Simple Summary
Gastrointestinal stromal tumors (GISTs) are malignant mesenchymal tumors classified primarily as soft tissue sarcomas and characteristically arise from the intestinal pacemaker cells of Cajal responsible for the gastrointestinal tract motility. They comprise a heterogenous group of tumors due to a variety of molecular alterations, mostly KIT (60–70%) or PDGFRA (10–15%) mutations, but also more rare alterations, including inactivation of the NF1 gene, mutations in the succinate dehydrogenase (SDH), BRAF, and RAS genes, and also gene fusions. Here, we review the most recent data on the molecular profile of GISTs and highlight systemic therapeutic implications according to distinct GIST molecular subtypes.
Abstract
Gastrointestinal stromal tumors (GISTs) are malignant mesenchymal tumors arising from the intestinal pacemaker cells of Cajal. They compose a heterogenous group of tumors due to a variety of molecular alterations. The most common gain-of-function mutations in GISTs are either in the KIT (60–70%) or platelet-derived growth factor receptor alpha (PDGFRA) genes (10–15%), which are mutually exclusive. However, a smaller subset, lacking KIT and PDGFRA mutations, is considered wild-type GISTs and presents distinct molecular findings with the activation of different proliferative pathways, structural chromosomal and epigenetic changes, such as inactivation of the NF1 gene, mutations in the succinate dehydrogenase (SDH), BRAF, and RAS genes, and also NTRK fusions. Currently, a molecular evaluation of GISTs is imperative in many scenarios, aiding in treatment decisions from the (neo)adjuvant to the metastatic setting. Here, we review the most recent data on the molecular profile of GISTs and highlight therapeutic implications according to distinct GIST molecular subtypes.
1. Introduction
Gastrointestinal stromal tumors (GISTs) are malignant mesenchymal tumors classified primarily as soft tissue sarcomas [1] and characteristically arise from the intestinal pacemaker cells of Cajal responsible for the gastrointestinal tract motility [2]. GISTs are the most frequent type of sarcomas and also the most frequent sarcoma of the gastrointestinal tract [3]. Most GISTs are sporadic, solitary tumors, and more are routinely found in the stomach or small intestines (60–65% and 20–35% of cases, respectively) [2]. Primary GISTs of the rectum can be found in 3–5% of cases, and an even smaller portion of cases can be found in the colon, esophagus, and peritoneal cavity. The most common metastatic sites are the liver and peritoneal cavity, mostly due to anatomic contiguity [4].
GISTs are more commonly diagnosed in an older subset of patients, and there is also a slight male predominance. A smaller portion of GISTs (approximately 2%) occur in pediatric patients [5] and can be associated with genetic syndromes such as neurofibromatiosis-1 (NF1), familial GISTs, the Carney Triad (CT) and Carney–Stratakis Syndrome (CSS) [4,6,7,8,9]. Pathological features of GISTs have already been demonstrated, and 95% of them express KIT (CD117) by immunohistochemistry [10] in addition to other markers such as DOG1 (95%), CD34 (60–70%), smooth muscle actin (SMA) (30–40%), and S100 protein (5%), which are more useful in diagnosing KIT-negative GISTs [11,12,13].
GISTs comprise a heterogenous group of tumors mostly due to a variety of molecular alterations. The most common gain-of-function mutations in GISTs are either in the KIT (60–70%) or platelet-derived growth factor receptor alpha (PDGFRA) genes (10–15%) [2], which are mutually exclusive [14]. However, a smaller subset, which lacks KIT and PDGFRA mutations, is considered wild-type GISTs and presents distinct molecular findings with activation of different proliferative pathways, structural chromosomal and epigenetic changes, such as inactivation of the NF1 gene, mutations in the succinate dehydrogenase (SDH), BRAF, and RAS genes, and also NTRK fusions [15]. Currently, molecular evaluation of GISTs is imperative in many scenarios.
Classically known to harbor resistance to cytotoxic chemotherapies, GISTs are considered a mark of the advances in precision medicine after the success of tyrosine kinase inhibitors (TKIs), such as imatinib, in the treatment of KIT-positive advanced GISTs in 2002 [16]. However, treatment resistance is still a pertinent issue. As novel mutations and further insights into the underlying molecular signature of GISTs were uncovered, new lines of therapy presented optimistic results with targeted therapy [17]. Nonetheless, there is still a portion of GISTs that present treatment challenges. Therefore, it is important to accurately evaluate tumor pathology, including cell morphology and immunohistochemistry (accessing positiveness for KIT and DOG1), and the molecular profile [3] in order to improve the prediction of the biological behavior of the tumor and define the prognosis, as well as treatment response. Here, we review the most recent data on the molecular profile of GISTs and the systemic therapeutic implications.
2. Molecular Classification of GISTs
Until recently, GISTs were classified into two main groups: KIT/PDGFRA mutated GISTs (85% of cases) and wild-type GISTs. However, rare mutations or fusions have been described among the latter, which made the “wild-type” description less precise. Amongst the wild-type KIT/PDGFRA GISTs, subgroups with well-defined molecular features have been described, including those harboring mutations in the BRAF, RAS, or NF1 genes [18,19]. GISTs with mutations in BRAF/RAS or NF1 were once referred to as the RAS-pathway (RAS-P) mutant GISTs. In addition, around a third of wild-type KIT/PDGFRA GISTs demonstrate the loss-of-function of the succinate dehydrogenase complex (SDH), manifested by the loss of subunit B (SDHB) protein expression [19]. In 2015, an Italian group proposed that KIT/PDGFRA/SDH/RAS-P wild-type GISTs should be designated as quadruple wild-type GISTs [19].
Currently, there has been an effort to subdivide GISTs into two major groups according to SDH competency by SDHB immunohistochemistry [7,20]. SDH is a heterotetrameric enzyme complex located in the inner mitochondrial membrane and participates in the Krebs cycle. Subunit A (SDHA) is responsible for converting succinate into fumarate, while subunit B (SDHB) participates in the electron transport chain for the oxidation of ubiquinone to ubiquinol. Subunits C and D (SDHC and SDHD) are membrane-anchoring subunits [7]. Loss of any SDH subunit renders the complex inactive and leads to loss of detectable SDHB by immunohistochemistry.
The SDH-competent GIST group is by far the most common one. It includes the large proportion of patients who harbor either KIT or PDGFRA mutations, but also GISTs with less frequent mutations (i.e., BRAF, NF1, HRAS, NRAS), with even more rare mutations (i.e., ARID1A, ARID1B, ATR CBL, FGFR1, KRAS, LTK, MEN1, PARK2, PIK3CA, SUFU, and ZNF217) and those with structural chromosomal changes (i.e., FGFR1-HOOK3, FGFR1-TACC1, ETV6–NTRK3, KIT-PDGFRA, and PRKAR1B-BRAF) [21].
Conversely, the group of SDH-deficient GISTs includes wild-type GISTs in association with CT, CSS, and sporadic pediatric and young adult GISTs.
