; Peyrot, V.; Barbier, P. Tau Interaction with Tubulin and Microtubules: From Purified Proteins to Cells. In Tau Protein: Methods and Protocols; Smet-Nocca, C., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 61–85. ISBN 978-1-4939-6598-4. [Google Scholar]
- Di Maïo, I.L.; Barbier, P.; Allegro, D.; Brault, C.; Peyrot, V. Quantitative Analysis of Tau-Microtubule Interaction Using FRET. Int. J. Mol. Sci. 2014, 15, 14697–14714. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear Magnetic Resonance Spectroscopy Characterization of Interaction of Tau with DNA and Its Regulation by Phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; León-Espinosa, G.; García, E.; García-Escudero, V.; Hernández, F.; DeFelipe, J. Tau Phosphorylation by GSK3 in Different Conditions. Int. J. Alzheimers Dis. 2012, 2012, e578373. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Song, X.; Nisbet, R.; Götz, J. Co-immunoprecipitation with Tau Isoform-specific Antibodies Reveals Distinct Protein Interactions and Highlights a Putative Role for 2N Tau in Disease. J. Biol. Chem. 2016, 291, 8173–8188. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of Phosphorylation of Tau by Cyclin-Dependent Kinase 5 and Glycogen Synthase Kinase-3 at Substrate Level. FEBS Lett. 2006, 580, 5925–5933. [Google Scholar] [CrossRef] [PubMed]
- Thiel, G. Synapsin I, Synapsin II, and Synaptophysin: Marker Proteins of Synaptic Vesicles. Brain Pathol. 1993, 3, 87–95. [Google Scholar] [CrossRef]
- Gong, C.-X.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K. Post-Translational Modifications of Tau Protein in Alzheimer’s Disease. J. Neural Transm. 2005, 112, 813–838. [Google Scholar] [CrossRef]
- Maas, T.; Eidenmüller, J.; Brandt, R. Interaction of Tau with the Neural Membrane Cortex Is Regulated by Phosphorylation at Sites That Are Modified in Paired Helical Filaments. J. Biol. Chem. 2000, 275, 15733–15740. [Google Scholar] [CrossRef]
- Yu, J.-Z.; Rasenick, M.M. Tau Associates with Actin in Differentiating PC12 Cells. FASEB J. 2006, 20, 1452–1461. [Google Scholar] [CrossRef]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The Proline-Rich Domain of Tau Plays a Role in Interactions with Actin. BMC Cell Biol. 2009, 10, 81. [Google Scholar] [CrossRef]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau Interacts with Src-Family Non-Receptor Tyrosine Kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [CrossRef] [PubMed]
- Devred, F.; Douillard, S.; Briand, C.; Peyrot, V. First Tau Repeat Domain Binding to Growing and Taxol-Stabilized Microtubules, and Serine 262 Residue Phosphorylation. FEBS Lett. 2002, 523, 247–251. [Google Scholar] [CrossRef]
- Goode, B.L.; Chau, M.; Denis, P.E.; Feinstein, S.C. Structural and Functional Differences between 3-Repeat and 4-Repeat Tau Isoforms. Implications for Normal Tau Function and the Onset of Neurodegenetative Disease. J. Biol. Chem. 2000, 275, 38182–38189. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Mederlyova, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Promotion of Hyperphosphorylation by Frontotemporal Dementia Tau Mutations. J. Biol. Chem. 2004, 279, 34873–34881. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kim, D.; Ju, S.; Shin, S.; Cho, I.; Park, S.-H.; Grailhe, R.; Lee, C.; Kim, Y.K. Glioblastoma-Secreted Soluble CD44 Activates Tau Pathology in the Brain. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, N.; Shao, G.; Qian, J.; Shen, D.; Fei, Y.; Mao, W.; Wu, D. Relationship between Gastric Cancer Tau Protein Expression and Paclitaxel Sensitivity. Pathol. Oncol. Res. 2013, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Breuzard, G.; Hubert, P.; Nouar, R.; Bessa, T.D.; Devred, F.; Barbier, P.; Sturgis, J.N.; Peyrot, V. Molecular Mechanisms of Tau Binding to Microtubules and Its Role in Microtubule Dynamics in Live Cells. J. Cell Sci. 2013, 126, 2810–2819. [Google Scholar] [CrossRef]
- Mimori, K.; Sadanaga, N.; Yoshikawa, Y.; Ishikawa, K.; Hashimoto, M.; Tanaka, F.; Sasaki, A.; Inoue, H.; Sugimachi, K.; Mori, M. Reduced Tau Expression in Gastric Cancer Can Identify Candidates for Successful Paclitaxel Treatment. Br. J. Cancer 2006, 94, 1894–1897. [Google Scholar] [CrossRef]
- Smoter, M.; Bodnar, L.; Grala, B.; Stec, R.; Zieniuk, K.; Kozlowski, W.; Szczylik, C. Tau Protein as a Potential Predictive Marker in Epithelial Ovarian Cancer Patients Treated with Paclitaxel/Platinum First-Line Chemotherapy. J. Exp. Clin. Cancer Res. 2013, 32, 25. [Google Scholar] [CrossRef]
- Cirak, Y.; Sarsik, B.; Cakar, B.; Sen, S.; Simsir, A.; Uslu, R. Predictive and Prognostic Values of Tau and BubR1 Protein in Prostate Cancer and Their Relationship to the Gleason Score. Med. Oncol. 2013, 30, 526. [Google Scholar] [CrossRef] [PubMed]
- Gargini, R.; Segura-Collar, B.; Herránz, B.; García-Escudero, V.; Romero-Bravo, A.; Núñez, F.J.; García-Pérez, D.; Gutiérrez-Guamán, J.; Ayuso-Sacido, A.; Seoane, J.; et al. The IDH-TAU-EGFR Triad Defines the Neovascular Landscape of Diffuse Gliomas. Sci. Transl. Med. 2020, 12, eaax1501. [Google Scholar] [CrossRef]
- Steffensen, K.D.; Smoter, M.; Waldstrøm, M.; Grala, B.; Bodnar, L.; Stec, R.; Szczylik, C.; Jakobsen, A. Resistance to First Line Platinum Paclitaxel Chemotherapy in Serous Epithelial Ovarian Cancer: The Prediction Value of ERCC1 and Tau Expression. Int. J. Oncol. 2014, 44, 1736–1744. [Google Scholar] [CrossRef]
- Bonneau, C.; Gurard-Levin, Z.A.; Andre, F.; Pusztai, L.; Rouzier, R. Predictive and Prognostic Value of the Tau Protein in Breast Cancer. Anticancer Res. 2015, 35, 5179–5184. [Google Scholar]
- Papin, S.; Paganetti, P. Emerging Evidences for an Implication of the Neurodegeneration-Associated Protein TAU in Cancer. Brain Sci. 2020, 10, 862. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, L.; Gotta, M.; Meraldi, P. The Elephant in the Room: The Role of Microtubules in Cancer. In Cell Division Machinery and Disease; Gotta, M., Meraldi, P., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 93–124. ISBN 978-3-319-57127-0. [Google Scholar]
- Langie, S.A.S.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.; Brunborg, G.; et al. Causes of Genome Instability: The Effect of Low Dose Chemical Exposures in Modern Society. Carcinogenesis 2015, 36, S61–S88. [Google Scholar] [CrossRef] [PubMed]
- Brady, R.M.; Zinkowski, R.P.; Binder, L.I. Presence of Tau in Isolated Nuclei from Human Brain. Neurobiol. Aging 1995, 16, 479–486. [Google Scholar] [CrossRef]
- Greenwood, J.A.; Johnson, G.V.W. Localization and in Situ Phosphorylation State of Nuclear Tau. Exp. Cell Res. 1995, 220, 332–337. [Google Scholar] [CrossRef]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau Promotes Neurodegeneration through Global Chromatin Relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef]
- Bukar Maina, M.; Al-Hilaly, Y.K.; Serpell, L.C. Nuclear Tau and Its Potential Role in Alzheimer’s Disease. Biomolecules 2016, 6, 9. [Google Scholar] [CrossRef]
- Cross, D.C.; Muñoz, J.P.; Hernández, P.; Maccioni, R.B. Nuclear and Cytoplasmic Tau Proteins from Human Nonneuronal Cells Share Common Structural and Functional Features with Brain Tau. J. Cell. Biochem. 2000, 78, 305–317. [Google Scholar] [CrossRef]
- Loomis, P.A.; Howard, T.H.; Castleberry, R.P.; Binder, L.I. Identification of Nuclear Tau Isoforms in Human Neuroblastoma Cells. Proc. Natl. Acad. Sci. USA 1990, 87, 8422–8426. [Google Scholar] [CrossRef] [PubMed]
- Thurston, V.C.; Zinkowski, R.P.; Binder, L.I. Tau as a Nucleolar Protein in Human Nonneural Cells in Vitro and in Vivo. Chromosoma 1996, 105, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Nesslany, F.; Violet, M.; Bégard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear Tau, a Key Player in Neuronal DNA Protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A Major Role for Tau in Neuronal DNA and RNA Protection in Vivo under Physiological and Hyperthermic Conditions. Front. Cell. Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Chouala, M.S.; Sultan, A.; Marciniak, E.; Humez, S.; Binder, L.; Kayed, R.; et al. Prefibrillar Tau Oligomers Alter the Nucleic Acid Protective Function of Tau in Hippocampal Neurons in Vivo. Neurobiol. Dis. 2015, 82, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Hua, Q.; He, R. Tau Could Protect DNA Double Helix Structure. Biochim. Biophys. Acta 2003, 1645, 205–211. [Google Scholar] [CrossRef]
