
DNA Repair Mechanisms, Protein Interactions and Therapeutic Targeting of the MRN Complex


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
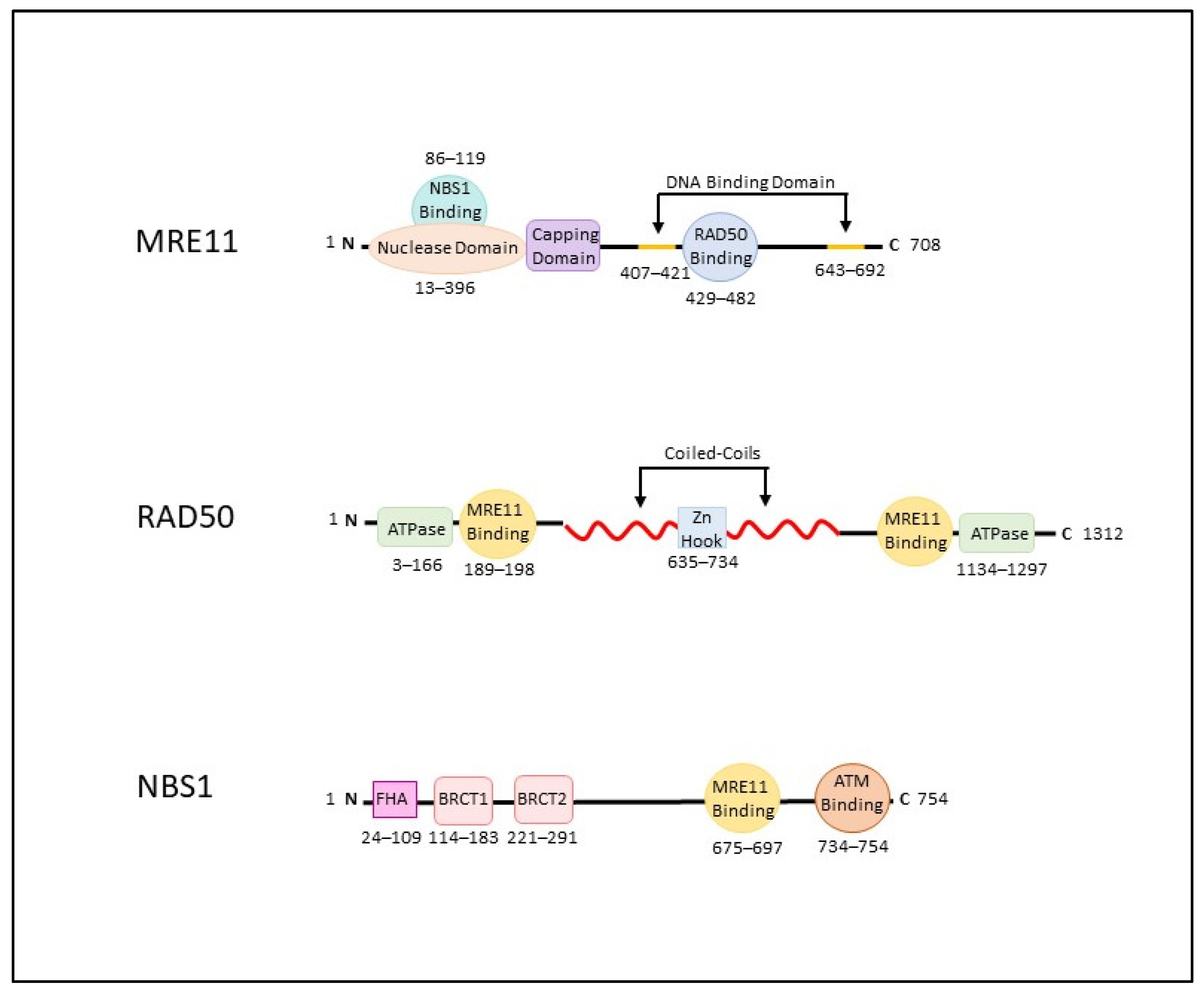
2. The MRN Complex (MRE11-RAD50-NBS1)
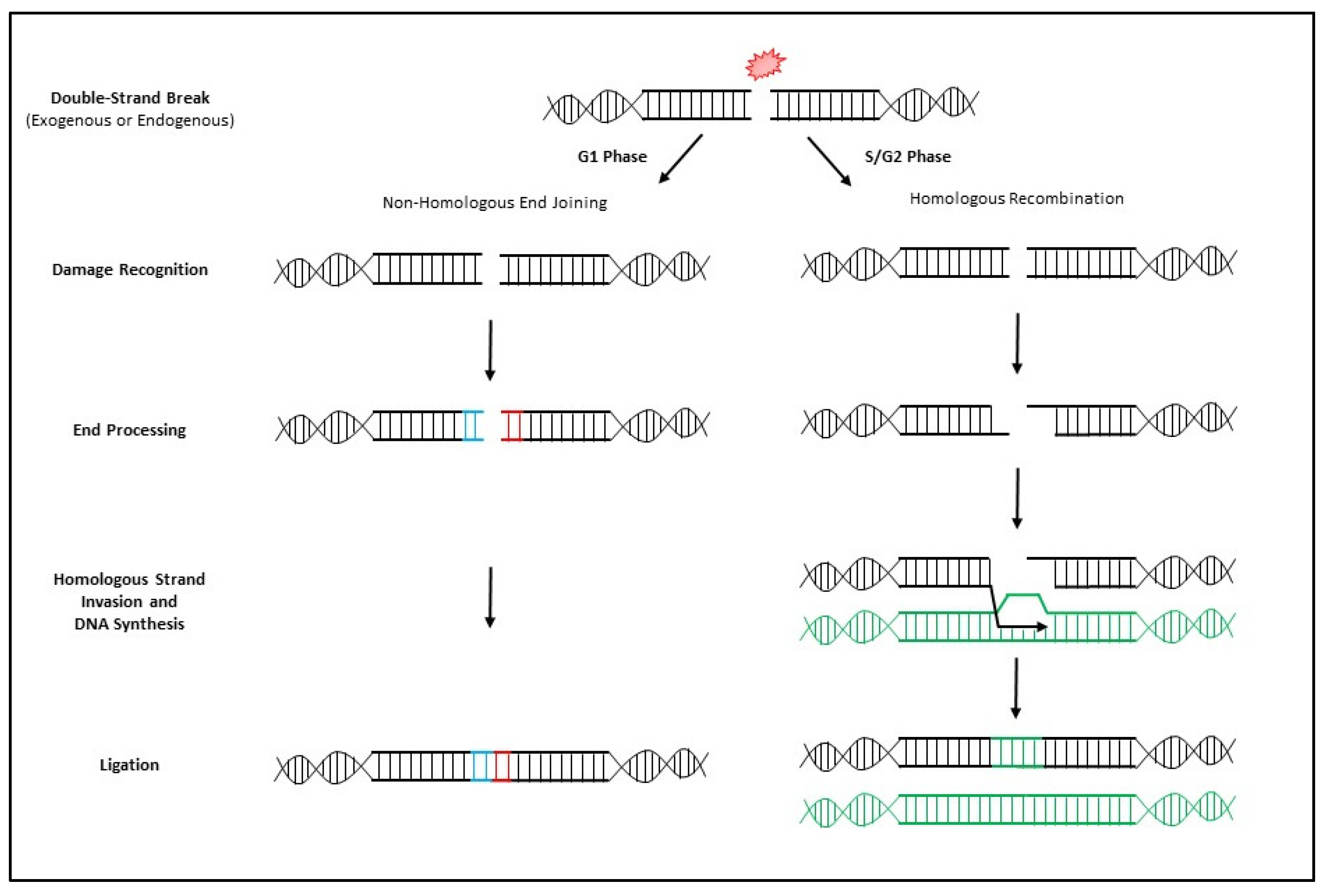
3. MRN Complex and Initiation of DNA Double-Strand Break Repair
4. Mutations within the MRN Complex and Human Disease
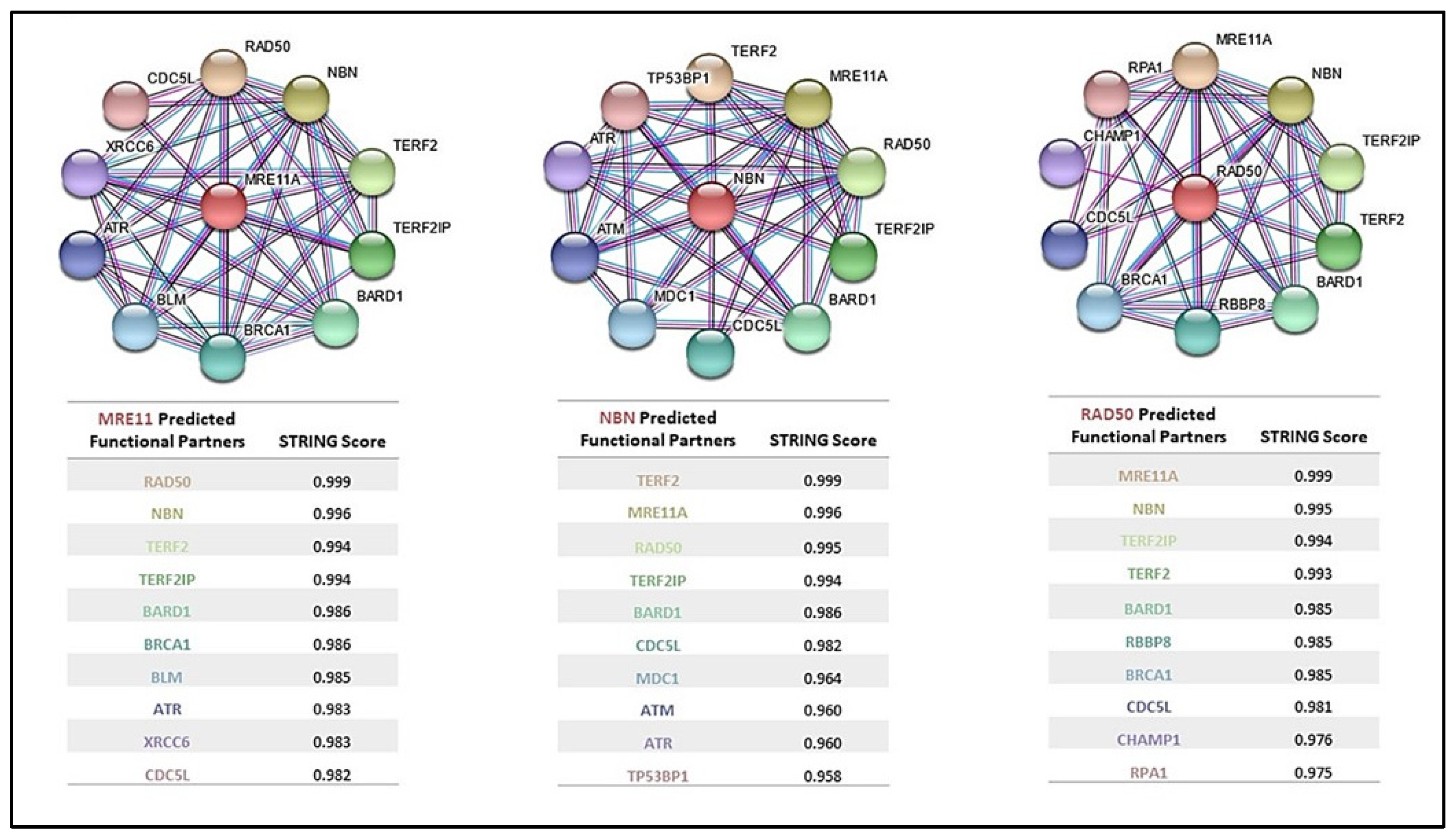
5. MRN Complex Interaction with Cancer-Predisposing Genes
6. Assessing Mutation Impact within the MRN Complex Using Functional Studies
7. MRN Targeted Therapies and Synthetic Lethality in Cancer Treatment
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA Damage, Repair, and Mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.K. DNA Damage, Mutagenesis and Cancer. Int. J. Mol. Sci. 2018, 19, 970. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-Damage Response in Human Biology and Disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA Damage and the Balance between Survival and Death in Cancer Biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- De Bont, R.; van Larebeke, N. Endogenous DNA Damage in Humans: A Review of Quantitative Data. Mutagenesis 2004, 19, 169–185. [Google Scholar] [CrossRef]
- Liu, P.; Carvalho, C.M.B.; Hastings, P.J.; Lupski, J.R. Mechanisms for Recurrent and Complex Human Genomic Rearrangements. Curr. Opin. Genet. Dev. 2012, 22, 211–220. [Google Scholar] [CrossRef]
- Casari, E.; Rinaldi, C.; Marsella, A.; Gnugnoli, M.; Colombo, C.V.; Bonetti, D.; Longhese, M.P. Processing of DNA Double-Strand Breaks by the MRX Complex in a Chromatin Context. Front. Mol. Biosci. 2019, 6, 43. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Lamarche, B.J.; Orazio, N.I.; Weitzman, M.D. The MRN Complex in Double-Strand Break Repair and Telomere Maintenance. FEBS Lett. 2010, 584, 3682–3695. [Google Scholar] [CrossRef]
- Salifou, K.; Burnard, C.; Basavarajaiah, P.; Grasso, G.; Helsmoortel, M.; Mac, V.; Depierre, D.; Franckhauser, C.; Beyne, E.; Contreras, X.; et al. Chromatin-Associated MRN Complex Protects Highly Transcribing Genes from Genomic Instability. Sci. Adv. 2021, 7, eabb2947. [Google Scholar] [CrossRef]
- Desmond, A.; Kurian, A.W.; Gabree, M.; Mills, M.A.; Anderson, M.J.; Kobayashi, Y.; Horick, N.; Yang, S.; Shannon, K.M.; Tung, N.; et al. Clinical Actionability of Multigene Panel Testing for Hereditary Breast and Ovarian Cancer Risk Assessment. JAMA Oncol. 2015, 1, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Tsaousis, G.N.; Papadopoulou, E.; Apessos, A.; Agiannitopoulos, K.; Pepe, G.; Kampouri, S.; Diamantopoulos, N.; Floros, T.; Iosifidou, R.; Katopodi, O.; et al. Analysis of Hereditary Cancer Syndromes by Using a Panel of Genes: Novel and Multiple Pathogenic Mutations. BMC Cancer 2019, 19, 535. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Kim, J.; Han, W. Multigene Panel Testing for Hereditary Cancer and Genetic Counseling. Adv. Exp. Med. Biol. 2021, 1187, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Graffeo, R.; Livraghi, L.; Pagani, O.; Goldhirsch, A.; Partridge, A.H.; Garber, J.E. Time to Incorporate Germline Multigene Panel Testing into Breast and Ovarian Cancer Patient Care. Breast Cancer Res. Treat. 2016, 160, 393–410. [Google Scholar] [CrossRef]
- Berkel, C.; Cacan, E. Involvement of ATMIN-DYNLL1-MRN Axis in the Progression and Aggressiveness of Serous Ovarian Cancer. Biochem. Biophys. Res. Commun. 2021, 570, 74–81. [Google Scholar] [CrossRef]
- Alblihy, A.; Shoqafi, A.; Toss, M.S.; Algethami, M.; Harris, A.E.; Jeyapalan, J.N.; Abdel-Fatah, T.; Servante, J.; Chan, S.Y.T.; Green, A.; et al. Untangling the Clinicopathological Significance of MRE11-RAD50-NBS1 Complex in Sporadic Breast Cancers. NPJ Breast Cancer 2021, 7, 14. [Google Scholar] [CrossRef]