Given the different subtypes of GISTs, we recommend the following stepwise fashion of testing (Figure 1) whenever systemic treatment is indicated and next-generation sequencing (NGS) is not promptly available.
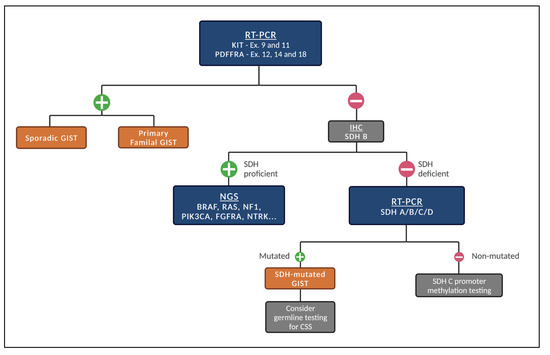
Figure 1.
Proposed stepwise molecular testing for GISTs (when NGS is not promptly available). Legend: Initial test for the presence of KIT and PDGFRA mutations. If negative, test for SDH status by IHC. If SDH-proficient, patients should be tested for other genetic alterations. If SDH-deficient, test for mutations in SDH A/B/C/D. When mutated, consider germline testing for CSS. If not mutated, consider SDH C promoter methylation testing.
2.1. SDH-Competent GIST with either KIT or PDGFRA Mutation
KIT codifies the c-Kit protein (also known as stem cell factor (SCF) receptor or CD117), a receptor tyrosine kinase (RTK) with five Ig-like extracellular (EC) domains (therefore, class III RTK) with 145 kDa and 976 amino acid residues in normal splicing [22]. In humans, four isoforms of c-Kit have been described so far, with the GNNK-negative isoform showing greater kinase activity and downstream signaling. The first three EC domains are essential for SCF binding and the other two for receptor homodimerization. The EC domains are connected by a single spanning transmembrane helix to the intra-cellular juxtamembrane (JM) domain, which works as a modulator of the signal transduction. The tyrosine kinase domain is split in two functional lobes (N-terminal and C-terminal), and in the enzyme inactive state, the JM domain inserts between the two lobes, sterically blocking the conformational changes needed for the activation loop of the catalytic unit to assume its active form. Finally, following the C-terminal lobe of the tyrosine kinase domain is the C-terminal intra-cellular tail. Upon dimeric SCF binding to adjacent c-Kit molecules, reorientation of D4-5 EC domains leads to receptor homodimerization. The following conformational changes lead to sequential trans-phosphorylations in the JM domain, activation loop, kinase insert region, and C-terminal tail [22]. These phosphorylated tyrosine residues serve as substrate docking sites for signal transduction. The following signaling pathways are activated by c-Kit: mitogen-activated protein kinase (MAP kinase), phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT), phospholipase C gamma (PLC-gamma), Janus kinase/signal transducer and activator of transcription (JAK/STAT), and Scr kinase pathways [22,23]. The specific phosphorylated tyrosine residues determine which signaling pathways are activated.
Primary KIT mutations occur in roughly 70% of GISTs [24]. These gain-of-function mutations lead to constitutive activation of the receptor and are commonly found in two hot spots (Figure 2): membrane-proximal EC domain (exon 9; 9–10% of all GISTs) and intra-cellular JM domain (exon 11; 60% of all GISTS). GISTs with exon 11 mutations can occur throughout the gastrointestinal tract, and those with exon 9 mutations arise primarily in the small or large bowel [2]. Importantly, the prognosis is dependent on the specific type of mutation within exon 11, with tumors displaying deletions in codons 557 and 558 presenting more aggressive clinical behavior. Primary mutations in exons 8 (membrane-proximal EC domain), 13 (ATP-binding site), and 17 (activation loop) are rare (<1% of all GISTs) [24].
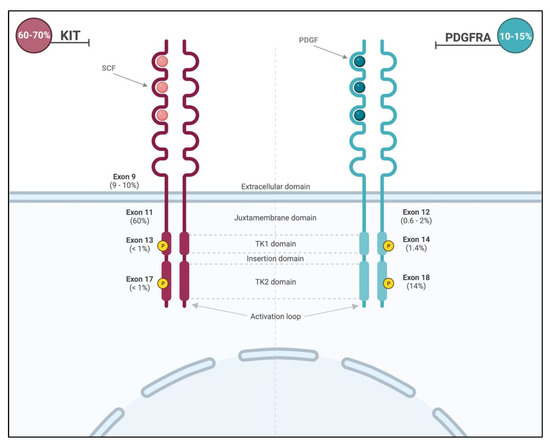
Figure 2.
Location and frequencies of most common KIT and PDGFRA mutations.
Another important and common molecular alteration and drive mutation in GISTs is PDFGA mutation. As the KIT gene, PDGFRA is located on chromosome 4, encodes a type III RTK, and is homologous to KIT, presenting a similar structure and downstream pathways, such as RAS/RAF/MAPK and PI3K [14,25,26]. Importantly, KIT and PDGFRA mutations are considered to be mutually exclusive. PDGFRA mutations, once present, trigger a ligand-independent phosphorylation and uninhibited cellular proliferation [15].
PDGFRA mutations are mainly encountered in exon 18 (activation loop of the receptor; 13–14%) and less frequently in exon 12 (JM domain; 0.6–2%) or exon 14 (ATP-binding site; 1.4%) [2] (Figure 2). The PDGFRA exon 18 D842V missense mutation can be found in around 50–70% of PDFRA-mutated GISTs and 8% of all GISTs [27] and promotes the stabilization of the kinase conformation in its active form, therefore conferring resistance of this molecular subtype to the most commonly used treatments [28,29]. The majority of PDFGRA-mutated GISTs arise from the stomach (15–18%) and the small intestine (5–7%). They classically have a more indolent behavior [2] and favorable prognosis compared to the more commonly encountered KIT mutations.
2.2. Therapeutic Implications of GIST with KIT or PDGFRA Mutations
The main therapeutic strategy to treat GISTs with either KIT or PDGFRA mutations has been the use of TKIs, especially imatinib. Initially developed to target the BCR-ABL translocation of chronic myelogenous leukemia [30], it was shown to be active against most GISTs [31]. Thus, imatinib is considered one of the first targeted therapies for the treatment of solid tumors.
The initial treatment of high-risk (>50% recurrence risk) GISTs in the adjuvant setting is considered to be a daily 400 mg dose of imatinib for a period of ≥3 years [32,33,34]. The recommended duration of treatment was defined based on evidence that, with 3 years compared with 1 year of therapy, recurrence-free survival (RFS) at 5 years was 71.1% versus 52.3% and overall survival (OS) was 91.9% versus 82.3%, respectively [32]. However, studies have been conducted aiming to define the appropriate duration of adjuvant treatment with imatinib and possibly expanding the duration for a period of 5 years [35], and yet, other studies are underway in order to determine the efficacy of extending the duration of adjuvant treatment (NCT02413736, NCT02260505).
