
Methylome Profiling of PD-L1-Expressing Glioblastomas Shows Enrichment of Post-Transcriptional and RNA-Associated Gene Regulation


Abstract
Simple Summary
Abstract
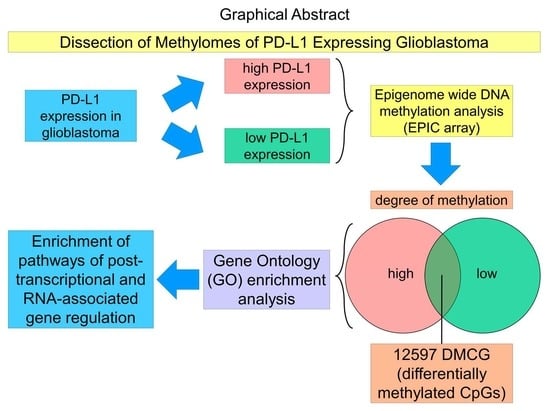
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Tissue Collection and Immunohistochemical Analysis
2.2. Molecular Genetic Analysis
2.3. Computational Data Analysis
3. Results
3.1. Glioblastoma IDH Wildtype CNS WHO Grade 4 Show Different Amount of PD-L1 Expression Quantified by Tumor Proportion Score (TPS)
3.2. Differential Methylation Analysis Shows PD-L1 Correlation with Methylation Signatures in Glioblastoma IDH Mutant CNS WHO Grade 4
3.3. Enrichment Analysis of DMCGs Reveals Distinct Altered Pathways Correlating with PD-L1 Status
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 who classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of o6-methylguanine methyltransferase (mgmt) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate mgmt activity. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar] [CrossRef]
- Hegi, M.E.; Sciuscio, D.; Murat, A.; Levivier, M.; Stupp, R. Epigenetic deregulation of DNA repair and its potential for therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5026–5031. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. Mgmt: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [PubMed]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. Mgmt gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Campesato, L.F.; Barroso-Sousa, R.; Jimenez, L.; Correa, B.R.; Sabbaga, J.; Hoff, P.M.; Reis, L.F.; Galante, P.A.; Camargo, A.A. Comprehensive cancer-gene panels can be used to estimate mutational load and predict clinical benefit to pd-1 blockade in clinical practice. Oncotarget 2015, 6, 34221–34227. [Google Scholar] [CrossRef]
- Gatalica, Z.; Snyder, C.; Maney, T.; Ghazalpour, A.; Holterman, D.A.; Xiao, N.; Overberg, P.; Rose, I.; Basu, G.D.; Vranic, S.; et al. Programmed cell death 1 (pd-1) and its ligand (pd-l1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2965–2970. [Google Scholar] [CrossRef]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the pd-1 and pd-l1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs cd4+ t cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef]
- Sun, C.; Mezzadra, R.; Schumacher, T.N. Regulation and function of the pd-l1 checkpoint. Immunity 2018, 48, 434–452. [Google Scholar] [CrossRef]
- Honda, Y.; Otsuka, A.; Ono, S.; Yamamoto, Y.; Seidel, J.A.; Morita, S.; Hirata, M.; Kataoka, T.R.; Takenouchi, T.; Fujii, K.; et al. Infiltration of pd-1-positive cells in combination with tumor site pd-l1 expression is a positive prognostic factor in cutaneous angiosarcoma. Oncoimmunology 2017, 6, e1253657. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Ogawa, S. Genetic biomarkers for pd-1/pd-l1 blockade therapy. Oncoscience 2016, 3, 311–312. [Google Scholar] [CrossRef]