- Chang, J.; Seng, S.; Yoo, J.; Equivel, P.; Lum, S.S. Clinical Management of Patients at Risk for Hereditary Breast Cancer with Variants of Uncertain Significance in the Era of Multigene Panel Testing. Ann. Surg. Oncol. 2019, 26, 3389–3396. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Eccles, B.K.; Copson, E.; Maishman, T.; Abraham, J.E.; Eccles, D.M. Understanding of BRCA VUS Genetic Results by Breast Cancer Specialists. BMC Cancer 2015, 15, 936. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Bai, Y.; Wang, W.; Li, S.; Zhan, J.; Li, H.; Zhao, M.; Zhou, X.A.; Li, S.; Li, X.; Huo, Y.; et al. C1QBP Promotes Homologous Recombination by Stabilizing MRE11 and Controlling the Assembly and Activation of MRE11/RAD50/NBS1 Complex. Mol. Cell 2019, 75, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Tankimanova, M.; Capper, R.; Letsolo, B.T.; Rowson, J.; Jones, R.E.; Britt-Compton, B.; Taylor, A.M.R.; Baird, D.M. Mre11 Modulates the Fidelity of Fusion between Short Telomeres in Human Cells. Nucleic Acids Res. 2012, 40, 2518. [Google Scholar] [CrossRef]
- Chen, L.; Nievera, C.J.; Lee, A.Y.L.; Wu, X. Cell Cycle-Dependent Complex Formation of BRCA1.CtIP.MRN Is Important for DNA Double-Strand Break Repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, X.; Ferguson, D.O.; Chang, S. Multiple Roles for MRE11 at Uncapped Telomeres. Nature 2009, 460, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Quennet, V.; Beucher, A.; Barton, O.; Takeda, S.; Löbrich, M. CtIP and MRN Promote Non-Homologous End-Joining of Etoposide-Induced DNA Double-Strand Breaks in G1. Nucleic Acids Res. 2011, 39, 2144–2152. [Google Scholar] [CrossRef]
- Buis, J.; Wu, Y.; Deng, Y.; Leddon, J.; Westfield, G.; Eckersdorff, M.; Sekiguchi, J.A.M.; Chang, S.; Ferguson, D.O. Mre11 Nuclease Activity Has Essential Roles in DNA Repair and Genomic Stability Distinct from ATM Activation. Cell 2008, 135, 85. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Lees-Miller, S.P.; Tainer, J.A. Mre11-Rad50-Nbs1 Conformations and the Control of Sensing, Signaling, and Effector Responses at DNA Double-Strand Breaks. DNA Repair 2010, 9, 1299–1306. [Google Scholar] [CrossRef]
- Damiola, F.; Pertesi, M.; Oliver, J.; Le Calvez-Kelm, F.; Voegele, C.; Young, E.L.; Robinot, N.; Forey, N.; Durand, G.; Vallée, M.P.; et al. Rare Key Functional Domain Missense Substitutions in MRE11A, RAD50, and NBN Contribute to Breast Cancer Susceptibility: Results from a Breast Cancer Family Registry Case-Control Mutation-Screening Study. Breast Cancer Res. 2014, 16, R58. [Google Scholar] [CrossRef]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal Structure of Human Mre11: Understanding Tumorigenic Mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Karcher, A.; Craig, L.; Woo, T.T.; Carney, J.P.; Tainer, J.A. Structural Biochemistry and Interaction Architecture of the DNA Double-Strand Break Repair Mre11 Nuclease and Rad50-ATPase. Cell 2001, 105, 473–485. [Google Scholar] [CrossRef]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Neale, M.J.; Pan, J.; Keeney, S. Endonucleolytic Processing of Covalent Protein-Linked DNA Double-Strand Breaks. Nature 2005, 436, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gottesman, M.; Gautier, J. MRN, CtIP, and BRCA1 Mediate Repair of Topoisomerase II-DNA Adducts. J. Cell Biol. 2016, 212, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Milman, N.; Higuchi, E.; Smith, G.R. Meiotic DNA Double-Strand Break Repair Requires Two Nucleases, MRN and Ctp1, to Produce a Single Size Class of Rec12 (Spo11)-Oligonucleotide Complexes. Mol. Cell. Biol. 2009, 29, 5998–6005. [Google Scholar] [CrossRef] [PubMed]
- Hartsuiker, E.; Neale, M.J.; Carr, A.M. Distinct Requirements for the Rad32(Mre11) Nuclease and Ctp1(CtIP) in the Removal of Covalently Bound Topoisomerase I and II from DNA. Mol. Cell 2009, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Stracker, T.H.; Petrini, J.H.J. The MRE11 Complex: Starting from the Ends. Nat. Rev. Mol. Cell Biol. 2011, 12, 90–103. [Google Scholar] [CrossRef]
- Hopfner, K.P.; Karcher, A.; Shin, D.S.; Craig, L.; Arthur, L.M.; Carney, J.P.; Tainer, J.A. Structural Biology of Rad50 ATPase: ATP-Driven Conformational Control in DNA Double-Strand Break Repair and the ABC-ATPase Superfamily. Cell 2000, 101, 789–800. [Google Scholar] [CrossRef]