In the advanced/metastatic disease setting, imatinib is started at a 400 mg daily dose, thus achieving response rates and progression-free survival (PFS) rates of up to 40 months and 70%, respectively, in patients with the most common KIT and PDGFRA mutations [36,37,38,39,40]. However, regardless of such outcomes with first-line imatinib, subsequent lines of therapy present only modest benefits, and wild-type KIT/PDGFRA GISTs typically present with early progression with imatinib [36,41].
Tumors with deletions of codons 557 and 558 present more aggressive clinical behavior than those with exon 11 missense mutations. However, such mutations display significant responses to imatinib. Furthermore, patients’ genetics seems to modify the chance of tumor response to imatinib. Among patients with exon 11 mutant GISTs, differences in clinical response to imatinib have been shown to be associated with single-nucleotide polymorphisms in genes related to the absorption, distribution, metabolism, and excretion (ADME) process [42]. GISTs with exon 9 typically present lower response to imatinib when compared to those with exon 11 mutations. Nonetheless, in the metastatic setting, this can be partially circumvented by doubling the dose of imatinib (800 mg/day). Data from randomized trials suggest that the higher dose of imatinib is associated with an improved objective response rate (47 vs. 21%) and PFS (HR = 0.57; p = 0.017) [39]. While some GISTs with primary exon 13 mutations are sensitive to imatinib, most primary exon 17 mutations confer primary resistance do imatinib [2].
Most PDGFRA mutations are sensitive to imatinib therapy (exons 12, 14, and 18). However, PDGFRA exon 18 D842V mutation can also present with primary resistance to imatinib [41], as well as other TKIs [14,24,43]. Yet, due to the more indolent nature of this GIST subtype, patients may present with stable disease while on therapy [40].
Of note, around 10% of patients diagnosed with advanced GIST can present with primary resistance to imatinib with disease progression within the first 6 months of treatment. This primary resistance to treatment is linked to the GIST genotype [36,38,39,43] and is found to be more frequent in KIT exon 9 mutations and in wild-type KIT/PDGFRA GISTs.
Acquired KIT and PDGFRA mutations occur in up to 90% of patients with metastatic GIST treated with imatinib and are the main cause of secondary resistance [44]. In most situations, imatinib therapy induces tumor volume reduction upon inducing cellular apoptosis. However, a fraction of tumor cells enters a quiescent, non-proliferative state and may acquire secondary genetic mutations, rendering such cells imatinib-resistant, leading to disease progression. Such mutations confer an evolutionary advantage under the selective pressure of imatinib exposure and cluster into two hot regions of the KIT gene: the ATP-binding pocket (exons 13 and 14 of KIT and exon 14 of PDGFRA) and the activation loop (exons 17 and 18 of KIT and exon 18 of PDGFRA) [2,45,46,47]. Therefore, treatment sequencing with other TKIs becomes necessary to overcome disease progression due to such mutations. c-Kit molecules with secondary mutations in the ATP-binding pocket are relatively sensitive to sunitinib [48], ponatinib [49], ripretinib [17,50], and avapritinib [51,52], but not to regorafenib or sorafenib [2], as shown in Table 1. Conversely, receptors carrying acquired mutations in the activation loop can be targeted with regorafenib [53], sorafenib [54], nilotinib [55], ponatinib [49], dasatinib (NCT01643278), ripretinib [50], or avapritinib [51]. The one exception is the D816V mutation in exon 17, which is resistant to all TKIs with the exception of ponatinib, ripretinib, and avapritinib [44,56,57].

Table 1.
Sensitivity profile of different molecular subtypes of KIT or PDGFRA GISTs.
The PDGFRA-mutant GISTs’ resistance mechanism profile is less detailed compared to KIT, and little is known beyond primary or secondary PDGFRA D482V mutations [58]. However, D842V has been shown to be homologues to the KIT exon 17 D816V mutation [24] and, as such, demonstrates a similar treatment sensitivity profile with resistance to the majority of the approved TKIs for the treatment of advanced GISTs, harboring a very poor prognosis. Yet, in 2020, the NAVIGATOR trial demonstrated the activity of avapritinib, a fourth-generation TKI designed specifically to inhibit PDGFRA D842V in advanced GISTs with such mutation with an objective response rate of 84% (7% complete responses) [51]. However, resistance mechanisms to avapritinib in PDGFRA D842V mutated GISTs have been demonstrated and cause avapritinib binding blockage due to substitutions in exons 13, 14, and 15 (V658A, N659K, Y676C, and G680R) [58].
2.3. SDH-Competent GIST without KIT and PDGFRA Mutation
NF1-Mutant GIST
Neurofibromatosis 1 (NF1) is an autosomal dominant disorder caused by inherited or de novo germline mutations within the NF1 tumor suppressor gene. It is clinically characterized by multiple café-au-lait macules, intertriginous freckling, multiple cutaneous neurofibromas, plexiform neurofibromas, malignant peripheral nerve sheath tumors and, sometimes, learning disability or behavioral problems [59,60]. The product of the NF1 gene is the large and multifunctional cytoplasmic protein neurofibromin, which belongs to a family of guanosine-triphosphate-hydrolase (GTPase)-activating proteins that stimulate intrinsic GTPase activity in the Ras family [59].
Patients with germline inactivation of the NF1 gene have a propensity to develop several tumors through acquired inactivation of the normal NF1 allele, and it has been estimated that 7% of individuals with NF1 develop GIST during their lifespan, which corresponds to a 34-fold higher risk than the average population [59,61]. NF1-associated GISTs, which correspond to 1% of all GISTs, are commonly multicentric and predominantly located in the small intestine [62,63] as shown in Table 2. Although most cases of NF1-related GISTs are germinative, somatic inactivating NF1 mutations may also occur as an oncogenic mechanism for a small subset of wild-type GISTs [62].
Table 2.
GIST molecular subtypes and characteristics.
NF1-related GISTs typically express KIT, DOG1, and SDHB by IHC [64]. They also frequently demonstrate loss of heterozygosity (LOH) at 14q and 22q, which may contribute to the early phase of tumor development [65]. In addition, genomic profiling studies of adult GISTs have revealed that NF1 alterations are enriched in GISTs located at the duodenal–jejunal flexure. These tumors may harbor concurrent activating KIT and/or inactivating Notch pathway mutations [63].
2.4. Therapeutic Implications of NF1-Mutant GISTs
Since most NF1-mutant GISTs do not harbor KIT mutations, they are typically insensitive to imatinib and surgery remains the mainstay therapy for these patients. Although, so far, there are no data on target-specific drugs for NF1-related GISTs, MEK inhibitors have pointed towards clinical efficacy in other NF1-associated tumors. Given the key role of abnormal RAF/MEK/ERK pathway signaling in neurofibromas, a phase I study of selumetinib (a MEK inhibitor) in children/young adults with NF1-related inoperable plexiform neurofibromas has demonstrated a median maximal decrease of 24% in the volume of the tumors [66]. Currently, a phase II trial with a different MEK inhibitor is ongoing for adolescents and young adults with NF1-associated plexiform neurofibromas (NCT02096471).