- Rossi, G.; Conconi, D.; Panzeri, E.; Redaelli, S.; Piccoli, E.; Paoletta, L.; Dalprà, L.; Tagliavini, F. Mutations in MAPT Gene Cause Chromosome Instability and Introduce Copy Number Variations Widely in the Genome. J. Alzheimers Dis. 2013, 33, 969–982. [Google Scholar] [CrossRef]
- Migliore, L.; Testa, A.; Scarpato, R.; Pavese, N.; Petrozzi, L.; Bonuccelli, U. Spontaneous and Induced Aneuploidy in Peripheral Blood Lymphocytes of Patients with Alzheimer’s Disease. Hum. Genet. 1997, 101, 299–305. [Google Scholar] [CrossRef]
- Mosch, B.; Morawski, M.; Mittag, A.; Lenz, D.; Tarnok, A.; Arendt, T. Aneuploidy and DNA Replication in the Normal Human Brain and Alzheimer’s Disease. J. Neurosci. 2007, 27, 6859–6867. [Google Scholar] [CrossRef]
- Iourov, I.Y.; Vorsanova, S.G.; Liehr, T.; Yurov, Y.B. Aneuploidy in the Normal, Alzheimer’s Disease and Ataxia-Telangiectasia Brain: Differential Expression and Pathological Meaning. Neurobiol. Dis. 2009, 34, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Yurov, Y.B.; Vorsanova, S.G.; Liehr, T.; Kolotii, A.D.; Iourov, I.Y. X Chromosome Aneuploidy in the Alzheimer’s Disease Brain. Mol. Cytogenet. 2014, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Pope, W.B.; Lambert, M.P.; Leypold, B.; Seupaul, R.; Sletten, L.; Krafft, G.; Klein, W.L. Microtubule-Associated Protein Tau Is Hyperphosphorylated during Mitosis in the Human Neuroblastoma Cell Line SH-SY5Y. Exp. Neurol. 1994, 126, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Preuss, U.; Döring, F.; Illenberger, S.; Mandelkow, E.M. Cell Cycle-Dependent Phosphorylation and Microtubule Binding of Tau Protein Stably Transfected into Chinese Hamster Ovary Cells. Mol. Biol. Cell. 1995, 6, 1397–1410. [Google Scholar] [CrossRef] [PubMed]
- Flores-Rodríguez, P.; Harrington, C.R.; Wischik, C.M.; Ibarra-Bracamontes, V.; Zarco, N.; Navarrete, A.; Martínez-Maldonado, A.; Guadarrama-Ortíz, P.; Villanueva-Fierro, I.; Ontiveros-Torres, M.A.; et al. Phospho-Tau Protein Expression in the Cell Cycle of SH-SY5Y Neuroblastoma Cells: A Morphological Study. J. Alzheimers Dis. 2019, 71, 631–645. [Google Scholar] [CrossRef]
- Bougé, A.-L.; Parmentier, M.-L. Tau Excess Impairs Mitosis and Kinesin-5 Function, Leading to Aneuploidy and Cell Death. Dis. Model. Mech. 2016, 9, 307–319. [Google Scholar] [CrossRef]
- Giam, M.; Rancati, G. Aneuploidy and Chromosomal Instability in Cancer: A Jackpot to Chaos. Cell Div. 2015, 10, 3. [Google Scholar] [CrossRef]
- Jordan, M.A.; Thrower, D.; Wilson, L. Mechanism of Inhibition of Cell Proliferation by Vinca Alkaloids1. Cancer Res. 1991, 51, 2212–2222. [Google Scholar]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef]
- Spicakova, T.; O’Brien, M.M.; Duran, G.E.; Sweet-Cordero, A.; Sikic, B.I. Expression and Silencing of the Microtubule-Associated Protein Tau in Breast Cancer Cells. Mol. Cancer Ther. 2010, 9, 2970–2981. [Google Scholar] [CrossRef][Green Version]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Muzi, P.; Mattei, V.; Bologna, M.; Angelucci, A. Tau Oligomers Accumulation Sensitizes Prostate Cancer Cells to Docetaxel Treatment. J. Cancer Res. Clin. Oncol. 2021, 147, 1957–1971. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Price, A.; Eberhart, C.; Morris, M. Increased Tau Expression Correlates with IDH Mutation in Infiltrating Gliomas and Impairs Cell Migration. J. Neuropathol. Exp. Neurol. 2020, 79, 493–499. [Google Scholar] [CrossRef]
- Yan, H.; Bigner, D.D.; Velculescu, V.; Parsons, D.W. Mutant Metabolic Enzymes Are at the Origin of Gliomas. Cancer Res. 2009, 69, 9157–9159. [Google Scholar] [CrossRef] [PubMed]
- Breuzard, G.; Pagano, A.; Bastonero, S.; Malesinski, S.; Parat, F.; Barbier, P.; Peyrot, V.; Kovacic, H. Tau Regulates the Microtubule-Dependent Migration of Glioblastoma Cells via the Rho-ROCK Signaling Pathway. J. Cell Sci. 2019, 132, jcs222851. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Lee, G. Tau Impacts on Growth-Factor-Stimulated Actin Remodeling. J. Cell Sci. 2007, 120, 748–757. [Google Scholar] [CrossRef]
- Griffith, L.M.; Pollard, T.D. Evidence for Actin Filament-Microtubule Interaction Mediated by Microtubule-Associated Proteins. J. Cell Biol. 1978, 78, 958–965. [Google Scholar] [CrossRef]
- Griffith, L.M.; Pollard, T.D. The Interaction of Actin Filaments with Microtubules and Microtubule-Associated Proteins. J. Biol. Chem. 1982, 257, 9143–9151. [Google Scholar] [CrossRef]
- Pollard, T.D.; Selden, S.C.; Maupin, P. Interaction of Actin Filaments with Microtubules. J. Cell Biol. 1984, 99, 33s–37s. [Google Scholar] [CrossRef]
- Selden, S.C.; Pollard, T.D. Interaction of Actin Filaments with Microtubules Is Mediated by Microtubule-Associated Proteins and Regulated by Phosphorylation. Ann. N. Y. Acad. Sci. 1986, 466, 803–812. [Google Scholar] [CrossRef]
- Farias, G.A.; Muñoz, J.P.; Garrido, J.; Maccioni, R.B. Tubulin, Actin, and Tau Protein Interactions and the Study of Their Macromolecular Assemblies. J. Cell. Biochem. 2002, 85, 315–324. [Google Scholar] [CrossRef]
- Kadowaki, T.; Fujita-Yamaguchi, Y.; Nishida, E.; Takaku, F.; Akiyama, T.; Kathuria, S.; Akanuma, Y.; Kasuga, M. Phosphorylation of Tubulin and Microtubule-Associated Proteins by the Purified Insulin Receptor Kinase. J. Biol. Chem. 1985, 260, 4016–4020. [Google Scholar] [CrossRef]
- Hoshi, M.; Nishida, E.; Miyata, Y.; Sakai, H.; Miyoshi, T.; Ogawara, H.; Akiyama, T. Protein Kinase C Phosphorylates Tau and Induces Its Functional Alterations. FEBS Lett. 1987, 217, 237–241. [Google Scholar] [CrossRef]
- Zhu, D.; Kosik, K.S.; Meigs, T.E.; Yanamadala, V.; Denker, B.M. Gα12 Directly Interacts with PP2A: Evidence for gα12-stimulated pp2a phosphatase activity and dephosphorylation of microtubule-associated protein, tau. J. Biol. Chem. 2004, 279, 54983–54986. [Google Scholar] [CrossRef]
- Siedlak, S.L.; Cras, P.; Kawai, M.; Richey, P.; Perry, G. Basic Fibroblast Growth Factor Binding Is a Marker for Extracellular Neurofibrillary Tangles in Alzheimer Disease. J. Histochem. Cytochem. 1991, 39, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.H.; Fimmen, B.A.; Thal, D.R.; Schluesener, H.J.; Meyermann, R. Aberrant Neuronal and Paracellular Deposition of Endostatin in Brains of Patients with Alzheimer’s Disease. J. Neurosci. 2002, 22, 10621–10626. [Google Scholar] [CrossRef]
- Ułamek-Kozioł, M.; Czuczwar, S.J.; Januszewski, S.; Pluta, R. Proteomic and Genomic Changes in Tau Protein, Which Are Associated with Alzheimer’s Disease after Ischemia-Reperfusion Brain Injury. Int. J. Mol. Sci. 2020, 21, 892. [Google Scholar] [CrossRef]
- Jeong, H.; Shin, J.Y.; Lee, K.; Lee, S.-J.; Chong, H.-J.; Jeong, H.; Jeon, Y.-E.; Shin, D.-S.; Jang, S.; Kim, K.H.; et al. Caffeoyl-Prolyl-Histidine Amide Inhibits Fyn and Alleviates Atopic Dermatitis-Like Phenotypes via Suppression of NF-ΚB Activation. Int. J. Mol. Sci. 2020, 21, 7160. [Google Scholar] [CrossRef]
- Merlini, M.; Wanner, D.; Nitsch, R.M. Tau Pathology-Dependent Remodelling of Cerebral Arteries Precedes Alzheimer’s Disease-Related Microvascular Cerebral Amyloid Angiopathy. Acta Neuropathol. 2016, 131, 737–752. [Google Scholar] [CrossRef]
- Castillo-Carranza, D.L.; Nilson, A.N.; Van Skike, C.E.; Jahrling, J.B.; Patel, K.; Garach, P.; Gerson, J.E.; Sengupta, U.; Abisambra, J.; Nelson, P.; et al. Cerebral Microvascular Accumulation of Tau Oligomers in Alzheimer’s Disease and Related Tauopathies. Aging Dis. 2017, 8, 257–266. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau Induces Blood Vessel Abnormalities and Angiogenesis-Related Gene Expression in P301L Transgenic Mice and Human Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Bennett, R.E.; Hu, M.; Fernandes, A.; Perez-Rando, M.; Robbins, A.; Kamath, T.; Dujardin, S.; Hyman, B.T. Tau Reduction in Aged Mice Does Not Impact Microangiopathy. Acta Neuropathol. Commun. 2020, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia Signalling in Cancer and Approaches to Enforce Tumour Regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and Emerging Mechanisms of the Angiogenic Switch during Tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Bergers, G.; Benjamin, L.E. Tumorigenesis and the Angiogenic Switch. Nat. Rev. Cancer 2003, 3, 401–410. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B.; Crivellato, E.; Roccaro, A.M.; Vacca, A. The History of the Angiogenic Switch Concept. Leukemia 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Jászai, J.; Schmidt, M.H.H. Trends and Challenges in Tumor Anti-Angiogenic Therapies. Cells 2019, 8, 1102. [Google Scholar] [CrossRef]