- Hohl, M.; Kochańczyk, T.; Tous, C.; Aguilera, A.; KrEzel, A.; Petrini, J.H.J. Interdependence of the Rad50 Hook and Globular Domain Functions. Mol. Cell 2015, 57, 479–491. [Google Scholar] [CrossRef]
- Hohl, M.; Kwon, Y.; Galván, S.M.; Xue, X.; Tous, C.; Aguilera, A.; Sung, P.; Petrini, J.H.J. The Rad50 Coiled-Coil Domain Is Indispensable for Mre11 Complex Functions. Nat. Struct. Mol. Biol. 2010, 18, 1124–1131. [Google Scholar] [CrossRef]
- Käshammer, L.; Saathoff, J.H.; Lammens, K.; Gut, F.; Bartho, J.; Alt, A.; Kessler, B.; Hopfner, K.P. Mechanism of DNA End Sensing and Processing by the Mre11-Rad50 Complex. Mol. Cell 2019, 76, 382–394. [Google Scholar] [CrossRef]
- Paull, T.T.; Gellert, M. Nbs1 Potentiates ATP-Driven DNA Unwinding and Endonuclease Cleavage by the Mre11/Rad50 Complex. Genes Dev. 1999, 13, 1276–1288. [Google Scholar] [CrossRef]
- Komatsu, K. NBS1 and Multiple Regulations of DNA Damage Response. J. Radiat.Res. 2016, 57, i11–i17. [Google Scholar] [CrossRef] [PubMed]
- Cilli, D.; Mirasole, C.; Pennisi, R.; Pallotta, V.; D’Alessandro, A.; Antoccia, A.; Zolla, L.; Ascenzi, P.; Di Masi, A. Identification of the Interactors of Human Nibrin (NBN) and of Its 26 KDa and 70 KDa Fragments Arising from the NBN 657del5 Founder Mutation. PLoS ONE 2014, 9, e0114651. [Google Scholar] [CrossRef] [PubMed]
- Carney, J.P.; Maser, R.S.; Olivares, H.; Davis, E.M.; Le Beau, M.; Yates, J.R.; Hays, L.; Morgan, W.F.; Petrini, J.H.J. The HMre11/HRad50 Protein Complex and Nijmegen Breakage Syndrome: Linkage of Double-Strand Break Repair to the Cellular DNA Damage Response. Cell 1998, 93, 477–486. [Google Scholar] [CrossRef]
- Anand, R.; Jasrotia, A.; Bundschuh, D.; Howard, S.M.; Ranjha, L.; Stucki, M.; Cejka, P. NBS1 Promotes the Endonuclease Activity of the MRE11-RAD50 Complex by Sensing CtIP Phosphorylation. EMBO J. 2019, 38, e101005. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Chahwan, C.; Bailis, J.; Hunter, T.; Russell, P. ATM Activation and Its Recruitment to Damaged DNA Require Binding to the C Terminus of Nbs1. Mol. Cell. Biol. 2005, 25, 5363–5379. [Google Scholar] [CrossRef]
- Lee, J.; Paull, T. Direct Activation of the ATM Protein Kinase by the Mre11/Rad50/Nbs1 Complex. Science 2004, 304, 93–96. [Google Scholar] [CrossRef]
- Lavin, M.F.; Kozlov, S.; Gatei, M.; Kijas, A.W. ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 2015, 5, 2877–2902. [Google Scholar] [CrossRef]
- Kurose, A.; Tanaka, T.; Huang, X.; Halicka, H.D.; Traganos, F.; Dai, W.; Darzynkiewicz, Z. Assessment of ATM Phosphorylation on Ser-1981 Induced by DNA Topoisomerase I and II Inhibitors in Relation to Ser-139-Histone H2AX Phosphorylation, Cell Cycle Phase, and Apoptosis. Cytom. Part A 2005, 68, 1–9. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; van Vugt, M.A.T.M.; Jørgensen, C.; Miron, I.M.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic Discovery of In Vivo Phosphorylation Networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Tosti, E.; Waldbaum, L.; Warshaw, G.; Gross, E.A.; Ruggieri, R. The Stress Kinase MRK Contributes to Regulation of DNA Damage Checkpoints through a P38γ-Independent Pathway. J. Biol. Chem. 2004, 279, 47652–47660. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.; Mun Tho, L.; Xu, N.; A. Gillespie, D. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. In Advances in Cancer Research; Academic Press Inc.: Cambridge, MA, USA, 2010; Volume 108, pp. 73–112. [Google Scholar]
- Leung-Pineda, V.; Ryan, C.E.; Piwnica-Worms, H. Phosphorylation of Chk1 by ATR Is Antagonized by a Chk1-Regulated Protein Phosphatase 2A Circuit. Mol. Cell. Biol. 2006, 26, 7529–7538. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline Mutations in RAD51D Confer Susceptibility to Ovarian Cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Gutiérrez-Enríquez, S.; Bonache, S.; Ruíz De Garibay, G.; Osorio, A.; Santamariña, M.; Ramõn, Y. Cajal, T.; Esteban-Cardeñosa, E.; Tenés, A.; Yanowsky, K.; et al. About 1% of the Breast and Ovarian Spanish Families Testing Negative for BRCA1 and BRCA2 Are Carriers of RAD51D Pathogenic Variants. Int. J. Cancer 2014, 134, 2088–2097. [Google Scholar] [CrossRef]