2.5. BRAF Mutated GIST
BRAF mutations at the exon 15 V600E hotspot are encountered in approximately 4–13% of adult wild-type GISTs and are almost mutually exclusive of KIT/PDGFRA mutations [67,68,69]. They are more frequently found in the small bowel and have variable clinical behavior [67]. Phenotypically and morphologically, BRAF-mutant GISTs are similar to KIT/PDGFRA positive GISTs [67,68,69]. More recently, BRAF fusions have been described in GISTs [70].
2.6. Therapeutic Implications of BRAF Mutant GISTs
BRAF V600E mutation in GISTs has been shown to confer resistance to imatinib and sunitinib [71]. Since BRAF V600E mutations are currently considered an agnostic genetic alteration and seem to respond to BRAF inhibitors regardless of the primary tumor type, we recommend a BRAF inhibitor with or without a MEK inhibitor to patients with unresectable or metastatic BRAF mutant GISTs. The combination of dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor) has recently received FDA approval for any BRAF mutant tumor, regardless of the origin site. The first case of a patient with a BRAF mutant GIST who responded during 8 months to dabrafenib was reported in 2013 [72]. This patient had already failed to receive benefit from imatinib, sunitinib, and a MEK inhibitor.
Interestingly, a complete radiological response with first-line regorafenib was reported in a 51-year-old woman with a BRAF-mutated GIST [73]. Regorafenib has a wide spectrum of action, with MAPK signaling pathway blockade at different levels. There is an ongoing phase II, single-arm, trial evaluating regorafenib in the first-line setting for advanced KIT/PDGFRA wild-type GISTs (NCT02638766).
The role of kinase inhibitors targeting BRAF is yet unknown in the adjuvant setting for this subset of patients. Therefore, we recommend no systemic therapy following R0 resections, even for those with high-risk BRAF mutant disease.
2.7. GISTs with NTRK Fusion
TRK fusions arise from aberrant rearrangements of neurotrophic receptor kinase (NTRK) genes 1–3 with several gene partners. They are constitutively active ligand-independent oncogenic drivers in different solid tumors, including GISTs. Until recently, only ETV6-NTRK3 gene fusions were described in GISTs. However, NTRK 1 fusion has been recently reported [74].
2.8. Therapeutic Implications of GISTs Harboring NTRK Fusions
NTRK fusions are considered an agnostic feature that renders tumors sensitive to TRK inhibitors independently of the primary cancer. In the metastatic setting, larotrectinib, a TRK inhibitor, demonstrated great activity against solid tumors harboring NTRK fusions [75]. In a pooled analysis from three phase I/II clinical trials, 159 patients with different tumor types harboring NTRK fusions were treated with larotrectinib. The objective response rate was 79%. Indeed, four patients with GISTs were included, and all of them achieved an objective response [75]. In another pooled analysis from three different studies of individuals with NTRK fusions, three patients with GIST were treated with larotrectinib and all responded, including one with complete response [76].
2.9. GISTs with FGFR Pathway Alterations
Fibroblast growth factor receptor (FGFR) fusions, mutations, and ligand overexpression represent the most common molecular alterations in quadruple WT GISTs, suggesting a possible pathogenetic role [77].
2.10. Therapeutic Implications of FGFR-Altered GISTs
Many drugs targeting FGFR are already in use in clinical practice (such as regorafenib) or have been tested in clinical trials for GISTs (dovitinib, masitinib, ponatinib, and pazopanib), but none so far specifically for FGFR-altered GISTs [75,78,79,80,81,82,83,84,85,86].
Although very promising, FGF/FGFR pathway inhibition in GISTs is still far from clinical practice.
2.11. Other Rare Mutations
KRAS-mutated GISTs are extremely rare, and the clinicopathologic features still lack complete comprehension [87]. Yet another extremely rare GIST subtype are the PIK3CA mutant GISTs. Currently, the most robust study involving imatinib-naïve GIST patients, 10 (mostly localized tumors) out of 529 GISTs harbored PIK3CA mutations, which were associated with larger and aggressive tumors [88] ]. Given the limited number of patients, further studies with longer follow up are required.
2.12. SDH-Deficient GIST
Wild-type GISTs are the primary form of GIST in children (85%) and only occasionally occur in adults (15%) [89]. They are often localized in the stomach, multifocal, with lymph node involvement, indolent, and primarily affect young females [90]. Approximately half of pediatric WT-GISTs are SDH-deficient, which have loss-of-function of the SDH complex subunit (SDHA, SDHB, SDHC, or SDHD). This loss of function occurs either due to a mutation in one of the SDHx genes or due to epigenetic silencing [91,92].
The SDH complex is a mitochondrial complex that participates in the Krebs cycle in the conversion of succinate to fumarate (succinate reductase). Loss of the mitochondrial SDH complex through mutations in SDHA, SDHB, SDHC, or SDHD causes the accumulation of succinate, resulting in the overexpression of hypoxia-inducible factor (HIF) proteins and increased transcription of HIF-1a-regulated genes. It also leads to decreased DNA demethylation. Hence, an increase in DNA methylation and activation of insulin-like growth factor 1 receptor (IGF1R) and vascular endothelial growth factor receptor (VEGFR) are the molecular characteristics of SDH-deficient GISTs [93].
SDH-deficient GISTs have different clinical courses, molecular characteristics, prognoses, and therapeutic responses when compared to other GISTs. SDHA is the most commonly mutated (30%) in all SDH-deficient GISTs [94]. Immunohistochemical analysis for SDHB is appropriate to diagnose SDH deficiency in KIT/PDGFRA wild-type GISTs, as the absence of SDHB can indicate the deficiency of any of the SDHx genes. However, not all SDHB-immunonegative GISTs harbor an SDH gene mutation; these tumors may have other epigenetic and genetic defects in the SDH pathway [8].
Approximately 5% of patients with GISTs have genetic syndromes associated with the development of these tumors, which include primary familial GIST syndrome (heritable mutations in either the KIT or PDGFRA genes), NF1, CSS (SDHx genes), and CT (hypermethylation of the SDHC promoter). CSS, also referred to as the Carney–Stratakis dyad, is a rare inherited predisposition syndrome caused by germline mutations in the SDHB/C or D subunits, which presents with the dyad of GISTs and paragangliomas [95,96]. The Carney triad is an extremely rare nonhereditary syndrome consisting of GISTs, paragangliomas, and pulmonary chondromas caused by epigenetic inactivation (hypermethylation) of the SDHC gene with functional impairment of the SDH complex [95,97,98].
Patients diagnosed with SDH-deficient GISTs and those who have NF1 mutations should be referred for genetic counseling and appropriate germline testing. Furthermore, patients with SDH-deficient GISTs, or known SDH mutations, could have paraganglioma associated (Carney–Stratakis dyad or CT), and therefore, serum/urine catecholamine/metanephrine testing should be considered prior to surgery [99].