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene Expression Profiling of Gliomas Strongly Predicts Survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular Subclasses of High-Grade Glioma Predict Prognosis, Delineate a Pattern of Disease Progression, and Resemble Stages in Neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Murakami, C.; Ikota, H.; Nobusawa, S.; Nakata, S.; Yamazaki, T.; Hashiba, Y.; Hirato, J.; Yokoo, H. Oligodendroglioma Showing Pleomorphic Xanthoastrocytoma-like Perivascular Microlesion: With IDH1, TERT Promoter Mutation and 1p/19q Codeletion Detected in Both Components. Pathol. Int. 2020, 70, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yu, Y.; Liu, W.; Liu, W.; Li, Z.; Wei, Z.; Jiang, R. Microtubule-Associated Protein Tau Is Associated with the Resistance to Docetaxel in Prostate Cancer Cell Lines. Res. Rep. Urol. 2017, 9, 71–77. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation Regulates Tau Interactions with Src Homology 3 Domains of Phosphatidylinositol 3-Kinase, Phospholipase Cγ1, Grb2, and Src Family Kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [PubMed]
- Pagano, A.; Breuzard, G.; Parat, F.; Tchoghandjian, A.; Figarella-Branger, D.; De Bessa, T.C.; Garrouste, F.; Douence, A.; Barbier, P.; Kovacic, H. Tau Regulates Glioblastoma Progression, 3D Cell Organization, Growth and Migration via the PI3K-AKT Axis. Cancers 2021, 13, 5818. [Google Scholar] [CrossRef]
- Wu, Y.-H.; Huang, Y.-F.; Chen, C.-C.; Chou, C.-Y. Akt Inhibitor SC66 Promotes Cell Sensitivity to Cisplatin in Chemoresistant Ovarian Cancer Cells through Inhibition of COL11A1 Expression. Cell Death Dis. 2019, 10, 322. [Google Scholar] [CrossRef]
- Sekino, Y.; Han, X.; Babasaki, T.; Goto, K.; Inoue, S.; Hayashi, T.; Teishima, J.; Shiota, M.; Takeshima, Y.; Yasui, W.; et al. Microtubule-Associated Protein Tau (MAPT) Promotes Bicalutamide Resistance and Is Associated with Survival in Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 795.e1–795.e8. [Google Scholar] [CrossRef]
- Giannakakou, P.; Sackett, D.L.; Ward, Y.; Webster, K.R.; Blagosklonny, M.V.; Fojo, T. P53 Is Associated with Cellular Microtubules and Is Transported to the Nucleus by Dynein. Nat. Cell Biol. 2000, 2, 709–717. [Google Scholar] [CrossRef]
- Sola, M.; Magrin, C.; Pedrioli, G.; Pinton, S.; Salvadè, A.; Papin, S.; Paganetti, P. Tau Affects P53 Function and Cell Fate during the DNA Damage Response. Commun. Biol. 2020, 3, 245. [Google Scholar] [CrossRef]
- Miyashita, K.; Kawakami, K.; Nakada, M.; Mai, W.; Shakoori, A.; Fujisawa, H.; Hayashi, Y.; Hamada, J.; Minamoto, T. Potential Therapeutic Effect of Glycogen Synthase Kinase 3beta Inhibition against Human Glioblastoma. Clin. Cancer Res. 2009, 15, 887–897. [Google Scholar] [CrossRef]
- Liang, S.; Shen, G.; Liu, Q.; Xu, Y.; Zhou, L.; Xiao, S.; Xu, Z.; Gong, F.; You, C.; Wei, Y. Isoform-Specific Expression and Characterization of 14-3-3 Proteins in Human Glioma Tissues Discovered by Stable Isotope Labeling with Amino Acids in Cell Culture-Based Proteomic Analysis. Proteom. Clin. Appl. 2009, 3, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xu, H.; Wang, Q.; Wang, S.; Xiong, N. 14-3-3ζ Promotes Gliomas Cells Invasion by Regulating Snail through the PI3K/AKT Signaling. Cancer Med. 2019, 8, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, M.; Sobue, K.; Paudel, H.K. 14-3-3ζ Is an Effector of Tau Protein Phosphorylation. J. Biol. Chem. 2000, 275, 25247–25254. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, H.Y.; Li, T.; MacDonald, R.; Cho, C.M.; Leclerc, N.; Paudel, H.K. Interaction of 14-3-3ζ with Microtubule-Associated Protein Tau within Alzheimer’s Disease Neurofibrillary Tangles. Biochemistry 2013, 52, 6445–6455. [Google Scholar] [CrossRef] [PubMed]
- Agarwal-Mawal, A.; Qureshi, H.Y.; Cafferty, P.W.; Yuan, Z.; Han, D.; Lin, R.; Paudel, H.K. 14-3-3 Connects Glycogen Synthase Kinase-3β to Tau within a Brain Microtubule-Associated Tau Phosphorylation Complex. J. Biol. Chem. 2003, 278, 12722–12728. [Google Scholar] [CrossRef]
- Yuan, Z.; Agarwal-Mawal, A.; Paudel, H.K. 14-3-3 Binds to and Mediates Phosphorylation of Microtubule-Associated Tau Protein by Ser9-Phosphorylated Glycogen Synthase Kinase 3beta in the Brain. J. Biol. Chem. 2004, 279, 26105–26114. [Google Scholar] [CrossRef]
- Martin, D.; Brown-Luedi, M.; Chiquet-Ehrismann, R. Tenascin-C Signaling through Induction of 14-3-3 Tau. J. Cell Biol. 2003, 160, 171–175. [Google Scholar] [CrossRef]
- Li, T.; Hawkes, C.; Qureshi, H.Y.; Kar, S.; Paudel, H.K. Cyclin-Dependent Protein Kinase 5 Primes Microtubule-Associated Protein Tau Site-Specifically for Glycogen Synthase Kinase 3β. Biochemistry 2006, 45, 3134–3145. [Google Scholar] [CrossRef]
- Noble, W.; Olm, V.; Takata, K.; Casey, E.; Mary, O.; Meyerson, J.; Gaynor, K.; LaFrancois, J.; Wang, L.; Kondo, T.; et al. Cdk5 Is a Key Factor in Tau Aggregation and Tangle Formation in Vivo. Neuron 2003, 38, 555–565. [Google Scholar] [CrossRef]
- Qin, A.; Musket, A.; Musich, P.R.; Schweitzer, J.B.; Xie, Q. Receptor Tyrosine Kinases as Druggable Targets in Glioblastoma: Do Signaling Pathways Matter? Neurooncol. Adv. 2021, 3, vdab133. [Google Scholar] [CrossRef]
- Bare, D.J.; Lauder, J.M.; Wilkie, M.B.; Maness, P.F. P59fyn in Rat Brain Is Localized in Developing Axonal Tracts and Subpopulations of Adult Neurons and Glia. Oncogene 1993, 8, 1429–1436. [Google Scholar] [PubMed]
- Kempf, M.; Clement, A.; Faissner, A.; Lee, G.; Brandt, R. Tau Binds to the Distal Axon Early in Development of Polarity in a Microtubule- and Microfilament-Dependent Manner. J. Neurosci. 1996, 16, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting Cellular Pathways in Glioblastoma Multiforme. Signal Transduct. Target. Ther. 2017, 2, 1–11. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Makino, Y.; Arakawa, Y.; Yoshioka, E.; Shofuda, T.; Minamiguchi, S.; Kawauchi, T.; Tanji, M.; Kanematsu, D.; Nonaka, M.; Okita, Y.; et al. Infrequent RAS Mutation Is Not Associated with Specific Histological Phenotype in Gliomas. BMC Cancer 2021, 21, 1025. [Google Scholar] [CrossRef]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP Kinase Signalling Pathways in Cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Cheng, J.; North, B.J.; Wei, W. Functional Analyses of Major Cancer-Related Signaling Pathways in Alzheimer’s Disease Etiology. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 341–358. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK Mitogen-Activated Protein Kinase Cascade for the Treatment of Cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- du Plessis, M.; Davis, T.; Loos, B.; Pretorius, E.; de Villiers, W.J.S.; Engelbrecht, A.M. Molecular Regulation of Autophagy in a Pro-Inflammatory Tumour Microenvironment: New Insight into the Role of Serum Amyloid A. Cytokine Growth Factor Rev. 2021, 59, 71–83. [Google Scholar] [CrossRef]
- Gan, H.K.; Kaye, A.H.; Luwor, R.B. The EGFRvIII Variant in Glioblastoma Multiforme. J. Clin. Neurosci. 2009, 16, 748–754. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal Growth Factor Receptor (EGFR) and EGFRvIII in Glioblastoma (GBM): Signaling Pathways and Targeted Therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Chen, S.; Yang, L.; Li, Z.; Zhuo, S.; Yan, B.; Zhang, Z.; Zhang, J.; Feng, H.; Yang, K. EGFR/EGFRvIII Partly Regulates the Tumourigenesis of Glioblastoma through the SOX9-GLUT3 Axis. Am. J. Transl. Res. 2021, 13, 6055–6065. [Google Scholar] [PubMed]
- Shibasaki, F.; Homma, Y.; Takenawa, T. Two Types of Phosphatidylinositol 3-Kinase from Bovine Thymus. Monomer and Heterodimer Form. J. Biol. Chem. 1991, 266, 8108–8114. [Google Scholar] [CrossRef]
- Koyama, S.; Yu, H.; Dalgarno, D.C.; Shin, T.B.; Zydowsky, L.D.; Schreiber, S.L. Structure of the Pl3K SH3 Domain and Analysis of the SH3 Family. Cell 1993, 72, 945–952. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion Blocks Apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef]
- Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and Inactivation of Glycogen Synthase Kinase 3 by Protein Kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef]
- Cai, Z.; Chen, G.; He, W.; Xiao, M.; Yan, L.-J. Activation of MTOR: A Culprit of Alzheimer’s Disease? Neuropsychiatr. Dis. Treat. 2015, 11, 1015–1030. [Google Scholar] [CrossRef]
- Morris, G.; Berk, M.; Maes, M.; Puri, B.K. Could Alzheimer’s Disease Originate in the Periphery and If So How So? Mol. Neurobiol. 2019, 56, 406–434. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Stokoe, D. PTEN. Curr. Biol. 2001, 11, R502. [Google Scholar] [CrossRef]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by P53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Suzuki, A.; de la Pompa, J.L.; Stambolic, V.; Elia, A.J.; Sasaki, T.; Barrantes, I.d.B.; Ho, A.; Wakeham, A.; Ltie, A.; Khoo, W.; et al. High Cancer Susceptibility and Embryonic Lethality Associated with Mutation of the PTEN Tumor Suppressor Gene in Mice. Curr. Biol. 1998, 8, 1169–1178. [Google Scholar] [CrossRef]
- Podsypanina, K.; Ellenson, L.H.; Nemes, A.; Gu, J.; Tamura, M.; Yamada, K.M.; Cordon-Cardo, C.; Catoretti, G.; Fisher, P.E.; Parsons, R. Mutation of Pten/Mmac1 in Mice Causes Neoplasia in Multiple Organ Systems. Proc. Natl. Acad. Sci. USA 1999, 96, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Bamias, A.; Karina, M.; Papakostas, P.; Kostopoulos, I.; Bobos, M.; Vourli, G.; Samantas, E.; Christodoulou, C.; Pentheroudakis, G.; Pectasides, D.; et al. A Randomized Phase III Study of Adjuvant Platinum/Docetaxel Chemotherapy with or without Radiation Therapy in Patients with Gastric Cancer. Cancer Chemother. Pharmacol. 2010, 65, 1009–1021. [Google Scholar] [CrossRef]
- Schroeder, C.; Grell, J.; Hube-Magg, C.; Kluth, M.; Lang, D.; Simon, R.; Höflmayer, D.; Minner, S.; Burandt, E.; Clauditz, T.S.; et al. Aberrant Expression of the Microtubule-Associated Protein Tau Is an Independent Prognostic Feature in Prostate Cancer. BMC Cancer 2019, 19, 193. [Google Scholar] [CrossRef]
- Koo, H.; Choi, S.W.; Cho, H.J.; Lee, I.-H.; Kong, D.-S.; Seol, H.J.; Lee, J.-I.; Choi, J.-W.; Sa, J.K.; Nam, D.-H. Ethnic Delineation of Primary Glioblastoma Genome. Cancer Med. 2020, 9, 7352–7359. [Google Scholar] [CrossRef]
- Bonneau, D.; Longy, M. Mutations of the Human PTEN Gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Leslie, N.R.; Downes, C.P. PTEN Function: How Normal Cells Control It and Tumour Cells Lose It. Biochem. J. 2004, 382, 1–11. [Google Scholar] [CrossRef]
- Lv, S.; Teugels, E.; Sadones, J.; De Brakeleer, S.; Duerinck, J.; Du Four, S.; Michotte, A.; De Grève, J.; Neyns, B. Correlation of EGFR, IDH1 and PTEN Status with the Outcome of Patients with Recurrent Glioblastoma Treated in a Phase II Clinical Trial with the EGFR-Blocking Monoclonal Antibody Cetuximab. Int. J. Oncol. 2012, 41, 1029–1035. [Google Scholar] [CrossRef]
- Choi, S.W.; Lee, Y.; Shin, K.; Koo, H.; Kim, D.; Sa, J.K.; Cho, H.J.; Shin, H.; Lee, S.J.; Kim, H.; et al. Mutation-Specific Non-Canonical Pathway of PTEN as a Distinct Therapeutic Target for Glioblastoma. Cell Death Dis. 2021, 12, 374. [Google Scholar] [CrossRef]
- Pyo, H.; Lovati, E.; Pasinetti, G.M.; Ksiezak-Reding, H. Phosphorylation of Tau at THR212 and SER214 in Human Neuronal and Glial Cultures: The Role of AKT. Neuroscience 2004, 127, 649–658. [Google Scholar] [CrossRef]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, Increased Phosphorylation of Akt Substrates and Loss and Altered Distribution of Akt and PTEN Are Features of Alzheimer’s Disease Pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kerr, F.; Rickle, A.; Nayeem, N.; Brandner, S.; Cowburn, R.F.; Lovestone, S. PTEN, a Negative Regulator of PI3 Kinase Signalling, Alters Tau Phosphorylation in Cells by Mechanisms Independent of GSK-3. FEBS Lett. 2006, 580, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, F.; Bulloj, A.; Zhang, Y.-W.; Tong, G.; Zhang, Z.; Liao, F.-F.; Xu, H. Tumor-Suppressor PTEN Affects Tau Phosphorylation, Aggregation, and Binding to Microtubules. FASEB J. 2006, 20, 1272–1274. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chen, B.; Xu, W.-F.; Liu, R.-F.; Yang, J.; Yu, C.-X. Effects of PTEN Inhibition on Regulation of Tau Phosphorylation in an Okadaic Acid-Induced Neurodegeneration Model. Int. J. Dev. Neurosci. 2012, 30, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.J.; Zaidi, T.; Grundke-Iqbal, I.; Iqbal, K. Modulation of GSK-3-Catalyzed Phosphorylation of Microtubule-Associated Protein Tau by Non-Proline-Dependent Protein Kinases. FEBS Lett. 1995, 358, 4–8. [Google Scholar] [CrossRef]
- Wagner, U.; Utton, M.; Gallo, J.M.; Miller, C.C. Cellular Phosphorylation of Tau by GSK-3 Beta Influences Tau Binding to Microtubules and Microtubule Organisation. J. Cell Sci. 1996, 109, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Zhang, J.Y.; Li, H.L.; Fang, Z.Y.; Wang, Q.; Deng, H.M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.Z. Tau Becomes a More Favorable Substrate for GSK-3 When It Is Prephosphorylated by PKA in Rat Brain. J. Biol. Chem. 2004, 279, 50078–50088. [Google Scholar] [CrossRef]
- Avila, J.; Hernández, F. GSK-3 Inhibitors for Alzheimer’s Disease. Expert Rev. Neurother. 2007, 7, 1527–1533. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular Functions Regulated by Src Family Kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src Family Kinases, Key Regulators of Signal Transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed]
- Portugal, C.C.; Almeida, T.O.; Socodato, R.; Relvas, J.B. Src Family Kinases (SFKs): Critical Regulators of Microglial Homeostatic Functions and Neurodegeneration in Parkinson’s and Alzheimer’s Diseases. FEBS J. 2021. [Google Scholar] [CrossRef] [PubMed]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA Related to the Transforming Gene(s) of Avian Sarcoma Viruses Is Present in Normal Avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Arias-Salgado, E.G.; Lizano, S.; Sarkar, S.; Brugge, J.S.; Ginsberg, M.H.; Shattil, S.J. Src Kinase Activation by Direct Interaction with the Integrin Beta Cytoplasmic Domain. Proc. Natl. Acad. Sci. USA 2003, 100, 13298–13302. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lee, F.Y.; Bhalla, K.N.; Wu, J. Potent Inhibition of Platelet-Derived Growth Factor-Induced Responses in Vascular Smooth Muscle Cells by BMS-354825 (Dasatinib). Mol. Pharmacol. 2006, 69, 1527–1533. [Google Scholar] [CrossRef]
- Schittenhelm, M.M.; Shiraga, S.; Schroeder, A.; Corbin, A.S.; Griffith, D.; Lee, F.Y.; Bokemeyer, C.; Deininger, M.W.N.; Druker, B.J.; Heinrich, M.C. Dasatinib (BMS-354825), a Dual SRC/ABL Kinase Inhibitor, Inhibits the Kinase Activity of Wild-Type, Juxtamembrane, and Activation Loop Mutant KIT Isoforms Associated with Human Malignancies. Cancer Res. 2006, 66, 473–481. [Google Scholar] [CrossRef]
- Lu, K.V.; Zhu, S.; Cvrljevic, A.; Huang, T.T.; Sarkaria, S.; Ahkavan, D.; Dang, J.; Dinca, E.B.; Plaisier, S.B.; Oderberg, I.; et al. Fyn and SRC Are Effectors of Oncogenic Epidermal Growth Factor Receptor Signaling in Glioblastoma Patients. Cancer Res. 2009, 69, 6889–6898. [Google Scholar] [CrossRef]
- Kleber, S.; Sancho-Martinez, I.; Wiestler, B.; Beisel, A.; Gieffers, C.; Hill, O.; Thiemann, M.; Mueller, W.; Sykora, J.; Kuhn, A.; et al. Yes and PI3K Bind CD95 to Signal Invasion of Glioblastoma. Cancer Cell 2008, 13, 235–248. [Google Scholar] [CrossRef]
- Stettner, M.R.; Wang, W.; Nabors, L.B.; Bharara, S.; Flynn, D.C.; Grammer, J.R.; Gillespie, G.Y.; Gladson, C.L. Lyn Kinase Activity Is the Predominant Cellular SRC Kinase Activity in Glioblastoma Tumor Cells. Cancer Res. 2005, 65, 5535–5543. [Google Scholar] [CrossRef]
- Liu, W.M.; Huang, P.; Kar, N.; Burgett, M.; Muller-Greven, G.; Nowacki, A.S.; Distelhorst, C.W.; Lathia, J.D.; Rich, J.N.; Kappes, J.C.; et al. Lyn Facilitates Glioblastoma Cell Survival under Conditions of Nutrient Deprivation by Promoting Autophagy. PLoS ONE 2013, 8, e70804. [Google Scholar] [CrossRef] [PubMed]