- Zhao, S.; Wong, Y.C.; Yuan, S.S.F.; Lin, Y.T.; Hsu, H.C.; Lin, S.C.J.; Gerbino, E.; Song, M.H.; Zdzlenlcka, M.Z.; Gatti, R.A.; et al. Functional Link between Ataxia-Telangiectasia and Nijmegen Breakage Syndrome Gene Products. Nature 2000, 405, 473–477. [Google Scholar] [CrossRef]
- Gatei, M.; Kijas, A.W.; Biard, D.; Dörk, T.; Lavin, M.F. RAD50 Phosphorylation Promotes ATR Downstream Signaling and DNA Restart Following Replication Stress. Hum. Mol. Genet. 2014, 23, 4232–4248. [Google Scholar] [CrossRef]
- Lee, J.H.; Mand, M.R.; Deshpande, R.A.; Kinoshita, E.; Yang, S.H.; Wyman, C.; Paull, T.T. Ataxia Telangiectasia-Mutated (ATM) Kinase Activity Is Regulated by ATP-Driven Conformational Changes in the Mre11/Rad50/Nbs1 (MRN) Complex. J. Biol. Chem. 2013, 288, 12840–12851. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Kawamura, K.; Yanagihara, H.; Kobayashi, J.; Zhang-Akiyama, Q.M. NBS1 Is Regulated by Two Kind of Mechanisms: ATM-Dependent Complex Formation with MRE11 and RAD50, and Cell Cycle-Dependent Degradation of Protein. J. Radiat. Res. 2017, 58, 487–494. [Google Scholar] [CrossRef]
- Park, S.; Kang, J.M.; Kim, S.J.; Kim, H.; Hong, S.; Lee, Y.J.; Kim, S.J. Smad7 Enhances ATM Activity by Facilitating the Interaction between ATM and Mre11-Rad50-Nbs1 Complex in DNA Double-Strand Break Repair. Cell. Mol. Life Sci. 2015, 72, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chehab, N.; Pavletich, N. Structure and Activation Mechanism of the CHK2 DNA Damage Checkpoint Kinase. Mol. Cell 2009, 35, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Mustofa, M.K.; Tanoue, Y.; Tateishi, C.; Vaziri, C.; Tateishi, S. Roles of Chk2/CHEK2 in Guarding against Environmentally Induced DNA Damage and Replication-Stress. Environ. Mol. Mutagen. 2020, 61, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Menolfi, D.; Zha, S. ATM, ATR and DNA-PKcs Kinases-the Lessons from the Mouse Models: Inhibition = Deletion. Cell Biosci. 2020, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Saldivar, J.C.; Cortez, D.; Cimprich, K.A. The Essential Kinase ATR: Ensuring Faithful Duplication of a Challenging Genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Fernandez-Capetillo, O.; Chen, H.T.; Celeste, A.; Ward, I.; Romanienko, P.J.; Morales, J.C.; Naka, K.; Xia, Z.; Camerini-Otero, R.D.; Motoyama, N.; et al. DNA Damage-Induced G2-M Checkpoint Activation by Histone H2AX and 53BP1. Nat. Cell Biol. 2002, 4, 993–997. [Google Scholar] [CrossRef]
- Allen, B.; Pezone, A.; Porcellini, A.; Muller, M.T.; Masternak, M.M. Non-Homologous End Joining Induced Alterations in DNA Methylation: A Source of Permanent Epigenetic Change. Oncotarget 2017, 8, 40359–40372. [Google Scholar] [CrossRef]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in Chromosomal Nonhomologous End Joining in Mammalian Cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef]
- Haber, J.E. DNA Repair: The Search for Homology. BioEssays 2018, 40, e1700229. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Amirifar, P.; Ranjouri, M.; Yazdani, R.; Abolhassani, H.; Aghamohammadi, A. Ataxia-Telangiectasia: A Review of Clinical Features and Molecular Pathology. Pediatr. Allergy Immunol. 2019, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.; Lang, A.E. Ataxia-Telangiectasia: A Review of Movement Disorders, Clinical Features, and Genotype Correlations. Mov. Disord. 2018, 33, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Duedal, S.; Kirner, J.; McGuffog, L.; Last, J.; Reiman, A.; Byrd, P.; Taylor, M.; Easton, D.F. Cancer Risks and Mortality in Heterozygous ATM Mutation Carriers. J. Natl. Cancer Inst. 2005, 97, 813–822. [Google Scholar] [CrossRef]