- Kataoka, K.; Shiraishi, Y.; Takeda, Y.; Sakata, S.; Matsumoto, M.; Nagano, S.; Maeda, T.; Nagata, Y.; Kitanaka, A.; Mizuno, S.; et al. Aberrant pd-l1 expression through 3′-utr disruption in multiple cancers. Nature 2016, 534, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Isaacsson Velho, P.; Antonarakis, E.S. Pd-1/pd-l1 pathway inhibitors in advanced prostate cancer. Expert Rev. Clin. Pharmacol. 2018, 11, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Che, X.; Qu, J.; Hou, K.; Wen, T.; Li, Z.; Li, C.; Wang, S.; Xu, L.; Liu, Y.; et al. Exosomal pd-l1 retains immunosuppressive activity and is associated with gastric cancer prognosis. Ann. Surg. Oncol. 2019, 26, 3745–3755. [Google Scholar] [CrossRef] [PubMed]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus chemotherapy for pd-l1-positive non-small-cell lung cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Li, X.; Wetherilt, C.S.; Krishnamurti, U.; Yang, J.; Ma, Y.; Styblo, T.M.; Meisel, J.L.; Peng, L.; Siddiqui, M.T.; Cohen, C.; et al. Stromal pd-l1 expression is associated with better disease-free survival in triple-negative breast cancer. Am. J. Clin. Pathol. 2016, 146, 496–502. [Google Scholar] [CrossRef]
- Fujita, Y.; Yagishita, S.; Hagiwara, K.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Tsuta, K.; Nokihara, H.; Tamura, T.; et al. The clinical relevance of the mir-197/cks1b/stat3-mediated pd-l1 network in chemoresistant non-small-cell lung cancer. Mol. Ther. 2015, 23, 717–727. [Google Scholar] [CrossRef]
- Holzl, D.; Hutarew, G.; Zellinger, B.; Schlicker, H.U.; Schwartz, C.; Winkler, P.A.; Sotlar, K.; Kraus, T.F.J. Integrated analysis of programmed cell death ligand 1 expression reveals increased levels in high-grade glioma. J. Cancer Res. Clin. Oncol. 2021, 147, 2271–2280. [Google Scholar] [CrossRef]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. Pd-l1 expression and prognostic impact in glioblastoma. Neuro-Oncology 2016, 18, 195–205. [Google Scholar] [CrossRef]
- Heiland, D.H.; Haaker, G.; Delev, D.; Mercas, B.; Masalha, W.; Heynckes, S.; Gabelein, A.; Pfeifer, D.; Carro, M.S.; Weyerbrock, A.; et al. Comprehensive analysis of pd-l1 expression in glioblastoma multiforme. Oncotarget 2017, 8, 42214–42225. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Chen, G.; Zhao, H.; Li, Y.; Chen, J.; Zhang, H.; Li, S.; Zhao, Y.; Chen, F.; Li, W.; et al. Pd-l1 expression in glioblastoma, the clinical and prognostic significance: A systematic literature review and meta-analysis. Front. Oncol. 2020, 10, 1015. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Huang, C.; Mok, T.S.; Zhuang, W.; Xu, H.; Miao, Q.; Fan, X.; Zhu, W.; Huang, Y.; Lin, X.; et al. Comparison of 22c3 pd-l1 expression between surgically resected specimens and paired tissue microarrays in non-small cell lung cancer. J. Thorac. Oncol. 2017, 12, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Neuman, T.; London, M.; Kania-Almog, J.; Litvin, A.; Zohar, Y.; Fridel, L.; Sandbank, J.; Barshak, I.; Vainer, G.W. A harmonization study for the use of 22c3 pd-l1 immunohistochemical staining on Ventana’s platform. J. Thorac. Oncol. 2016, 11, 1863–1868. [Google Scholar] [CrossRef] [PubMed]
- Roge, R.; Vyberg, M.; Nielsen, S. Accurate pd-l1 protocols for non-small cell lung cancer can be developed for automated staining platforms with clone 22c3. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kraus, T.F.J.; Machegger, L.; Poppe, J.; Zellinger, B.; Dovjak, E.; Schlicker, H.U.; Schwartz, C.; Ladisich, B.; Spendel, M.; Kral, M.; et al. Diffuse midline glioma of the cervical spinal cord with h3 k27m genotype phenotypically mimicking anaplastic ganglioglioma: A case report and review of the literature. Brain Tumor Pathol. 2020, 37, 89–94. [Google Scholar] [CrossRef]