2.13. Therapeutic Implications of SDH-Deficient GISTs
An observational study evaluated the clinical and molecular features of KIT/PDGFRA WT-GISTs in a cohort (patients < 19 y with GIST or > 19 y with WT-GIST), which was characterized by IHC for SDHB, sequencing of SDH genes, determination of SDHC promoter methylation, as well as germline testing of SDH genes for some of them (consented). Among the 95 patients analyzed (70% female; median age: 23 years; 19% with syndromic GIST), three molecular subtypes were defined: 11 had SDH competency (which included mutations in NF1; BRAF; other rare mutations/fusions; or no identified), and 84 had SDH deficiency (75% due to SDH mutations and 25% due to SDHC promoter hypermethylation). SDH mutations were often germline (82%) among 38 tested patients. In this cohort, despite the non-standardized retrospective assessment of treatment, only 1 of 49 patients treated with imatinib and 4 of 38 patients treated with sunitinib had an objective response [7].
SDH-deficient GIST is an indolent disease, and most patients have a good median survival of approximately 10 years even though with disease progression [100]. This subtype of GISTs is known to be generally unresponsive to TKIs. In the analysis of molecular alterations associated with the treatment benefit of adjuvant imatinib in the ACOSOG Z9001 study, KIT/PDGFRA WT tumors were uncommon (9 in the placebo and 6 in the imatinib group). As a result, the therapeutic impact of adjuvant imatinib could not be considered definitive in this subset and remains controversial [101]. The National Comprehensive Cancer Network (NCCN) guidelines version 2.2022 [99] and the European Society for Medical Oncology (ESMO) guidelines 2021 [3] do not support routine clinical use of imatinib adjuvant for WT-GISTs. In the advanced scenario, the best way to treat these patients is not established, and personalized treatment is recommended [3]. Although refractory to imatinib, they may have some responsiveness to sunitinib [102], regorafenib [103], Pazopanib [85], Nilotinib, and Linsitinib [104]. However, the data on TKIs in patients with SDH-deficient GISTs is extremely limited. Referral to a clinical trial is a reasonable consideration for them.
Molecular characterization demonstrated that temozolomide (TMZ) elicits DNA damage and apoptosis in SDH-deficient GISTs with a 40% rate of objective responses among five patients treated with this alkylating agent [105]. Based on this early efficacy data, a phase II study of single-agent TMZ (NCT03556384) in patients with SDH-mutant GISTs is ongoing.
Other agents are under study in clinical trials, including rogaratinib (potent and selective pan-FGFR inhibitor/NCT04595747), vandetanib (potent VEGFR inhibitor/NCT02015065), and guadecitabine (DNA methyl transferase inhibitor/NCT03165721). In addition, DNA hypomethylating agents are being evaluated in SDH-deficient GISTs, which may be associated with DNA hypermethylation [106].
3. Conclusions
Recent advances in molecular profiling of GISTs brought significant value to prognostication and therapeutic decisions in clinical practice. New drugs have been approved for advanced GISTs with either the KIT or PDGFRA mutation. Nonetheless, there are significant unmet needs for the subset of patients with KIT/PDGFRA wild-type GISTs.
Author Contributions
Conceptualization, R.D.P. and M.C.M.-M.; writing—original draft preparation, M.C.M.-M., V.H.F.d.J., L.J.d.C.O., M.N. and R.D.P.; writing—review and editing, all authors; supervision, R.D.P.; project administration, R.D.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- The WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours, 5th ed.; IARC Press: Lyon, France, 2020; Volume 3, Available online: https://publications.iarc.fr/Book-And-Report-Series/Who-Classification-Of-Tumours/Soft-Tissue-And-Bone-Tumours-2020 (accessed on 11 August 2022).
- Blay, J.Y.; Kang, Y.K.; Nishida, T.; von Mehren, M. Gastrointestinal stromal tumours. Nat. Rev. Dis. Primers 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Abecassis, N.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; Broto, J.M.; et al. Gastrointestinal stromal tumours: ESMO–EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Miceli, R.; Messerini, L.; Bearzi, I.; Mazzoleni, G.; Capella, C.; Arrigoni, G.; Sonzogni, A.; Sidoni, A.; Toffolatti, L.; et al. Natural history of imatinib-naive GISTs: A retrospective analysis of 929 cases with long-term follow-up and development of a survival nomogram based on mitotic index and size as continuous variables. Am. J. Surg. Pathol. 2011, 35, 1646–1656. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Hirota, S.; Taniguchi, M.; Hashimoto, K.; Isozaki, K.; Nakamura, H.; Kanakura, Y.; Tanaka, T.; Takabayashi, A.; Matsuda, H.; et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat. Genet. 1998, 19, 323–324. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; Von Mehren, M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef]
- Søreide, K.; Sandvik, O.M.; Søreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef]
- Sarlomo-Rikala, M.; Kovatich, A.J.; Barusevicius, A.; Miettinen, M. CD117: A sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod. Pathol. 1998, 11, 728–734. [Google Scholar]
- Espinosa, I.; Lee, C.-H.; Kim, M.K.; Rouse, B.-T.; Subramanian, S.; Montgomery, K.; Varma, S.; Corless, C.L.; Heinrich, M.; Smith, K.S.; et al. A Novel Monoclonal Antibody Against DOG1 is a Sensitive and Specific Marker for Gastrointestinal Stromal Tumors. Am. J. Surg. Pathol. 2008, 32, 210–218. [Google Scholar] [CrossRef]
- Liegl, B.; Hornick, J.L.; Corless, C.L.; Fletcher, C.D.M. Monoclonal Antibody DOG1.1 Shows Higher Sensitivity Than KIT in the Diagnosis of Gastrointestinal Stromal Tumors, Including Unusual Subtypes. Am. J. Surg. Pathol. 2009, 33, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Wang, Z.F.; Lasota, J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: A study of 1840 cases. Am. J. Surg. Pathol. 2009, 33, 1401–1408. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.-J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Brčić, I.; Argyropoulos, A.; Liegl-Atzwanger, B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics 2021, 11, 194. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Von Mehren, M.; Blanke, C.D.; van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Lostes-Bardaji, M.J.; García-Illescas, D.; Valverde, C.; Serrano, C. Ripretinib in gastrointestinal stromal tumor: The long-awaited step forward. Ther. Adv. Med Oncol. 2021, 13, 175883592098649. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstößer, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Kays, J.K.; Sohn, J.D.; Kim, B.J.; Goze, K.; Koniaris, L.G. Approach to wild-type gastrointestinal stromal tumors. Transl. Gastroenterol. Hepatol. 2018, 3, 92. [Google Scholar] [CrossRef]