- Zepecki, J.P.; Snyder, K.M.; Moreno, M.M.; Fajardo, E.; Fiser, A.; Ness, J.; Sarkar, A.; Toms, S.A.; Tapinos, N. Regulation of Human Glioma Cell Migration, Tumor Growth, and Stemness Gene Expression Using a Lck Targeted Inhibitor. Oncogene 2019, 38, 1734–1750. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Ying, C.; Zhang, A.; Xu, H.; Jiang, Y.; Lou, M. HCK Promotes Glioblastoma Progression by TGFβ Signaling. Biosci. Rep. 2020, 40, BSR20200975. [Google Scholar] [CrossRef]
- Lund, C.V.; Nguyen, M.T.N.; Owens, G.C.; Pakchoian, A.J.; Shaterian, A.; Kruse, C.A.; Eliceiri, B.P. Reduced Glioma Infiltration in Src-Deficient Mice. J. Neurooncol. 2006, 78, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC Kinase in Glioblastoma: News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef]
- Bjorge, J.D.; Jakymiw, A.; Fujita, D.J. Selected Glimpses into the Activation and Function of Src Kinase. Oncogene 2000, 19, 5620–5635. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, M.S.; de Groot, J.; Liu, W.; Gladson, C.L. Targeting SRC in Glioblastoma Tumors and Brain Metastases: Rationale and Preclinical Studies. Cancer Lett. 2010, 298, 139–149. [Google Scholar] [CrossRef]
- Bhaskar, K.; Yen, S.-H.; Lee, G. Disease-Related Modifications in Tau Affect the Interaction between Fyn and Tau. J. Biol. Chem. 2005, 280, 35119–35125. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-Mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422. [Google Scholar] [CrossRef]
- Comba, A.; Dunn, P.J.; Argento, A.E.; Kadiyala, P.; Ventosa, M.; Patel, P.; Zamler, D.B.; Núñez, F.J.; Zhao, L.; Castro, M.G.; et al. Fyn Tyrosine Kinase, a Downstream Target of Receptor Tyrosine Kinases, Modulates Antiglioma Immune Responses. Neuro-Oncology 2020, 22, 806–818. [Google Scholar] [CrossRef]
- Gordon-Weeks, P.R.; Gary Mansfield, S.; Curran, I. Direct Visualisation of the Soluble Pool of Tubulin in the Neuronal Growth Cone: Immunofluorescence Studies Following Taxol Polymerisation. Dev. Brain Res. 1989, 49, 305–310. [Google Scholar] [CrossRef]
- DiTella, M.; Feiguin, F.; Morfini, G.; Cáceres, A. Microfilament-associated growth cone component depends upon Tau for its intracellular localization. Cell Motil. 1994, 29, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Helmke, S.; Pfenninger, K.H. Growth Cone Enrichment and Cytoskeletal Association of Non-Receptor Tyrosine Kinases. Cell Motil. Cytoskeleton 1995, 30, 194–207. [Google Scholar] [CrossRef] [PubMed]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional Implications for the Microtubule-Associated Protein Tau: Localization in Oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef]
- Umemori, H.; Satot, S.; Yagi, T.; Aizawal, S.; Yamamoto, T. Initial Events of Myelination Involve Fyn Tyrosine Kinase Signalling. Nature 1994, 367, 572–576. [Google Scholar] [CrossRef]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; Van Meir, E.G. Frequent Co-Alterations of TP53, P16/CDKN2A, P14ARF, PTEN Tumor Suppressor Genes in Human Glioma Cell Lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, C.; Gibert, M.; Cruickshanks, N.; Wang, B.; Coughlan, M.; Yang, Y.; Setiady, I.; Deveau, C.; Saoud, K.; et al. The P53 Pathway in Glioblastoma. Cancers 2018, 10, 297. [Google Scholar] [CrossRef]
- Baquero, J.; Varriano, S.; Ordonez, M.; Kuczaj, P.; Murphy, M.R.; Aruggoda, G.; Lundine, D.; Morozova, V.; Makki, A.E.; Alonso, A.D.C.; et al. Nuclear Tau, P53 and Pin1 Regulate PARN-Mediated Deadenylation and Gene Expression. Front. Mol. Neurosci. 2019, 12, 242. [Google Scholar] [CrossRef]
- Chou, P.-Y.; Lin, S.-R.; Lee, M.-H.; Schultz, L.; Sze, C.-I.; Chang, N.-S. A P53/TIAF1/WWOX Triad Exerts Cancer Suppression but May Cause Brain Protein Aggregation Due to P53/WWOX Functional Antagonism. Cell Commun. Signal. 2019, 17, 76. [Google Scholar] [CrossRef]
- Farmer, K.M.; Ghag, G.; Puangmalai, N.; Montalbano, M.; Bhatt, N.; Kayed, R. P53 Aggregation, Interactions with Tau, and Impaired DNA Damage Response in Alzheimer’s Disease. Acta Neuropathol. Commun. 2020, 8, 132. [Google Scholar] [CrossRef]
- Montalbano, M.; McAllen, S.; Cascio, F.L.; Sengupta, U.; Garcia, S.; Bhatt, N.; Ellsworth, A.; Heidelman, E.A.; Johnson, O.D.; Doskocil, S.; et al. TDP-43 and Tau Oligomers in Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol. Dis. 2020, 146, 105130. [Google Scholar] [CrossRef] [PubMed]
- Jazvinšćak Jembrek, M.; Slade, N.; Hof, P.R.; Šimić, G. The Interactions of P53 with Tau and Aß as Potential Therapeutic Targets for Alzheimer’s Disease. Prog. Neurobiol. 2018, 168, 104–127. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3 β: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, e930710. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.-P.; Anderton, B.H. Glycogen Synthase Kinase-3 Induces Alzheimer’s Disease-like Phosphorylation of Tau: Generation of Paired Helical Filament Epitopes and Neuronal Localisation of the Kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Mandelkow, E.M.; Drewes, G.; Biernat, J.; Gustke, N.; Van Lint, J.; Vandenheede, J.R.; Mandelkow, E. Glycogen Synthase Kinase-3 and the Alzheimer-like State of Microtubule-Associated Protein Tau. FEBS Lett. 1992, 314, 315–321. [Google Scholar] [CrossRef]
- Sun, W.; Qureshi, H.Y.; Cafferty, P.W.; Sobue, K.; Agarwal-Mawal, A.; Neufield, K.D.; Paudel, H.K. Glycogen Synthase Kinase-3beta Is Complexed with Tau Protein in Brain Microtubules. J. Biol. Chem. 2002, 277, 11933–11940. [Google Scholar] [CrossRef]
- Korur, S.; Huber, R.M.; Sivasankaran, B.; Petrich, M.; Jr, P.M.; Hemmings, B.A.; Merlo, A.; Lino, M.M. GSK3β Regulates Differentiation and Growth Arrest in Glioblastoma. PLoS ONE 2009, 4, e7443. [Google Scholar] [CrossRef]
- Kotliarova, S.; Pastorino, S.; Kovell, L.C.; Kotliarov, Y.; Song, H.; Zhang, W.; Bailey, R.; Maric, D.; Zenklusen, J.C.; Lee, J.; et al. Glycogen Synthase Kinase 3 Inhibition Induces Glioma Cell Death through C-MYC, NF-ΚB and Glucose Regulation. Cancer Res. 2008, 68, 6643–6651. [Google Scholar] [CrossRef]
- Nowicki, M.O.; Dmitrieva, N.; Stein, A.M.; Cutter, J.L.; Godlewski, J.; Saeki, Y.; Nita, M.; Berens, M.E.; Sander, L.M.; Newton, H.B.; et al. Lithium Inhibits Invasion of Glioma Cells; Possible Involvement of Glycogen Synthase Kinase-3. Neuro-Oncology 2008, 10, 690–699. [Google Scholar] [CrossRef]
- Aitken, A.; Collinge, D.B.; van Heusden, B.P.; Isobe, T.; Roseboom, P.H.; Rosenfeld, G.; Soll, J. 14-3-3 Proteins: A Highly Conserved, Widespread Family of Eukaryotic Proteins. Trends Biochem. Sci. 1992, 17, 498–501. [Google Scholar] [CrossRef]
- Joo, Y.; Schumacher, B.; Landrieu, I.; Bartel, M.; Smet-Nocca, C.; Jang, A.; Choi, H.S.; Jeon, N.L.; Chang, K.-A.; Kim, H.-S.; et al. Involvement of 14-3-3 in Tubulin Instability and Impaired Axon Development Is Mediated by Tau. FASEB J. 2015, 29, 4133–4144. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D. The Cell Cycle: Principles of Control; New Science Press: London, UK, 2006; 297p. [Google Scholar]
- Lagace, D.C.; Benavides, D.R.; Kansy, J.W.; Mapelli, M.; Greengard, P.; Bibb, J.A.; Eisch, A.J. Cdk5 Is Essential for Adult Hippocampal Neurogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 18567–18571. [Google Scholar] [CrossRef] [PubMed]
- Tan, T.C.; Valova, V.A.; Malladi, C.S.; Graham, M.E.; Berven, L.A.; Jupp, O.J.; Hansra, G.; McClure, S.J.; Sarcevic, B.; Boadle, R.A.; et al. Cdk5 Is Essential for Synaptic Vesicle Endocytosis. Nat. Cell Biol. 2003, 5, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Dhariwala, F.A.; Rajadhyaksha, M.S. An Unusual Member of the Cdk Family: Cdk5. Cell. Mol. Neurobiol. 2008, 28, 351–369. [Google Scholar] [CrossRef]
- Liebl, J.; Weitensteiner, S.B.; Vereb, G.; Takács, L.; Fürst, R.; Vollmar, A.M.; Zahler, S. Cyclin-Dependent Kinase 5 Regulates Endothelial Cell Migration and Angiogenesis. J. Biol. Chem. 2010, 285, 35932–35943. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-S.; Alexander, K.; Santiago, P.; Hinds, P.W. ERM Proteins and Cdk5 in Cellular Senescence. Cell Cycle Georget. Tex. 2003, 2, 517–520. [Google Scholar] [CrossRef]
- Lazaro, J.B.; Kitzmann, M.; Poul, M.A.; Vandromme, M.; Lamb, N.J.; Fernandez, A. Cyclin Dependent Kinase 5, Cdk5, Is a Positive Regulator of Myogenesis in Mouse C2 Cells. J. Cell Sci. 1997, 110, 1251–1260. [Google Scholar] [CrossRef]