- Kraus, T.F.J.; Schwartz, C.; Machegger, L.; Zellinger, B.; Holzl, D.; Schlicker, H.U.; Poppe, J.; Ladisich, B.; Spendel, M.; Kral, M.; et al. A patient with two gliomas with independent oligodendroglioma and glioblastoma biology proved by DNA-methylation profiling: A case report and review of the literature. Brain Tumor Pathol. 2022, 39, 111–119. [Google Scholar] [CrossRef]
- Holzl, D.; Hutarew, G.; Zellinger, B.; Alinger-Scharinger, B.; Schlicker, H.U.; Schwartz, C.; Sotlar, K.; Kraus, T.F.J. Egfr amplification is a phenomenon of idh wildtype and tert mutated high-grade glioma: An integrated analysis using fluorescence in situ hybridization and DNA methylome profiling. Biomedicines 2022, 10, 794. [Google Scholar] [CrossRef]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef]
- Muller, F.; Scherer, M.; Assenov, Y.; Lutsik, P.; Walter, J.; Lengauer, T.; Bock, C. Rnbeads 2.0: Comprehensive analysis of DNA methylation data. Genome Biol. 2019, 20, 55. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ank1, bin1, rhbdf2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic profiling implicates cortical deregulation of ank1 in Alzheimer’s disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Ilie, M.; Khambata-Ford, S.; Copie-Bergman, C.; Huang, L.; Juco, J.; Hofman, V.; Hofman, P. Use of the 22c3 anti-pd-l1 antibody to determine pd-l1 expression in multiple automated immunohistochemistry platforms. PLoS ONE 2017, 12, e0183023. [Google Scholar]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kim, T.M.; Frenel, J.-S.; Simonelli, M.; Lopez, J.; Subramaniam, D.S.; Siu, L.L.; Wang, H.; Krishnan, S.; Stein, K.; et al. Treatment with Pembrolizumab in Programmed Death Ligand 1–Positive Recurrent Glioblastoma: Results from the Multicohort Phase 1 KEYNOTE-028 Trial. Cancer 2021, 127, 1620–1629. [Google Scholar] [CrossRef]
- Jiang, F.; Lang, X.; Chen, N.; Jin, L.; Liu, L.; Wei, X.; Pan, J.; Yu, F.; Blake, A.; Xiao, S. A novel hnrnph1::Erg rearrangement in aggressive acute myeloid leukemia. Genes Chromosomes Cancer 2022, 61, 503–508. [Google Scholar] [CrossRef]
- Liu, M.; Yang, L.; Liu, X.; Nie, Z.; Zhang, X.; Lu, Y.; Pan, Y.; Wang, X.; Luo, J. Hnrnph1 is a novel regulator of cellular proliferation and disease progression in chronic myeloid leukemia. Front. Oncol. 2021, 11, 682859. [Google Scholar] [CrossRef]
- Aoki, T.; Miyamoto, T.; Yoshida, S.; Yamamoto, A.; Yamauchi, T.; Yoshimoto, G.; Mori, Y.; Kamezaki, K.; Iwasaki, H.; Takenaka, K.; et al. Additional acquisition of t(1;21) (p32;q22) in a patient relapsing with acute myelogenous leukemia with nup98-hoxa9. Int. J. Hematol. 2008, 88, 571–574. [Google Scholar] [CrossRef]
- Miyamoto, R.; Kanai, A.; Okuda, H.; Komata, Y.; Takahashi, S.; Matsui, H.; Inaba, T.; Yokoyama, A. Hoxa9 promotes myc-mediated leukemogenesis by maintaining gene expression for multiple anti-apoptotic pathways. Elife 2021, 10, e64148. [Google Scholar] [CrossRef]