- Gill, A.J.; Chou, A.; Vilain, R.; Clarkson, A.; Lui, M.; Jin, R.; Tobias, V.; Samra, J.; Goldstein, D.; Smith, C.; et al. Immunohistochemistry for SDHB Divides Gastrointestinal Stromal Tumors (GISTs) into 2 Distinct Types. Am. J. Surg. Pathol. 2010, 34, 636–644. [Google Scholar] [CrossRef]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and Significance of c-KIT Receptor Tyrosine Kinase in Cancer: A Review. Bosn. J. Basic Med Sci. 2022, 22, 683–698. Available online: https://www.bjbms.org/ojs/index.php/bjbms/article/view/7399 (accessed on 11 September 2022). [CrossRef] [PubMed]
- Pathania, S.; Pentikäinen, O.T.; Singh, P.K. A holistic view on c-Kit in cancer: Structure, signaling, pathophysiology and its inhibitors. Biochim. Biophys. Acta (BBA) Rev. Cancer 2021, 1876, 188631. [Google Scholar] [CrossRef] [PubMed]
- Klug, L.R.; Khosroyani, H.M.; Kent, J.D.; Heinrich, M.C. New treatment strategies for advanced-stage gastrointestinal stromal tumours. Nat. Rev. Clin. Oncol. 2022, 19, 328–341. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Sainz, L.G.; Chi, P. The management of metastatic GIST: Current standard and investigational therapeutics. J. Hematol. Oncol. 2021, 14, 2. [Google Scholar] [CrossRef]
- Verschoor, A.J.; Bovée, J.V.M.G.; Overbeek, L.I.H.; Hogendoorn, P.C.W.; Gelderblom, H. The incidence, mutational status, risk classification and referral pattern of gastro-intestinal stromal tumours in the Netherlands: A nationwide pathology registry (PALGA) study. Virchows Arch. 2018, 472, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Kawanowa, K.; Sakuma, Y.; Sakurai, S.; Hishima, T.; Iwasaki, Y.; Saito, K.; Hosoya, Y.; Nakajima, T.; Funata, N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum. Pathol. 2006, 37, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Joensuu, H.; Roberts, P.J.; Sarlomo-Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.L.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the Tyrosine Kinase Inhibitor STI571 in a Patient with a Metastatic Gastrointestinal Stromal Tumor. New Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef]
- Joensuu, H.; Eriksson, M.; Hall, K.S.; Reichardt, A.; Hermes, B.; Schütte, J.; Cameron, S.; Hohenberger, P.; Jost, P.J.; Al-Batran, S.E.; et al. Survival Outcomes Associated With 3 Years vs 1 Year of Adjuvant Imatinib for Patients with High-Risk Gastrointestinal Stromal Tumors: An Analysis of a Randomized Clinical Trial After 10-Year Follow-up. JAMA Oncol. 2020, 6, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Eriksson, M.; Hall, K.S.; Hartmann, J.T.; Pink, D.; Schütte, J.; Ramadori, G.; Hohenberger, P.; Duyster, J.; Al-Batran, S.E.; et al. One vs. Three Years of Adjuvant Imatinib for Operable Gastrointestinal Stromal Tumor: A Randomized Trial. JAMA 2012, 307, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Hall, K.S.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients with Gastrointestinal Stromal Tumors Treated with Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Raut, C.P.; Espat, N.J.; Maki, R.G.; Araujo, D.M.; Trent, J.; Williams, T.F.; Purkayastha, D.D.; DeMatteo, R.P. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients with Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018, 4, e184060. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.-Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of Kinase Genotype and Clinical Outcome in the North American Intergroup Phase III Trial of Imatinib Mesylate for Treatment of Advanced Gastrointestinal Stromal Tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar]
- Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of Two Doses of Imatinib for the Treatment of Unresectable or Metastatic Gastrointestinal Stromal Tumors: A Meta-Analysis of 1640 Patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar] [CrossRef]
- Cassier, P.A.; Fumagalli, E.; Rutkowski, P.; Schöffski, P.; van Glabbeke, M.; Debiec-Rychter, M.; Emile, J.-F.; Duffaud, F.; Martin-Broto, J.; Landi, B.; et al. Outcome of Patients with Platelet-Derived Growth Factor Receptor Alpha–Mutated Gastrointestinal Stromal Tumors in the Tyrosine Kinase Inhibitor Era. Clin. Cancer Res. 2012, 18, 4458–4464. [Google Scholar] [CrossRef]
- Cassier, P.A.; Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Scoazec, J.-Y.; Bringuier, P.-P.; Decouvelaere, A.-V.; Méeus, P.; Cellier, D.; Blay, J.-Y.; et al. A prospective epidemiological study of new incident GISTs during two consecutive years in Rhône Alpes region: Incidence and molecular distribution of GIST in a European region. Br. J. Cancer 2010, 103, 165–170. [Google Scholar] [CrossRef]
- Ravegnini, G.; Urbini, M.; Simeon, V.; Genovese, C.; Astolfi, A.; Nannini, M.; Gatto, L.; Saponara, M.; Ianni, M.; Indio, V.; et al. An exploratory study by DMET array identifies a germline signature associated with imatinib response in gastrointestinal stromal tumor. Pharm. J. 2018, 19, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; Von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S.; Valverde, C.; Olivares, D.; García-Valverde, A.; Suárez, C.; Morales-Barrera, R.; Carles, J. Novel Insights into the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors. Target. Oncol. 2017, 12, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate with the Biological and Clinical Activity of Sunitinib in Imatinib-Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef] [PubMed]
- Nishida, T.; Kanda, T.; Nishitani, A.; Takahashi, T.; Nakajima, K.; Ishikawa, T.; Hirota, S. Secondary mutations in the kinase domain of the KIT gene are predominant in imatinib-resistant gastrointestinal stromal tumor. Cancer Sci. 2008, 99, 799–804. [Google Scholar] [CrossRef]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Vonmehren, M.; Demetri, G.D.; Fletcher, J.A.; Sun, J.; Hodgson, J.G.; Rivera, V.M.; Turner, C.D.; George, S. A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: Initial report. J. Clin. Oncol. 2014, 32 (Suppl. 15), 10506. [Google Scholar] [CrossRef]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.-K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Kang, Y.-K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; Von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Marino-Enriquez, A.; Presnell, A.; Donsky, R.S.; Griffith, D.J.; McKinley, A.; Patterson, J.; Taguchi, T.; Liang, C.-W.; Fletcher, J.A. Sorafenib Inhibits Many Kinase Mutations Associated with Drug-Resistant Gastrointestinal Stromal Tumors. Mol. Cancer Ther. 2012, 11, 1770–1780. [Google Scholar] [CrossRef]