- Chapman, D.E.; Reddy, B.J.N.; Huy, B.; Bovyn, M.J.; Cruz, S.J.S.; Al-Shammari, Z.M.; Han, H.; Wang, W.; Smith, D.S.; Gross, S.P. Regulation of in Vivo Dynein Force Production by CDK5 and 14-3-3ε and KIAA0528. Nat. Commun. 2019, 10, 228. [Google Scholar] [CrossRef]
- Contreras-Vallejos, E.; Utreras, E.; Gonzalez-Billault, C. Going out of the Brain: Non-Nervous System Physiological and Pathological Functions of Cdk5. Cell. Signal. 2012, 24, 44–52. [Google Scholar] [CrossRef]
- Do, P.A.; Lee, C.H. The Role of CDK5 in Tumours and Tumour Microenvironments. Cancers 2020, 13, 101. [Google Scholar] [CrossRef]
- Yushan, R.; Wenjie, C.; Suning, H.; Yiwu, D.; Tengfei, Z.; Madushi, W.M.; Feifei, L.; Changwen, Z.; Xin, W.; Roodrajeetsing, G.; et al. Insights into the Clinical Value of Cyclin-Dependent Kinase 5 in Glioma: A Retrospective Study. World J. Surg. Oncol. 2015, 13, 223. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mukherjee, S.; Tucker-Burden, C.; Kaissi, E.; Newsam, A.; Duggireddy, H.; Chau, M.; Zhang, C.; Diwedi, B.; Rupji, M.; Seby, S.; et al. CDK5 Inhibition Resolves PKA/CAMP-Independent Activation of CREB1 Signaling in Glioma Stem Cells. Cell Rep. 2018, 23, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, X.; Lv, P.; Yu, H.; Jiang, X. CDK5 Knockdown Inhibits Proliferation and Induces Apoptosis and Cell Cycle Arrest in Human Glioblastoma. J. Cancer 2021, 12, 3958–3966. [Google Scholar] [CrossRef] [PubMed]
- Huda, M.N.; Kim, D.H.; Erdene-Ochir, E.; Kim, Y.S.; Pan, C.-H. Expression, Phosphorylation, Localization, and Microtubule Binding of Tau in Colorectal Cell Lines. Appl. Biol. Chem. 2016, 59, 807–812. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized In Vitro and In Vivo Cancer Models to Guide Precision Medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Nowak, J.A.; Camarda, N.D.; Moffitt, R.A.; Ghazani, A.A.; Hazar-Rethinam, M.; Raghavan, S.; Kim, J.; Brais, L.K.; Ragon, D.; et al. Real-Time Genomic Characterization of Advanced Pancreatic Cancer to Enable Precision Medicine. Cancer Discov. 2018, 8, 1096–1111. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming Cancer Therapeutic Bottleneck by Drug Repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G. ESMO Guidelines Working Group High-Grade Glioma: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef]
- Moffat, J.G.; Rudolph, J.; Bailey, D. Phenotypic Screening in Cancer Drug Discovery—Past, Present and Future. Nat. Rev. Drug Discov. 2014, 13, 588–602. [Google Scholar] [CrossRef]
- Chen, H.I.; Song, H.; Ming, G.-L. Applications of Human Brain Organoids to Clinical Problems. Dev. Dyn. 2019, 248, 53–64. [Google Scholar] [CrossRef]
- Loong, H.H.-F.; Wong, A.M.; Chan, D.T.-M.; Cheung, M.S.-H.; Chow, C.; Ding, X.; Chan, A.K.-Y.; Johnston, P.A.; Lau, J.Y.-W.; Poon, W.S.; et al. Patient-Derived Tumor Organoid Predicts Drugs Response in Glioblastoma: A Step Forward in Personalized Cancer Therapy? J. Clin. Neurosci. 2020, 78, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Klein, E.; Hau, A.-C.; Oudin, A.; Golebiewska, A.; Niclou, S.P. Glioblastoma Organoids: Pre-Clinical Applications and Challenges in the Context of Immunotherapy. Front. Oncol. 2020, 10, 604121. [Google Scholar] [CrossRef] [PubMed]
- Vincent, F.; Nueda, A.; Lee, J.; Schenone, M.; Prunotto, M.; Mercola, M. Phenotypic Drug Discovery: Recent Successes, Lessons Learned and New Directions. Nat. Rev. Drug Discov. 2022, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Martín, B.; Medina, M.Á. Advances in the Knowledge of the Molecular Biology of Glioblastoma and Its Impact in Patient Diagnosis, Stratification, and Treatment. Adv. Sci. 2020, 7, 1902971. [Google Scholar] [CrossRef]
- Bougnaud, S.; Golebiewska, A.; Oudin, A.; Keunen, O.; Harter, P.N.; Mäder, L.; Azuaje, F.; Fritah, S.; Stieber, D.; Kaoma, T.; et al. Molecular Crosstalk between Tumour and Brain Parenchyma Instructs Histopathological Features in Glioblastoma. Oncotarget 2016, 7, 31955–31971. [Google Scholar] [CrossRef]
- Perrin, S.L.; Samuel, M.S.; Koszyca, B.; Brown, M.P.; Ebert, L.M.; Oksdath, M.; Gomez, G.A. Glioblastoma Heterogeneity and the Tumour Microenvironment: Implications for Preclinical Research and Development of New Treatments. Biochem. Soc. Trans. 2019, 47, 625–638. [Google Scholar] [CrossRef]
- Mitusova, K.; Peltek, O.O.; Karpov, T.E.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Overcoming the Blood-Brain Barrier for the Therapy of Malignant Brain Tumor: Current Status and Prospects of Drug Delivery Approaches. J. Nanobiotechnol. 2022, 20, 412. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Sigurdsson, E.M. Tau-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol. 2018, 14, 399–415. [Google Scholar] [CrossRef]
- Jadhav, S.; Avila, J.; Schöll, M.; Kovacs, G.G.; Kövari, E.; Skrabana, R.; Evans, L.D.; Kontsekova, E.; Malawska, B.; de Silva, R.; et al. A Walk through Tau Therapeutic Strategies. Acta Neuropathol. Commun. 2019, 7, 22. [Google Scholar] [CrossRef]
- Yu, T.-W.; Lane, H.-Y.; Lin, C.-H. Novel Therapeutic Approaches for Alzheimer’s Disease: An Updated Review. Int. J. Mol. Sci. 2021, 22, 8208. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Matsumoto, M.; Kamura, T.; Murayama, M.; Chui, D.-H.; Planel, E.; Takahashi, R.; Nakayama, K.I.; Takashima, A. U-Box Protein Carboxyl Terminus of Hsc70-Interacting Protein (CHIP) Mediates Poly-Ubiquitylation Preferentially on Four-Repeat Tau and Is Involved in Neurodegeneration of Tauopathy. J. Neurochem. 2004, 91, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 Regulate Tau Ubiquitination, Degradation and Aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, H.; Xie, J.; Meng, D.; Wang, X.; Ke, D.; Zeng, J.; Liu, R. Protein Phosphatase 2A as a Drug Target in the Treatment of Cancer and Alzheimer’s Disease. Curr. Med. Sci. 2020, 40, 1–8. [Google Scholar] [CrossRef]
- Ding, H.; Dolan, P.J.; Johnson, G.V.W. Histone Deacetylase 6 Interacts with the Microtubule-Associated Protein Tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Yun, H.S.; Lee, J.; Kil, W.J.; Kramp, T.R.; Tofilon, P.J.; Camphausen, K. The Radiosensitizing Effect of AZD0530 in Glioblastoma and Glioblastoma Stem-Like Cells. Mol. Cancer Ther. 2021, 20, 1672–1679. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.S.; Hamid, H. Natural and Synthetic Bioactive Inhibitors of Glycogen Synthase Kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Happold, C.; Gorlia, T.; Chinot, O.; Gilbert, M.R.; Nabors, L.B.; Wick, W.; Pugh, S.L.; Hegi, M.; Cloughesy, T.; Roth, P.; et al. Does Valproic Acid or Levetiracetam Improve Survival in Glioblastoma? A Pooled Analysis of Prospective Clinical Trials in Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2016, 34, 731–739. [Google Scholar] [CrossRef]
- Fay, M.F.; Head, R.; Sminia, P.; Dowson, N.; Cosgrove, L.; Rose, S.E.; Martin, J.H. Valproate in Adjuvant Glioblastoma Treatment. J. Clin. Oncol. 2016, 34, 3105–3107. [Google Scholar] [CrossRef][Green Version]
- Hu, J.-P.; Xie, J.-W.; Wang, C.-Y.; Wang, T.; Wang, X.; Wang, S.-L.; Teng, W.-P.; Wang, Z.-Y. Valproate Reduces Tau Phosphorylation via Cyclin-Dependent Kinase 5 and Glycogen Synthase Kinase 3 Signaling Pathways. Brain Res. Bull. 2011, 85, 194–200. [Google Scholar] [CrossRef]
- Bahmad, H.F.; Chalhoub, R.M.; Harati, H.; Bou-Gharios, J.; Assi, S.; Ballout, F.; Monzer, A.; Msheik, H.; Araji, T.; Elajami, M.K.; et al. Tideglusib Attenuates Growth of Neuroblastoma Cancer Stem/Progenitor Cells in Vitro and in Vivo by Specifically Targeting GSK-3β. Pharmacol. Rep. 2021, 73, 211–226. [Google Scholar] [CrossRef]
- Wei, D.; Zhu, X.; Li, S.; Liu, G.; Wang, Y.; Wang, W.; Zhang, Q.; Jiang, S. Tideglusib Suppresses Stem-Cell-like Features and Progression of Osteosarcoma by Inhibiting GSK-3β/NOTCH1 Signaling. Biochem. Biophys. Res. Commun. 2021, 554, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Bou-Gharios, J.; Assi, S.; Bahmad, H.F.; Kharroubi, H.; Araji, T.; Chalhoub, R.M.; Ballout, F.; Harati, H.; Fares, Y.; Abou-Kheir, W. The Potential Use of Tideglusib as an Adjuvant Radio-Therapeutic Treatment for Glioblastoma Multiforme Cancer Stem-like Cells. Pharmacol. Rep. 2021, 73, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Stankunas, E.; Levy, J.; Meskinyte, I.; Stankevicius, V.; Kaupinis, A.; Valius, M. Roscovitine in Cancer and Other Diseases. Ann. Transl. Med. 2015, 3, 6. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and Safety of Dinaciclib vs Ofatumumab in Patients with Relapsed/Refractory Chronic Lymphocytic Leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef]