- Roeb, W.; Higgins, J.; King, M.C. Response to DNA Damage of CHEK2 Missense Mutations in Familial Breast Cancer. Hum. Mol. Genet. 2012, 21, 2738–2744. [Google Scholar] [CrossRef]
- Ceppi, I.; Howard, S.M.; Kasaciunaite, K.; Pinto, C.; Anand, R.; Seidel, R.; Cejka, P. CtIP Promotes the Motor Activity of DNA2 to Accelerate Long-Range DNA End Resection. Proc. Natl. Acad. Sci. USA 2020, 117, 8859–8869. [Google Scholar] [CrossRef]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-Factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef]
- Deshpande, R.A.; Myler, L.R.; Soniat, M.M.; Makharashvili, N.; Lee, L.; Lees-Miller, S.P.; Finkelstein, I.J.; Paull, T.T. DNA-Dependent Protein Kinase Promotes DNA End Processing by MRN and CtIP. Sci. Adv. 2020, 6, eaay0922. [Google Scholar] [CrossRef]
- Feng, L.; Fong, K.W.; Wang, J.; Wang, W.; Chen, J. RIF1 Counteracts BRCA1-Mediated End Resection during DNA Repair. J. Biol. Chem. 2013, 288, 11135–11143. [Google Scholar] [CrossRef]
- Feng, L.; Li, N.; Li, Y.; Wang, J.; Gao, M.; Wang, W.; Chen, J. Cell Cycle-Dependent Inhibition of 53BP1 Signaling by BRCA1. Cell Discov. 2015, 1, 15019. [Google Scholar] [CrossRef] [PubMed]
- Escribano-Díaz, C.; Orthwein, A.; Fradet-Turcotte, A.; Xing, M.; Young, J.T.F.; Tkáč, J.; Cook, M.A.; Rosebrock, A.P.; Munro, M.; Canny, M.D.; et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol. Cell 2013, 49, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.K.; Longo, D.L. PALB2 Mutations and Breast-Cancer Risk. N. Engl. J. Med. 2014, 371, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 Loss Rescues BRCA1 Deficiency and Is Associated with Triple-Negative and BRCA-Mutated Breast Cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Easton, D.F.; Ford, D.; Bishop, D.T. Breast and Ovarian Cancer Incidence in BRCA1-Mutation Carriers. Am. J. Hum. Genet. 1995, 56, 265–271. [Google Scholar]
- Antoniou, A.C.; Casadei, S.; Heikkinen, T.; Barrowdale, D.; Pylkäs, K.; Roberts, J.; Lee, A.; Subramanian, D.; De Leeneer, K.; Fostira, F.; et al. Breast-Cancer Risk in Families with Mutations in PALB2. N. Engl. J. Med. 2014, 371, 497–506. [Google Scholar] [CrossRef]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated with Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.J.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Gachechiladze, M.; Škarda, J.; Soltermann, A.; Joerger, M. RAD51 as a Potential Surrogate Marker for DNA Repair Capacity in Solid Malignancies. Int. J. Cancer 2017, 141, 1286–1294. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting Amino Acid Changes That Affect Protein Function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, A.; Bellanger, D.; Valence, S.; Mobuchon, L.; Afenjar, A.; Giuliano, F.; Dubois d’Enghien, C.; Parfait, B.; Pedespan, J.; Auger, N.; et al. Three New Cases of Ataxia-Telangiectasia-like Disorder: No Impairment of the ATM Pathway, but S-Phase Checkpoint Defect. Hum. Mutat. 2019, 40, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Maser, R.; Zinkel, R.; Petrini, J. An Alternative Mode of Translation Permits Production of a Variant NBS1 Protein from the Common Nijmegen Breakage Syndrome Allele. Nat. Genet. 2001, 27, 417–421. [Google Scholar] [CrossRef]
- Seemanova, E.; Varon, R.; Vejvalka, J.; Jarolim, P.; Seeman, P.; Chrzanowska, K.H.; Digweed, M.; Resnick, I.; Kremensky, I.; Saar, K.; et al. The Slavic NBN Founder Mutation: A Role for Reproductive Fitness? PLoS ONE 2016, 11, e0167984. [Google Scholar] [CrossRef]
- Varon, R.; Vissinga, C.; Platzer, M.; Cerosaletti, K.; Chrzanowska, K.; Saar, K.; Beckmann, G.; Seemanová, E.; Cooper, P.; Nowak, N.; et al. Nibrin, a Novel DNA Double-Strand Break Repair Protein, Is Mutated in Nijmegen Breakage Syndrome. Cell 1998, 93, 467–476. [Google Scholar] [CrossRef]