- Reichardt, P.; Blay, J.-Y.; Gelderblom, H.; Schlemmer, M.; Demetri, G.; Bui-Nguyen, B.; McArthur, G.; Yazji, S.; Hsu, Y.; Galetic, I.; et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann. Oncol. 2012, 23, 1680–1687. [Google Scholar] [CrossRef]
- Gardino, A.K.; Evans, E.K.; Kim, J.L.; Brooijmans, N.; Hodous, B.L.; Wolf, B.; Lengauer, C. Targeting kinases with precision. Mol. Cell. Oncol. 2018, 5, e1435183. [Google Scholar] [CrossRef]
- Zalcberg, J.R. Ripretinib for the treatment of advanced gastrointestinal stromal tumor. Ther. Adv. Gastroenterol. 2021, 14, 17562848211008176. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, S.; Klug, L.R.; Mühlenberg, T.; Lategahn, J.; Falkenhorst, J.; Town, A.; Ehrt, C.; Wardelmann, E.; Hartmann, W.; Schildhaus, H.-U.; et al. Resistance to Avapritinib in PDGFRA-Driven GIST Is Caused by Secondary Mutations in the PDGFRA Kinase Domain. Cancer Discov. 2021, 11, 108–125. [Google Scholar] [CrossRef] [PubMed]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, e2365. [Google Scholar] [CrossRef]
- Karajannis, M.A.; Ferner, R.E. Neurofibromatosis-related tumors: Emerging biology and therapies. Curr. Opin. Pediatr. 2015, 27, 26–33. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.M.; De Siena, M.; Alkhuziem, M.; Tang, C.-M.; Medina, B.; Fanta, P.T.; Belinsky, M.G.; von Mehren, M.; Thorson, J.A.; Madlensky, L.; et al. Duodenal-Jejunal Flexure GI Stromal Tumor Frequently Heralds Somatic NF1 and Notch Pathway Mutations. JCO Precis. Oncol. 2017, 1, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gaal, J.; Stratakis, C.A.; Carney, J.A.; Ball, E.R.; Korpershoek, E.; Lodish, M.B.; Levy, I.; Xekouki, P.; van Nederveen, F.H.; Bakker, M.A.D.; et al. SDHB immunohistochemistry: A useful tool in the diagnosis of Carney–Stratakis and Carney triad gastrointestinal stromal tumors. Mod. Pathol. 2011, 24, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Tobo, T.; Nakamori, M.; Imamura, M.; Kojima, A.; Oda, Y.; Nakamura, N.; Takahira, T.; Yao, T.; Tsuneyoshi, M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J. Cancer Res. Clin. Oncol. 2009, 135, 791–798. [Google Scholar] [CrossRef]
- Widemann, B.C.; Marcus, L.J.; Fisher, M.J.; Weiss, B.D.; Kim, A.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Martin, S.; Gillespie, A.; et al. Phase I study of the MEK1/2 inhibitor selumetinib (AZD6244) hydrogen sulfate in children and young adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PNs). JCO 2014, 32 (Suppl. 15), 10018. [Google Scholar] [CrossRef]
- Huss, S.; Pasternack, H.; Ihle, M.A.; Merkelbach-Bruse, S.; Heitkötter, B.; Hartmann, W.; Trautmann, M.; Gevensleben, H.; Büttner, R.; Schildhaus, H.-U.; et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum. Pathol. 2017, 62, 206–214. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Jašek, K.; Váňová, B.; Grendár, M.; Štanclová, A.; Szépe, P.; Hornáková, A.; Holubeková, V.; Plank, L.; Lasabová, Z. BRAF mutations in KIT/PDGFRA positive gastrointestinal stromal tumours (GISTs): Is their frequency underestimated? Pathol. Res. Pract. 2020, 216, 153171. [Google Scholar] [CrossRef]
- Torrence, D.; Xie, Z.; Zhang, L.; Chi, P.; Antonescu, C.R. Gastrointestinal stromal tumors with BRAF gene fusions. A report of two cases showing low or absent KIT expression resulting in diagnostic pitfalls. Genes Chromosom. Cancer 2021, 60, 789–795. [Google Scholar] [CrossRef]
- Maki, R.; Keedy, V. Molecular Profiling of Gastrointestinal Stromal Tumor (GIST). Cancer Genome 2015. Available online: https://www.mycancergenome.org/content/disease/gist/ (accessed on 19 September 2022).
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF mutant gastrointestinal stromal tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Nannini, M.; Valerio, D.S.; Gruppioni, E.; Altimari, A.; Chiusole, B.; Saponara, M.; Pantaleo, M.A.; Brunello, A. Complete radiological response to first-line regorafenib in a patient with abdominal relapse of BRAF V600E mutated GIST. Ther. Adv. Gastroenterol. 2020, 13, 1756284820927305. [Google Scholar] [CrossRef] [PubMed]
- D’Alpino, P.R.; Medeiros, B.A.; Cronemberger, E.H. Resected High-Risk Rectal GIST Harboring NTRK1 Fusion: A Case Report and Review of the Literature. J. Gastrointest. Cancer 2021, 52, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Nathenson, M.; Demetri, G.; Lassen, U.; Hong, D.; Boni, V.; Deeken, J.; Dowlati, A.; Cox, M.; Ku, N.; Cruickshank, S.; et al. Activity of larotrectinib in patients with TRK fusion GI malignancies. Ann. Oncol. 2018, 29, v107. [Google Scholar] [CrossRef]
- Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int. J. Mol. Sci. 2020, 21, 3313. [Google Scholar] [CrossRef] [PubMed]
- Urbini, M.; Indio, V.; Tarantino, G.; Ravegnini, G.; Angelini, S.; Nannini, M.; Saponara, M.; Santini, D.; Ceccarelli, C.; Fiorentino, M.; et al. Gain of FGF4 is a frequent event in KIT/PDGFRA/SDH/RAS-P WT GIST. Genes Chromosom. Cancer 2019, 58, 636–642. [Google Scholar] [CrossRef]
- Flavahan, W.A.; Drier, Y.; Johnstone, S.E.; Hemming, M.L.; Tarjan, D.R.; Hegazi, E.; Shareef, S.J.; Javed, N.M.; Raut, C.P.; Eschle, B.K.; et al. Altered chromosomal topology drives oncogenic programs in SDH-deficient GISTs. Nature 2019, 575, 229–233. [Google Scholar] [CrossRef]
- Joensuu, H.; Blay, J.-Y.; Comandone, A.; Martin-Broto, J.; Fumagalli, E.; Grignani, G.; Del Muro, X.G.; Adenis, A.; Valverde, C.; Pousa, A.L.; et al. Dovitinib in patients with gastrointestinal stromal tumour refractory and/or intolerant to imatinib. Br. J. Cancer 2017, 117, 1278–1285. [Google Scholar] [CrossRef]