- Murphy, A.G.; Zahurak, M.; Shah, M.; Weekes, C.D.; Hansen, A.; Siu, L.L.; Spreafico, A.; LoConte, N.; Anders, N.M.; Miles, T.; et al. A Phase I Study of Dinaciclib in Combination With MK-2206 in Patients With Advanced Pancreatic Cancer. Clin. Transl. Sci. 2020, 13, 1178–1188. [Google Scholar] [CrossRef]
- Meijer, L.; Hery-Arnaud, G.; Leven, C.; Nowak, E.; Hillion, S.; Renaudineau, Y.; Durieu, I.; Chiron, R.; Prevotat, A.; Fajac, I.; et al. Safety and Pharmacokinetics of Roscovitine (Seliciclib) in Cystic Fibrosis Patients Chronically Infected with Pseudomonas Aeruginosa, a Randomized, Placebo-Controlled Study. J. Cyst. Fibros. 2021, 21, 529–536. [Google Scholar] [CrossRef]
- Sangodkar, J.; Perl, A.; Tohme, R.; Kiselar, J.; Kastrinsky, D.B.; Zaware, N.; Izadmehr, S.; Mazhar, S.; Wiredja, D.D.; O’Connor, C.M.; et al. Activation of Tumor Suppressor Protein PP2A Inhibits KRAS-Driven Tumor Growth. J. Clin. Investig. 2017, 127, 2081–2090. [Google Scholar] [CrossRef]
- Wei, H.; Zhang, H.-L.; Wang, X.-C.; Xie, J.-Z.; An, D.-D.; Wan, L.; Wang, J.-Z.; Zeng, Y.; Shu, X.-J.; Westermarck, J.; et al. Direct Activation of Protein Phosphatase 2A (PP2A) by Tricyclic Sulfonamides Ameliorates Alzheimer’s Disease Pathogenesis in Cell and Animal Models. Neurother. J. Am. Soc. Exp. Neurother. 2020, 17, 1087–1103. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. HATs and HDACs: From Structure, Function and Regulation to Novel Strategies for Therapy and Prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Lv, D.; Jia, F.; Hou, Y.; Sang, Y.; Alvarez, A.A.; Zhang, W.; Gao, W.-Q.; Hu, B.; Cheng, S.-Y.; Ge, J.; et al. Histone Acetyltransferase KAT6A Upregulates PI3K/AKT Signaling through TRIM24 Binding. Cancer Res. 2017, 77, 6190–6201. [Google Scholar] [CrossRef] [PubMed]
- Rokudai, S.; Laptenko, O.; Arnal, S.M.; Taya, Y.; Kitabayashi, I.; Prives, C. MOZ Increases P53 Acetylation and Premature Senescence through Its Complex Formation with PML. Proc. Natl. Acad. Sci. USA 2013, 110, 3895–3900. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Leaver, D.J.; Hermans, S.J.; Kelly, G.L.; Brennan, M.S.; Downer, N.L.; Nguyen, N.; Wichmann, J.; McRae, H.M.; Yang, Y.; et al. Inhibitors of Histone Acetyltransferases KAT6A/B Induce Senescence and Arrest Tumour Growth. Nature 2018, 560, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhang, M.; Zhou, Y.; Guo, W.; Yi, M.; Zhang, Z.; Ding, Y.; Wang, Y. The Application of Histone Deacetylases Inhibitors in Glioblastoma. J. Exp. Clin. Cancer Res. 2020, 39, 138. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhang, H.; Jiao, M.; Han, B.; Zhang, Z.; Li, J.; Zhang, Q. Histone Deacetylase 6 Inhibitors with Blood-Brain Barrier Penetration as a Potential Strategy for CNS-Disorders Therapy. Eur. J. Med. Chem. 2022, 229, 114090. [Google Scholar] [CrossRef] [PubMed]
- Perez, T.; Bergès, R.; Maccario, H.; Oddoux, S.; Honoré, S. Low Concentrations of Vorinostat Decrease EB1 Expression in GBM Cells and Affect Microtubule Dynamics, Cell Survival and Migration. Oncotarget 2021, 12, 304–315. [Google Scholar] [CrossRef]
- Balmik, A.A.; Chidambaram, H.; Dangi, A.; Marelli, U.K.; Chinnathambi, S. HDAC6 ZnF UBP as the Modifier of Tau Structure and Function. Biochemistry 2020, 59, 4546–4562. [Google Scholar] [CrossRef]
- Min, S.-W.; Cho, S.-H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of Tau Inhibits Its Degradation and Contributes to Tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Caballero, B.; Bourdenx, M.; Luengo, E.; Diaz, A.; Sohn, P.D.; Chen, X.; Wang, C.; Juste, Y.R.; Wegmann, S.; Patel, B.; et al. Acetylated Tau Inhibits Chaperone-Mediated Autophagy and Promotes Tau Pathology Propagation in Mice. Nat. Commun. 2021, 12, 2238. [Google Scholar] [CrossRef]
- Hastings, N.B.; Wang, X.; Song, L.; Butts, B.D.; Grotz, D.; Hargreaves, R.; Fred Hess, J.; Hong, K.-L.K.; Huang, C.R.-R.; Hyde, L.; et al. Inhibition of O-GlcNAcase Leads to Elevation of O-GlcNAc Tau and Reduction of Tauopathy and Cerebrospinal Fluid Tau in RTg4510 Mice. Mol. Neurodegener. 2017, 12, 39. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A Potent Mechanism-Inspired O-GlcNAcase Inhibitor That Blocks Phosphorylation of Tau in Vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- DeVos, S.L.; Miller, T.M. Antisense Oligonucleotides: Treating Neurodegeneration at the Level of RNA. Neurother. J. Am. Soc. Exp. Neurother. 2013, 10, 486–497. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Miller, R.L.; Schoch, K.M.; Holmes, B.B.; Kebodeaux, C.S.; Wegener, A.J.; Chen, G.; Shen, T.; Tran, H.; Nichols, B.; et al. Tau Reduction Prevents Neuronal Loss and Reverses Pathological Tau Deposition and Seeding in Mice with Tauopathy. Sci. Transl. Med. 2017, 9, eaag0481. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Layfield, R.; Serpell, L.; Narain, Y.; Goedert, M.; Spillantini, M.G. Proteasomal Degradation of Tau Protein. J. Neurochem. 2002, 83, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Dolan, P.J.; Johnson, G.V.W. A Caspase Cleaved Form of Tau Is Preferentially Degraded through the Autophagy Pathway. J. Biol. Chem. 2010, 285, 21978–21987. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Martinez-Vicente, M.; Krüger, U.; Kaushik, S.; Wong, E.; Mandelkow, E.-M.; Cuervo, A.M.; Mandelkow, E. Synergy and Antagonism of Macroautophagy and Chaperone-Mediated Autophagy in a Cell Model of Pathological Tau Aggregation. Autophagy 2010, 6, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Degradation of Tau Protein by Autophagy and Proteasomal Pathways. Biochem. Soc. Trans. 2012, 40, 644–652. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau Degradation: The Ubiquitin–Proteasome System versus the Autophagy-Lysosome System. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Caballero, B.; Wang, Y.; Diaz, A.; Tasset, I.; Juste, Y.R.; Stiller, B.; Mandelkow, E.-M.; Mandelkow, E.; Cuervo, A.M. Interplay of Pathogenic Forms of Human Tau with Different Autophagic Pathways. Aging Cell. 2018, 17, e12692. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Matheu, A. Impact of Chaperone-Mediated Autophagy in Brain Aging: Neurodegenerative Diseases and Glioblastoma. Front. Aging Neurosci. 2021, 12, 509. [Google Scholar] [CrossRef]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of Chaperone-Mediated Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; O’Leary, J.C.; Borysov, S.I.; Jones, J.R.; Li, Q.; Koren, J.; Abisambra, J.F.; Vestal, G.D.; Lawson, L.Y.; Johnson, A.G.; et al. Hsc70 Rapidly Engages Tau after Microtubule Destabilization. J. Biol. Chem. 2010, 285, 16798–16805. [Google Scholar] [CrossRef]
- Bi, X.; Haque, T.S.; Zhou, J.; Skillman, A.G.; Lin, B.; Lee, C.E.; Kuntz, I.D.; Ellman, J.A.; Lynch, G. Novel Cathepsin D Inhibitors Block the Formation of Hyperphosphorylated Tau Fragments in Hippocampus. J. Neurochem. 2000, 74, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Motoi, Y.; Ishiguro, K.; Kambe, T.; Matsumoto, S.; Itaya, M.; Kunichika, M.; Mori, H.; Shinohara, A.; Chiba, M.; et al. Long-Term Oral Lithium Treatment Attenuates Motor Disturbance in Tauopathy Model Mice: Implications of Autophagy Promotion. Neurobiol. Dis. 2012, 46, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, V.; Goedert, M. Stimulation of Autophagy Is Neuroprotective in a Mouse Model of Human Tauopathy. Autophagy 2012, 8, 1686–1687. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.-T.; Zhu, X.-C.; Zhang, Q.-Q.; Cao, L.; Wang, H.-F.; Tan, M.-S.; Gao, Q.; Qin, H.; Zhang, Y.-D.; et al. Temsirolimus Attenuates Tauopathy in Vitro and in Vivo by Targeting Tau Hyperphosphorylation and Autophagic Clearance. Neuropharmacology 2014, 85, 121–130. [Google Scholar] [CrossRef]
- Hebron, M.L.; Javidnia, M.; Moussa, C.E.-H. Tau Clearance Improves Astrocytic Function and Brain Glutamate-Glutamine Cycle. J. Neurol. Sci. 2018, 391, 90–99. [Google Scholar] [CrossRef]