- Barbi, G.; Scheres, J.M.J.C.; Schindler, D.; Taalman, R.D.F.M.; Rodens, K.; Mehnert, K.; Müller, M.; Seyschab, H. Chromosome Instability and X-Ray Hypersensitivity in a Microcephalic and Growth-Retarded Child. Am. J. Med. Genet. 1991, 40, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Waltes, R.; Kalb, R.; Gatei, M.; Kijas, A.W.; Stumm, M.; Sobeck, A.; Wieland, B.; Varon, R.; Lerenthal, Y.; Lavin, M.F.; et al. Human RAD50 Deficiency in a Nijmegen Breakage Syndrome-like Disorder. Am. J. Hum. Genet. 2009, 84, 605. [Google Scholar] [CrossRef] [PubMed]
- Ragamin, A.; Yigit, G.; Bousset, K.; Beleggia, F.; Verheijen, F.; de Wit, M.; Strom, T.; Dörk, T.; Wollnik, B.; Mancini, G. Human RAD50 Deficiency: Confirmation of a Distinctive Phenotype. Am. J. Med. Genet. A 2020, 182, 1378–1386. [Google Scholar] [CrossRef]
- Swift, M.; Morrell, D.; Massey, R.B.; Chase, C.L. Incidence of Cancer in 161 Families Affected by Ataxia–Telangiectasia. N. Engl. J. Med. 1991, 325, 1831–1836. [Google Scholar] [CrossRef]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.; Smith, S.; Uziel, T.; Sfez, S.; et al. A Single Ataxia Telangiectasia Gene with a Product Similar to PI-3 Kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef]
- Gatti, R.; Berkel, I.; Boder, E.; Braedt, G.; Charmley, P.; Concannon, P.; Ersoy, F.; Foroud, T.; Jaspers, N.; Lange, K. Localization of an Ataxia-Telangiectasia Gene to Chromosome 11q22-23. Nature 1988, 336, 577–580. [Google Scholar] [CrossRef]
- Stewart, G.; Maser, R.; Stankovic, T.; Bressan, D.; Kaplan, M.; Jaspers, N.; Raams, A.; Byrd, P.; Petrini, J.; Taylor, A. The DNA Double-Strand Break Repair Gene HMRE11 Is Mutated in Individuals with an Ataxia-Telangiectasia-like Disorder. Cell 1999, 99, 577–587. [Google Scholar] [CrossRef]
- Hernandez, D.; McConville, C.; Stacey, M.; Woods, C.; Brown, M.; Shutt, P.; Rysiecki, G.; Taylor, A.M. A Family Showing No Evidence of Linkage between the Ataxia Telangiectasia Gene and Chromosome 11q22-23. J. Med. Genet. 1993, 30, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Regal, J.A.; Festerling, T.A.; Buis, J.M.; Ferguson, D.O. Disease-Associated MRE11 Mutants Impact ATM/ATR DNA Damage Signaling by Distinct Mechanisms. Hum. Mol. Genet. 2013, 22, 5146. [Google Scholar] [CrossRef] [PubMed]
- Kleibl, Z.; Kristensen, V.N. Women at High Risk of Breast Cancer: Molecular Characteristics, Clinical Presentation and Management. Breast 2016, 28, 136–144. [Google Scholar] [CrossRef]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 Complex Alterations and DNA Damage Response: Implications for Cancer Treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic Variants Associated with Breast-Cancer Risk: Comprehensive Research Synopsis, Meta-Analysis, and Epidemiological Evidence. Lancet Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef] [PubMed]
- Rosen, E.M. BRCA1 in the DNA Damage Response and at Telomeres. Front. Genet. 2013, 4, 85. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Wilson, R.; Chatterjee, N.; Hudson, C.; Ruhl, R.; Hipfinger, C.; Helms, E.; Khan, O.F.; Anderson, D.G.; Anand, S. MicroRNA Regulation of the MRN Complex Impacts DNA Damage, Cellular Senescence, and Angiogenic Signaling Article. Cell Death Dis. 2018, 9, 632. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed]
- Brandt, S.; Samartzis, E.P.; Zimmermann, A.K.; Fink, D.; Moch, H.; Noske, A.; Dedes, K.J. Lack of MRE11-RAD50-NBS1 (MRN) Complex Detection Occurs Frequently in Low-Grade Epithelial Ovarian Cancer. BMC Cancer 2017, 17, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Y.; Zhou, K.; Zhou, C.; Zhou, R.; Cheng, C.; Wei, Q.; Lu, D.; Zhou, L. Genetic Variations in the Homologous Recombination Repair Pathway Genes Modify Risk of Glioma. J. Neurooncol. 2016, 126, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, W.H.; Wei, Y.; Zhu, Y.; Qin, X.J.; Zhang, H.L.; Ye, D.W. Elevated MRE11 Expression Associated with Progression and Poor Outcome in Prostate Cancer. J. Cancer 2019, 10, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Kavitha, C.V.; Choudhary, B.; Raghavan, S.C.; Muniyappa, K. Differential Regulation of MRN (Mre11-Rad50-Nbs1) Complex Subunits and Telomerase Activity in Cancer Cells. Biochem. Biophys. Res. Commun. 2010, 399, 575–580. [Google Scholar] [CrossRef]