- Choong, L.Y.; Lim, S.; Chong, P.K.; Wong, C.Y.; Shah, N.; Lim, Y.P. Proteome-wide profiling of the mcf10at breast cancer progression model. PLoS ONE 2010, 5, e11030. [Google Scholar] [CrossRef]
- Li, Y.; Wei, Z.; Huang, S.; Yang, B. Mrna expression and DNA methylation analysis of the inhibitory mechanism of h2o2 on the proliferation of a549 cells. Oncol. Lett. 2020, 20, 288. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, J.; Liu, Z.; Zhang, Y. High expression of mir-196b predicts poor prognosis in patients with ovarian cancer. OncoTargets Ther. 2020, 13, 9797–9806. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.G.; Davies, M.; Lowery, A.J.; Miller, N.; Kerin, M.J. The role of microrna as clinical biomarkers for breast cancer surgery and treatment. Int. J. Mol. Sci. 2021, 22, 8290. [Google Scholar] [CrossRef]
- Liuksiala, T.; Teittinen, K.J.; Granberg, K.; Heinaniemi, M.; Annala, M.; Maki, M.; Nykter, M.; Lohi, O. Overexpression of snord114-3 marks acute promyelocytic leukemia. Leukemia 2014, 28, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wen, J.; Huang, Z.; Chen, X.P.; Zhang, B.X.; Chu, L. Small nucleolar rnas: Insight into their function in cancer. Front. Oncol. 2019, 9, 587. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncrna in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar]
- Li, Y.; Li, W.; Liang, B.; Li, L.; Wang, L.; Huang, H.; Guo, S.; Wang, Y.; He, Y.; Chen, L.; et al. Identification of cancer risk lncrnas and cancer risk pathways regulated by cancer risk lncrnas based on genome sequencing data in human cancers. Sci. Rep. 2016, 6, 39294. [Google Scholar] [CrossRef]
- Ma, R.; Yan, W.; Zhang, G.; Lv, H.; Liu, Z.; Fang, F.; Zhang, W.; Zhang, J.; Tao, T.; You, Y.; et al. Upregulation of miR-196b confers a poor prognosis in glioblastoma patients via inducing a proliferative phenotype. PLoS ONE 2012, 7, e38096. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutarew, G.; Hölzl, D.; Schiefer, T.; Langwieder, C.K.; Alinger-Scharinger, B.; Schlicker, H.U.; Schwartz, C.; Sotlar, K.; Kraus, T.F.J. Methylome Profiling of PD-L1-Expressing Glioblastomas Shows Enrichment of Post-Transcriptional and RNA-Associated Gene Regulation. Cancers 2022, 14, 5375. https://doi.org/10.3390/cancers14215375
Hutarew G, Hölzl D, Schiefer T, Langwieder CK, Alinger-Scharinger B, Schlicker HU, Schwartz C, Sotlar K, Kraus TFJ. Methylome Profiling of PD-L1-Expressing Glioblastomas Shows Enrichment of Post-Transcriptional and RNA-Associated Gene Regulation. Cancers. 2022; 14(21):5375. https://doi.org/10.3390/cancers14215375
Chicago/Turabian StyleHutarew, Georg, Dorothee Hölzl, Tanja Schiefer, Celina K. Langwieder, Beate Alinger-Scharinger, Hans U. Schlicker, Christoph Schwartz, Karl Sotlar, and Theo F. J. Kraus. 2022. "Methylome Profiling of PD-L1-Expressing Glioblastomas Shows Enrichment of Post-Transcriptional and RNA-Associated Gene Regulation" Cancers 14, no. 21: 5375. https://doi.org/10.3390/cancers14215375
APA StyleHutarew, G., Hölzl, D., Schiefer, T., Langwieder, C. K., Alinger-Scharinger, B., Schlicker, H. U., Schwartz, C., Sotlar, K., & Kraus, T. F. J. (2022). Methylome Profiling of PD-L1-Expressing Glioblastomas Shows Enrichment of Post-Transcriptional and RNA-Associated Gene Regulation. Cancers, 14(21), 5375. https://doi.org/10.3390/cancers14215375