- Soria, J.; Massard, C.; Magné, N.; Bader, T.; Mansfield, C.; Blay, J.; Bui, B.; Moussy, A.; Hermine, O.; Armand, J. Phase 1 dose-escalation study of oral tyrosine kinase inhibitor masitinib in advanced and/or metastatic solid cancers. Eur. J. Cancer 2009, 45, 2333–2341. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.Y.; Bui, B.N.; Bouché, O.; Adenis, A.; Domont, J.; Cioffi, A.; Ray-Coquard, I.; Lassau, N.; Bonvalot, S.; et al. Phase II study of oral masitinib mesilate in imatinib-naïve patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur. J. Cancer 2010, 46, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Adenis, A.; Blay, J.-Y.; Bui-Nguyen, B.; Bouché, O.; Bertucci, F.; Isambert, N.; Bompas, E.; Chaigneau, L.; Domont, J.; Ray-Coquard, I.; et al. Masitinib in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib: A randomized controlled open-label trial. Ann. Oncol. 2014, 25, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Garner, A.P.; Gozgit, J.M.; Anjum, R.; Vodala, S.; Schrock, A.; Zhou, T.; Serrano, C.; Eilers, G.; Zhu, M.; Ketzer, J.; et al. Ponatinib Inhibits Polyclonal Drug-Resistant KIT Oncoproteins and Shows Therapeutic Potential in Heavily Pretreated Gastrointestinal Stromal Tumor (GIST) Patients. Clin. Cancer Res. 2014, 20, 5745–5755. [Google Scholar] [CrossRef] [PubMed]
- Ganjoo, K.N.; Villalobos, V.M.; Kamaya, A.; Fisher, G.A.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; D’Adamo, D.; McMillan, A.; Demetri, G.D.; et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann. Oncol. 2014, 25, 236–240. [Google Scholar] [CrossRef]
- Mir, O.; Cropet, C.; Toulmonde, M.; Le Cesne, A.; Molimard, M.; Bompas, E.; Cassier, P.; Ray-Coquard, I.; Rios, M.; Adenis, A.; et al. Pazopanib Plus Best Supportive Care Versus Best Supportive Care Alone in Advanced Gastrointestinal Stromal Tumours Resistant to Imatinib and Sunitinib (PAZOGIST): A Randomised, Multicentre, Open-Label Phase 2 Trial. Lancet Oncol. 2016, 17, 632–641. Available online: https://pubmed.ncbi.nlm.nih.gov/27068858/ (accessed on 25 October 2022). [CrossRef]
- 87. Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 1769–1776. [Google Scholar] [CrossRef]
- 88. Lasota, J.; Felisiak-Golabek, A.; Wasag, B.; Kowalik, A.; Zięba, S.; Chłopek, M.; Wang, Z.F.; Coates, T.; Kopczynski, J.; Gozdz, S.; et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: Molecular genetic study of 529 cases. Mod. Pathol. 2016, 29, 275–282. [Google Scholar] [CrossRef]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric KIT–Wild-Type and Platelet-Derived Growth Factor Receptor α–Wild-Type Gastrointestinal Stromal Tumors Share KIT Activation but not Mechanisms of Genetic Progression with Adult Gastrointestinal Stromal Tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef]
- Pappo, A.S.; Janeway, K.A. Pediatric Gastrointestinal Stromal Tumors. Hematol. Clin. N. Am. 2009, 23, 15–34. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-Function Mutations of c- kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; Chopra, S. Succinate Dehydrogenase–Deficient Gastrointestinal Stromal Tumors. Arch. Pathol. Lab. Med. 2020, 144, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Killian, J.K.; Wang, Z.-F.; Lasota, J.; Lau, C.; Jones, L.E.; Walker, R.; Pineda, M.; Zhu, Y.J.; Kim, S.Y.; et al. Immunohistochemical Loss of Succinate Dehydrogenase Subunit A (SDHA) in Gastrointestinal Stromal Tumors (GISTs) Signals SDHA Germline Mutation. Am. J. Surg. Pathol. 2013, 37, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney–Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Pasini, B.; McWhinney, S.R.; Bei, T.; Matyakhina, L.; Stergiopoulos, S.; Muchow, M.; Boikos, S.A.; Ferrando, B.; Pacak, K.; Assie, G.; et al. Clinical and molecular genetics of patients with the Carney–Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur. J. Hum. Genet. 2008, 16, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Haller, F.; Moskalev, E.A.; Faucz, F.R.; Barthelmeß, S.; Wiemann, S.; Bieg, M.; Assie, G.; Bertherat, J.; Schaefer, I.M.; Otto, C.; et al. Aberrant DNA hypermethylation of SDHC: A novel mechanism of tumor development in Carney triad. Endocr. Relat. Cancer 2014, 21, 567–577. [Google Scholar] [CrossRef]
- Matyakhina, L.; Bei, T.A.; McWhinney, S.R.; Pasini, B.; Cameron, S.; Gunawan, B.; Stergiopoulos, S.G.; Boikos, S.; Muchow, M.; Dutra, A.; et al. Genetics of carney triad: Recurrent losses at chromosome 1 but lack of germline mutations in genes associated with paragangliomas and gastrointestinal stromal tumors. J. Clin. Endocrinol. Metab. 2007, 92, 2938–2943. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Gastrointestinal Stromal Tumors (GISTs). 2022. Available online: https://www.nccn.org/professionals/physician_gls/pdf/gist.pdf (accessed on 4 September 2022).
- Weldon, C.B.; Madenci, A.L.; Boikos, S.A.; Janeway, K.A.; George, S.; von Mehren, M.; Pappo, A.S.; Schiffman, J.D.; Wright, J.; Trent, J.C.; et al. Surgical Management of Wild-Type Gastrointestinal Stromal Tumors: A Report from the National Institutes of Health Pediatric and Wildtype GIST Clinic. J. Clin. Oncol. 2017, 35, 523–528. [Google Scholar] [CrossRef]
- Corless, C.L.; Ballman, K.V.; Antonescu, C.R.; Kolesnikova, V.; Maki, R.G.; Pisters, P.W.T.; Blackstein, M.E.; Blanke, C.D.; Demetri, G.D.; Heinrich, M.C.; et al. Pathologic and Molecular Features Correlate with Long-Term Outcome After Adjuvant Therapy of Resected Primary GI Stromal Tumor: The ACOSOG Z9001 Trial. JCO 2014, 32, 1563–1570. [Google Scholar] [CrossRef]
- Janeway, K.A.; Albritton, K.H.; van den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatric Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Ben-Ami, E.; Barysauskas, C.M.; von Mehren, M.; Heinrich, M.C.; Corless, C.L.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; Choy, E.; Yap, J.T.; et al. Long-term follow-up results of the multicenter phase II trial of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of standard tyrosine kinase inhibitor therapy. Ann. Oncol. 2016, 27, 1794–1799. [Google Scholar] [CrossRef] [PubMed]
- von Mehren, M.; George, S.; Heinrich, M.C.; Schuetze, S.M.; Yap, J.T.; Yu, J.Q.; Abbott, A.; Litwin, S.; Crowley, J.; Belinsky, M.; et al. Linsitinib (OSI-906) for the Treatment of Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors, a SARC Phase II Study. Clin. Cancer Res. 2020, 26, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Yebra, M.; Bhargava, S.; Kumar, A.; Burgoyne, A.M.; Tang, C.M.; Yoon, H.; Banerjee, S.; Aguilera, J.; Cordes, T.; Sheth, V.; et al. Establishment of Patient-Derived Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumor Models for Predicting Therapeutic Response. Clin. Cancer Res. 2022, 28, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Jasek, K.; Kasubova, I.; Holubekova, V.; Stanclova, A.; Plank, L.; Lasabova, Z. Epigenetics: An alternative pathway in GISTs tumorigenesis. Neoplasma 2018, 65, 477–493. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).