- Takemura, H.; Rao, V.A.; Sordet, O.; Furuta, T.; Miao, Z.H.; Meng, L.H.; Zhang, H.; Pommier, Y. Defective Mre11-Dependent Activation of Chk2 by Ataxia Telangiectasia Mutated in Colorectal Carcinoma Cells in Response to Replication-Dependent DNA Double Strand Breaks. J. Biol. Chem. 2006, 281, 30814–30823. [Google Scholar] [CrossRef]
- Situ, Y.; Chung, L.; Lee, C.S.; Ho, V. MRN (MRE11-RAD50-NBS1) Complex in Human Cancer and Prognostic Implications in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 816. [Google Scholar] [CrossRef] [PubMed]
- Ho, V.; Chung, L.; Singh, A.; Lea, V.; Abubakar, A.; Lim, S.H.; Ng, W.; Lee, M.; de Souza, P.; Shin, J.S.; et al. Overexpression of the MRE11-RAD50-NBS1 (MRN) Complex in Rectal Cancer Correlates with Poor Response to Neoadjuvant Radiotherapy and Prognosis. BMC Cancer 2018, 18, 869. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Wokołorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojć, B.; Dȩbniak, T.; Górski, B.; Blecharz, P.; et al. Risk of Breast Cancer in Women with a CHEK2 Mutation with and without a Family History of Breast Cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.; Andreassen, P.R. PALB2 Functionally Connects the Breast Cancer Susceptibility Proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef]
- Antoniou, A.; Pharoah, P.D.P.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, Å.; et al. Average Risks of Breast and Ovarian Cancer Associated with BRCA1 or BRCA2 Mutations Detected in Case Series Unselected for Family History: A Combined Analysis of 22 Studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and Somatic Mutations in Homologous Recombination Genes Predict Platinum Response and Survival in Ovarian, Fallopian Tube, and Peritoneal Carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Ford, D.; Easton, D.F.; Bishop, D.T.; Narod, S.A.; Goldgar, D.E. Risks of Cancer in BRCA1-Mutation Carriers. Breast Cancer Linkage Consortium. Lancet 1994, 343, 692–695. [Google Scholar] [CrossRef]
- Ahmed, M.; Rahman, N. ATM and Breast Cancer Susceptibility. Oncogene 2006, 25, 5906–5911. [Google Scholar] [CrossRef]
- Tanaka, A.; Weinel, S.; Nagy, N.; O’Driscoll, M.; Lai-Cheong, J.E.; Kulp-Shorten, C.L.; Knable, A.; Carpenter, G.; Fisher, S.A.; Hiragun, M.; et al. Germline Mutation in ATR in Autosomal- Dominant Oropharyngeal Cancer Syndrome. Am. J. Hum. Genet. 2012, 90, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.Y.; Brock, I.W.; Connley, D.; Cramp, H.; Tucker, R.; Slate, J.; Reed, M.W.R.; Balasubramanian, S.P.; Cannon-Albright, L.A.; Camp, N.J.; et al. Associations of ATR and CHEK1 Single Nucleotide Polymorphisms with Breast Cancer. PLoS ONE 2013, 8, e0068578. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2018, 1–13. [Google Scholar] [CrossRef]
- Bertoni, F.; Codegoni, A.M.; Furlan, D.; Tibiletti, M.G.; Capella, C.; Broggini, M. CHK1 Frameshift Mutations in Genetically Unstable Colorectal and Endometrial Cancers. Genes Chromosomes Cancer 1999, 26, 176–180. [Google Scholar] [CrossRef]
- Menoyo, A.; Alazzouzi, H.; Espin, E.; Armengol, M.; Yamaoto, H.; Schwartz Jr, S. Somatic Mutations in the DNA Damage-Response Genes ATR and CHK1 in Sporadic Stomach Tumors with Microsatellite Instability–PubMed. Cancer Res. 2001, 61, 7727–7730. [Google Scholar]
- Vassileva, V.; Millar, A.; Briollais, L.; Chapman, W.; Bapat, B. Genes Involved in DNA Repair Are Mutational Targets in Endometrial Cancers with Microsatellite Instability–PubMed. Cancer Res. 2002, 62, 4095–4099. [Google Scholar]
- Jividen, K.; Kedzierska, K.Z.; Yang, C.S.; Szlachta, K.; Ratan, A.; Paschal, B.M. Genomic Analysis of DNA Repair Genes and Androgen Signaling in Prostate Cancer. BMC Cancer 2018, 18, 960. [Google Scholar] [CrossRef] [PubMed]
- Fadaka, A.O.; Bakare, O.O.; Sibuyi, N.R.S.; Klein, A. Gene Expression Alterations and Molecular Analysis of CHEK1 in Solid Tumors. Cancers 2020, 12, 662. [Google Scholar] [CrossRef] [PubMed]