- Hernandez, I.; Luna, G.; Rauch, J.N.; Reis, S.A.; Giroux, M.; Karch, C.M.; Boctor, D.; Sibih, Y.E.; Storm, N.J.; Diaz, A.; et al. A Farnesyltransferase Inhibitor Activates Lysosomes and Reduces Tau Pathology in Mice with Tauopathy. Sci. Transl. Med. 2019, 11, eaat3005. [Google Scholar] [CrossRef]
- Song, J.-X.; Malampati, S.; Zeng, Y.; Durairajan, S.S.K.; Yang, C.-B.; Tong, B.C.-K.; Iyaswamy, A.; Shang, W.-B.; Sreenivasmurthy, S.G.; Zhu, Z.; et al. A Small Molecule Transcription Factor EB Activator Ameliorates Beta-Amyloid Precursor Protein and Tau Pathology in Alzheimer’s Disease Models. Aging Cell 2020, 19, e13069. [Google Scholar] [CrossRef]
- Currais, A.; Prior, M.; Dargusch, R.; Armando, A.; Ehren, J.; Schubert, D.; Quehenberger, O.; Maher, P. Modulation of P25 and Inflammatory Pathways by Fisetin Maintains Cognitive Function in Alzheimer’s Disease Transgenic Mice. Aging Cell 2014, 13, 379–390. [Google Scholar] [CrossRef]
- Spagnuolo, P.A.; Hu, J.; Hurren, R.; Wang, X.; Gronda, M.; Sukhai, M.A.; Di Meo, A.; Boss, J.; Ashali, I.; Beheshti Zavareh, R.; et al. The Antihelmintic Flubendazole Inhibits Microtubule Function through a Mechanism Distinct from Vinca Alkaloids and Displays Preclinical Activity in Leukemia and Myeloma. Blood 2010, 115, 4824–4833. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.-J.; Luo, X.; Zhang, W.; Peng, F.; Cui, B.; Wu, S.-J.; Zheng, F.-M.; Xu, J.; Xu, L.-Z.; Long, Z.-J.; et al. Flubendazole, FDA-Approved Anthelmintic, Targets Breast Cancer Stem-like Cells. Oncotarget 2015, 6, 6326–6340. [Google Scholar] [CrossRef]
- Chauhan, S.; Ahmed, Z.; Bradfute, S.B.; Arko-Mensah, J.; Mandell, M.A.; Won Choi, S.; Kimura, T.; Blanchet, F.; Waller, A.; Mudd, M.H.; et al. Pharmaceutical Screen Identifies Novel Target Processes for Activation of Autophagy with a Broad Translational Potential. Nat. Commun. 2015, 6, 8620. [Google Scholar] [CrossRef] [PubMed]
- Cai, N.; Chen, J.; Bi, D.; Gu, L.; Yao, L.; Li, X.; Li, H.; Xu, H.; Hu, Z.; Liu, Q.; et al. Specific Degradation of Endogenous Tau Protein and Inhibition of Tau Fibrillation by Tanshinone IIA through the Ubiquitin–Proteasome Pathway. J. Agric. Food Chem. 2020, 68, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Bhaskar, K. Degradation and Transmission of Tau by Autophagic-Endolysosomal Networks and Potential Therapeutic Targets for Tauopathy. Front. Mol. Neurosci. 2020, 13, 199. [Google Scholar] [CrossRef]
- Gandini, A.; Bartolini, M.; Tedesco, D.; Martinez-Gonzalez, L.; Roca, C.; Campillo, N.E.; Zaldivar-Diez, J.; Perez, C.; Zuccheri, G.; Miti, A.; et al. Tau-Centric Multitarget Approach for Alzheimer’s Disease: Development of First-in-Class Dual Glycogen Synthase Kinase 3β and Tau-Aggregation Inhibitors. J. Med. Chem. 2018, 61, 7640–7656. [Google Scholar] [CrossRef]
- Cisek, K.; Cooper, G.L.; Huseby, C.J.; Kuret, J. Structure and Mechanism of Action of Tau Aggregation Inhibitors. Curr. Alzheimer Res. 2014, 11, 918–927. [Google Scholar] [CrossRef]
- Akoury, E.; Pickhardt, M.; Gajda, M.; Biernat, J.; Mandelkow, E.; Zweckstetter, M. Mechanistic Basis of Phenothiazine-Driven Inhibition of Tau Aggregation. Angew. Chem. Int. Ed. 2013, 52, 3511–3515. [Google Scholar] [CrossRef]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Wu, X.; Nakayama, S.; Sakata, Y.; Noguchi, Y.; Ogo, M.; Akasofu, S.; Ito, Y.; et al. PE859, a Novel Tau Aggregation Inhibitor, Reduces Aggregated Tau and Prevents Onset and Progression of Neural Dysfunction In Vivo. PLoS ONE 2015, 10, e0117511. [Google Scholar] [CrossRef]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Teruya, T.; Kawakami, H.; Takahashi, T.; Sugimoto, H. Design and Synthesis of Curcumin Derivatives as Tau and Amyloid β Dual Aggregation Inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5024–5028. [Google Scholar] [CrossRef]
- Sandusky-Beltran, L.A.; Sigurdsson, E.M. Tau Immunotherapies: Lessons Learned, Current Status and Future Considerations. Neuropharmacology 2020, 175, 108104. [Google Scholar] [CrossRef]
- Ji, C.; Sigurdsson, E.M. Current Status of Clinical Trials on Tau Immunotherapies. Drugs 2021, 81, 1135–1152. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, S.K.; Joly-Amado, A.; Gordon, M.N.; Morgan, D. Tau-Directed Immunotherapy: A Promising Strategy for Treating Alzheimer’s Disease and Other Tauopathies. J. Neuroimmune Pharmacol. 2016, 11, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Funk, K.E.; Mirbaha, H.; Jiang, H.; Holtzman, D.M.; Diamond, M.I. Distinct Therapeutic Mechanisms of Tau Antibodies: Promoting Microglial Clearance Versus Blocking Neuronal Uptake. J. Biol. Chem. 2015, 290, 21652–21662. [Google Scholar] [CrossRef]
- Luo, W.; Liu, W.; Hu, X.; Hanna, M.; Caravaca, A.; Paul, S.M. Microglial Internalization and Degradation of Pathological Tau Is Enhanced by an Anti-Tau Monoclonal Antibody. Sci. Rep. 2015, 5, 11161. [Google Scholar] [CrossRef] [PubMed]
- Congdon, E.E.; Lin, Y.; Rajamohamedsait, H.B.; Shamir, D.B.; Krishnaswamy, S.; Rajamohamedsait, W.J.; Rasool, S.; Gonzalez, V.; Levenga, J.; Gu, J.; et al. Affinity of Tau Antibodies for Solubilized Pathological Tau Species but Not Their Immunogen or Insoluble Tau Aggregates Predicts in Vivo and Ex Vivo Efficacy. Mol. Neurodegener. 2016, 11, 62. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Deng, Y.; Sigurdsson, E. Mechanistic Studies of Antibody-Mediated Clearance of Tau Aggregates Using an Ex Vivo Brain Slice Model. Front. Psychiatry 2011, 2, 59. [Google Scholar] [CrossRef]
- Gu, J.; Congdon, E.E.; Sigurdsson, E.M. Two Novel Tau Antibodies Targeting the 396/404 Region Are Primarily Taken up by Neurons and Reduce Tau Protein Pathology. J. Biol. Chem. 2013, 288, 33081–33095. [Google Scholar] [CrossRef]
- McEwan, W.A.; Falcon, B.; Vaysburd, M.; Clift, D.; Oblak, A.L.; Ghetti, B.; Goedert, M.; James, L.C. Cytosolic Fc Receptor TRIM21 Inhibits Seeded Tau Aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 574–579. [Google Scholar] [CrossRef]
- Shahpasand, K.; Sepehri Shamloo, A.; Nabavi, S.M.; Ping Lu, K.; Zhen Zhou, X. Tau Immunotherapy: Hopes and Hindrances. Hum. Vaccines Immunother. 2017, 14, 277–284. [Google Scholar] [CrossRef]





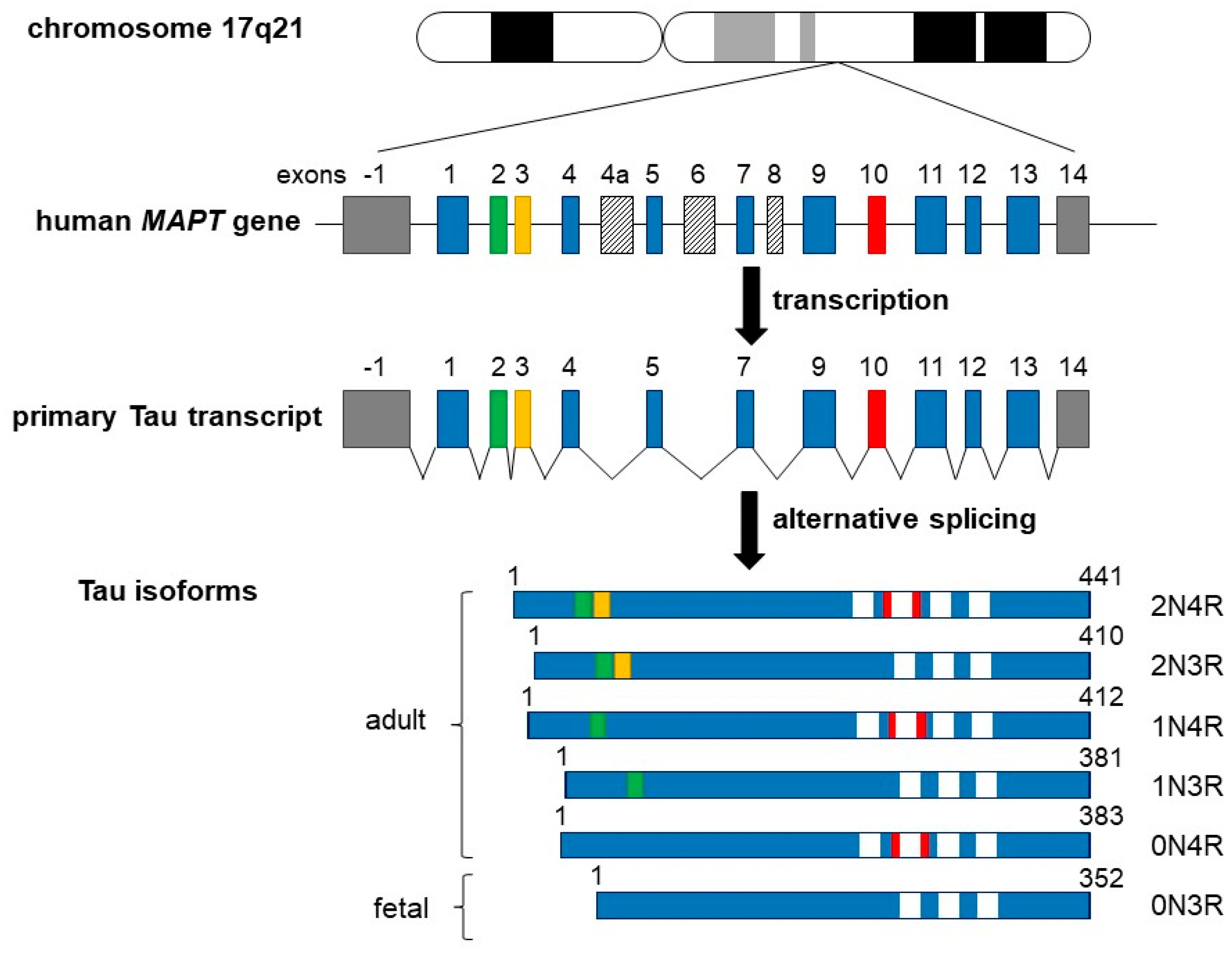{kind=link}
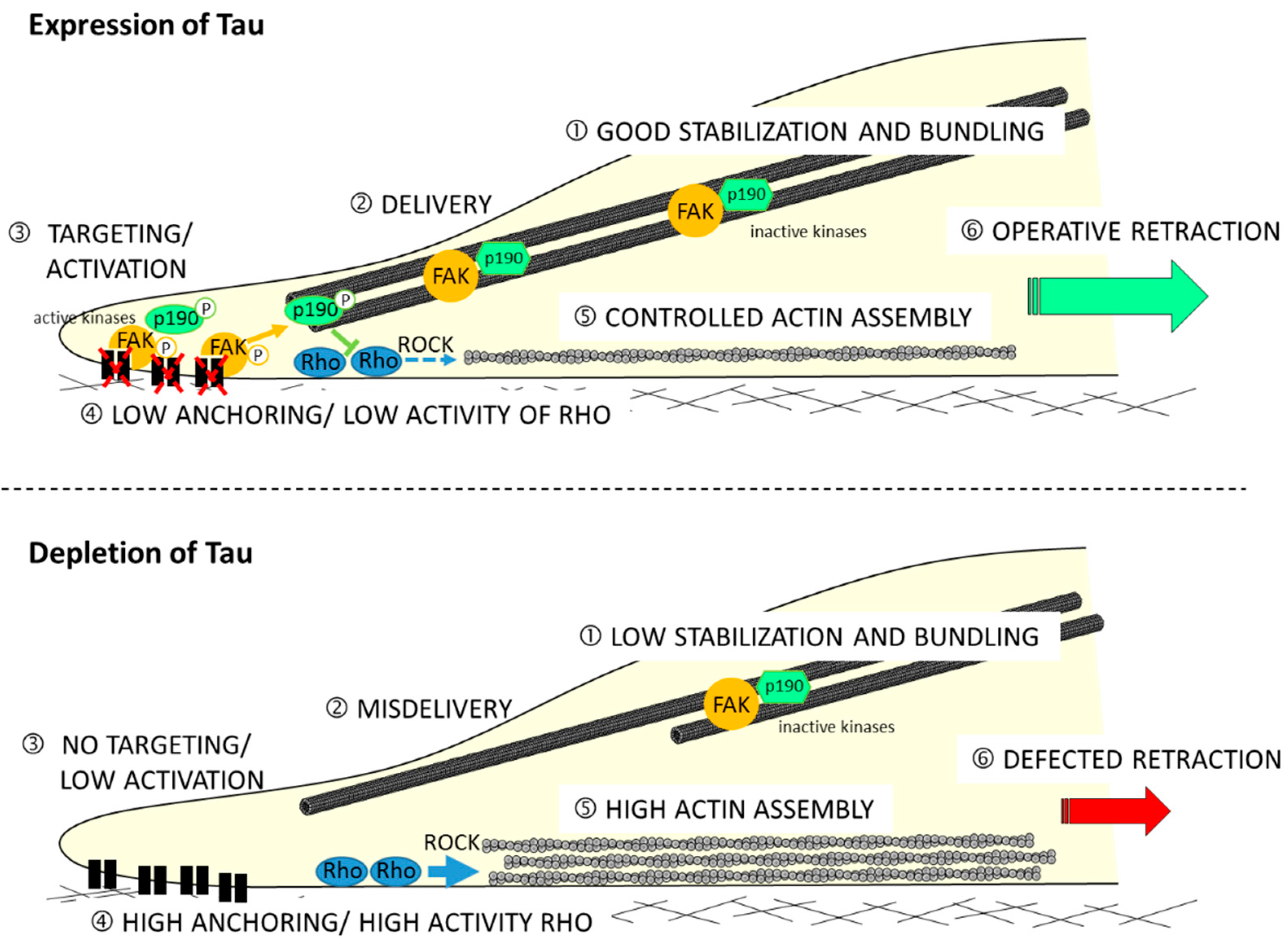{kind=link}
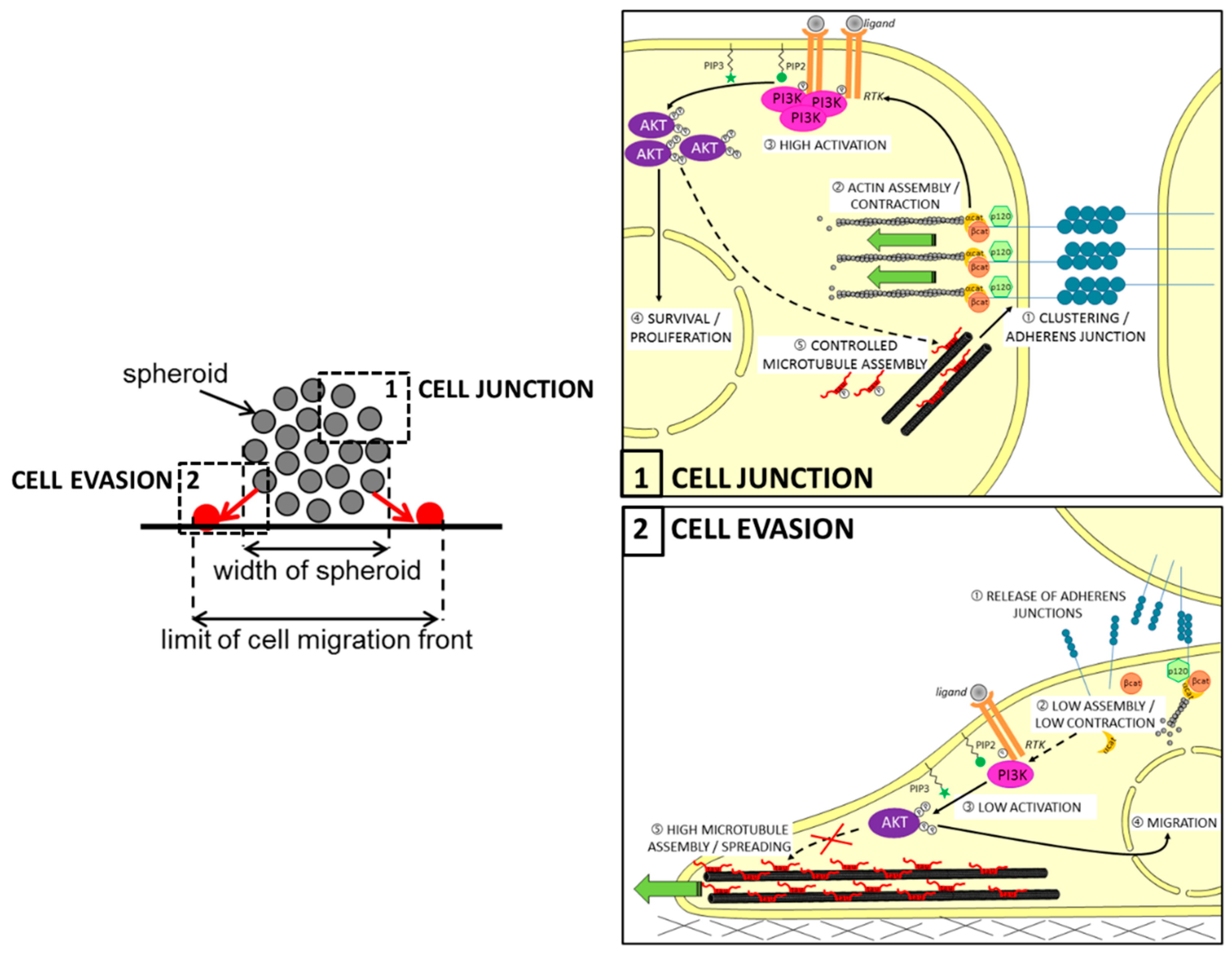{kind=link}
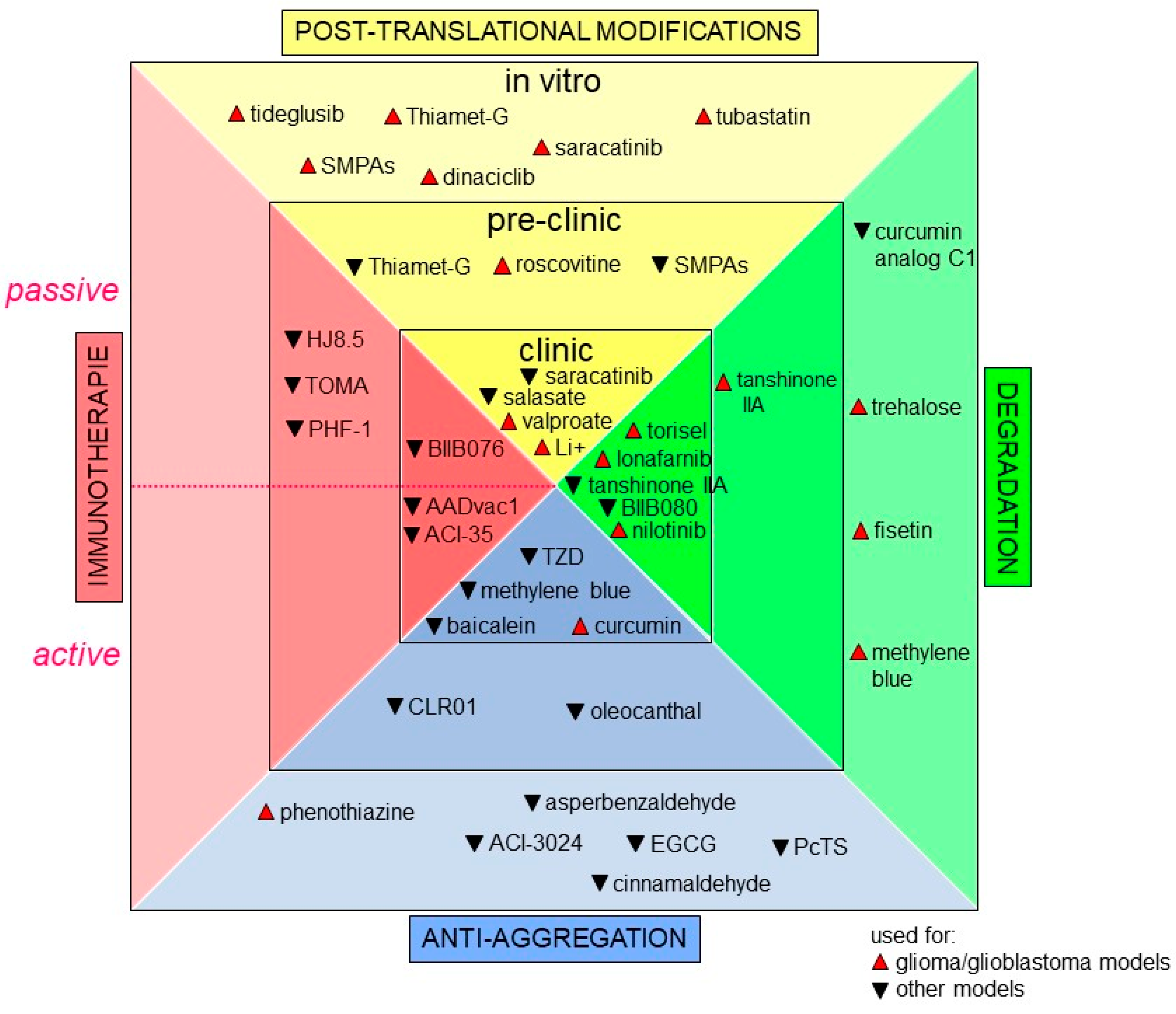{kind=link}
| Altered Pathways | Reported Targets | Tau Status | Cell Responses | References | |
|---|---|---|---|---|---|
| RTK pathway: | EGFR/Ras/MAPK | IDH | n.d. | Survival, proliferation | [65,96,126] |
| EGFR/Ras/MAPK | NFκB/TAZ | Upstream | Survival, angiogenesis | [65] | |
| PI3K/AKT ? | mTOR | Upstream ? | Drug resistance | [127] | |
| PI3K | - | n.a. | Interaction (in vitro) | [14,128] | |
| PI3K/AKT | n.d. | Upstream | Migration, adhesion, proliferation | [129] | |
| PTEN (deletion, mutation) | PI3K/AKT | Upstream | Survival, proliferation | [129,130,131] | |
| EGFR/PI3K/AKT ? | p53 | Upstream ? | Survival | [132,133] | |
| PI3K/AKT ? | GSK3β, 14-3-3 | Downstream | Survival, proliferation | [134,135,136,137,138,139,140,141] | |
| PI3K/AKT | CDK5, GSK3β | Downstream | Survival, proliferation, migration | [142,143] | |
| SKF pathway: | Src | RTK (PDGFR) | Upstream | Survival, migration | [99] |
| Fyn | RTK (EGFR, PDGFR, c-MET) ? | Upstream | Survival, proliferation | [144,145,146] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hedna, R.; Kovacic, H.; Pagano, A.; Peyrot, V.; Robin, M.; Devred, F.; Breuzard, G. Tau Protein as Therapeutic Target for Cancer? Focus on Glioblastoma. Cancers 2022, 14, 5386. https://doi.org/10.3390/cancers14215386
Hedna R, Kovacic H, Pagano A, Peyrot V, Robin M, Devred F, Breuzard G. Tau Protein as Therapeutic Target for Cancer? Focus on Glioblastoma. Cancers. 2022; 14(21):5386. https://doi.org/10.3390/cancers14215386
Chicago/Turabian StyleHedna, Rayane, Hervé Kovacic, Alessandra Pagano, Vincent Peyrot, Maxime Robin, François Devred, and Gilles Breuzard. 2022. "Tau Protein as Therapeutic Target for Cancer? Focus on Glioblastoma" Cancers 14, no. 21: 5386. https://doi.org/10.3390/cancers14215386
APA StyleHedna, R., Kovacic, H., Pagano, A., Peyrot, V., Robin, M., Devred, F., & Breuzard, G. (2022). Tau Protein as Therapeutic Target for Cancer? Focus on Glioblastoma. Cancers, 14(21), 5386. https://doi.org/10.3390/cancers14215386