- Malkin, D.; Li, F.P.; Strong, L.C.; Fraumeni, J.F.; Nelson, C.E.; Kim, D.H.; Kassel, J.; Gryka, M.A.; Bischoff, F.Z.; Tainsky, M.A.; et al. Germ Line P53 Mutations in a Familial Syndrome of Breast Cancer, Sarcomas, and Other Neoplasms. Science 1990, 250, 1233–1238. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA J. Am. Med. Assoc. 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 Foci as a Functional Biomarker of Homologous Recombination Repair and PARP Inhibitor Resistance in Germline BRCA-Mutated Breast Cancer. Ann. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ford, J.M. Multigene Panel Testing in Oncology Practice: How Should We Respond? JAMA Oncol. 2015, 1, 277–278. [Google Scholar] [CrossRef]
- Flanagan, S.; Patch, A.; Ellard, S. Using SIFT and PolyPhen to Predict Loss-of-Function and Gain-of-Function Mutations. Genet. Test. Mol. Biomark. 2010, 14, 533–537. [Google Scholar] [CrossRef]
- Pshennikova, V.G.; Barashkov, N.A.; Romanov, G.P.; Teryutin, F.M.; Solov’ev, A.V.; Gotovtsev, N.N.; Nikanorova, A.A.; Nakhodkin, S.S.; Sazonov, N.N.; Morozov, I.V.; et al. Comparison of Predictive in Silico Tools on Missense Variants in GJB2, GJB6, and GJB3 Genes Associated with Autosomal Recessive Deafness 1A (DFNB1A). Sci. World J. 2019, 2019, 5198931. [Google Scholar] [CrossRef]
- Leong, I.U.S.; Stuckey, A.; Lai, D.; Skinner, J.R.; Love, D.R. Assessment of the Predictive Accuracy of Five in Silico Prediction Tools, Alone or in Combination, and Two Metaservers to Classify Long QT Syndrome Gene Mutations. BMC Med. Genet. 2015, 16, 1–13. [Google Scholar] [CrossRef]
- Araki, K.; Yamashita, T.; Reddy, N.; Wang, H.; Abuzeid, W.M.; Khan, K.; O’Malley, B.W.; Li, D. Molecular Disruption of NBS1 with Targeted Gene Delivery Enhances Chemosensitisation in Head and Neck Cancer. Br. J. Cancer 2010, 103, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Lajud, S.A.; Nagda, D.A.; Yamashita, T.; Zheng, J.; Tanaka, N.; Abuzeid, W.M.; Civantos, A.; Bezpalko, O.; O’Malley, B.W.; Li, D. Dual Disruption of DNA Repair and Telomere Maintenance for the Treatment of Head and Neck Cancer. Clin. Cancer Res. 2014, 20, 6465–6478. [Google Scholar] [CrossRef] [PubMed]
- Koppensteiner, R.; Samartzis, E.P.; Noske, A.; Von Teichman, A.; Dedes, I.; Gwerder, M.; Imesch, P.; Ikenberg, K.; Moch, H.; Fink, D.; et al. Effect of MRE11 Loss on PARP-Inhibitor Sensitivity in Endometrial Cancer In Vitro. PLoS ONE 2014, 9, e0100041. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11-Rad50-Nbs1 Complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Berte, N.; Piée-Staffa, A.; Piecha, N.; Wang, M.; Borgmann, K.; Kaina, B.; Nikolova, T. Targeting Homologous Recombination by Pharmacological Inhibitors Enhances the Killing Response of Glioblastoma Cells Treated with Alkylating Drugs. Mol. Cancer Ther. 2016, 15, 2665–2678. [Google Scholar] [CrossRef]
- Petroni, M.; Sardina, F.; Infante, P.; Bartolazzi, A.; Locatelli, E.; Fabretti, F.; Di Giulio, S.; Capalbo, C.; Cardinali, B.; Coppa, A.; et al. MRE11 Inhibition Highlights a Replication Stress-Dependent Vulnerability of MYCN-Driven Tumors. Cell Death Dis. 2018, 9, 895. [Google Scholar] [CrossRef]
- Flores-Pérez, A.; Rafaelli, L.E.; Ramírez-Torres, N.; Aréchaga-Ocampo, E.; Frías, S.; Sánchez, S.; Marchat, L.A.; Hidalgo-Miranda, A.; Quintanar-Jurado, V.; Rodríguez-Cuevas, S.; et al. RAD50 Targeting Impairs DNA Damage Response and Sensitizes Human Breast Cancer Cells to Cisplatin Therapy. Cancer Biol. Ther. 2014, 15, 777–788. [Google Scholar] [CrossRef]
- Abuzeid, W.M.; Jiang, X.; Shi, G.; Wang, H.; Paulson, D.; Araki, K.; Jungreis, D.; Carney, J.; O’Malley, B.W.; Li, D. Molecular Disruption of RAD50 Sensitizes Human Tumor Cells to Cisplatin-Based Chemotherapy. J. Clin. Investig. 2009, 119, 1974–1985. [Google Scholar] [CrossRef]
- Christian Reinhardt, H.; Jiang, H.; Hemann, M.T.; Yaffe, M.B. Exploiting Synthetic Lethal Interactions for Targeted Cancer Therapy. Cell Cycle 2009, 8, 3112–3119. [Google Scholar] [CrossRef]
- Ashworth, A.; Lord, C.J.; Reis-Filho, J.S. Erratum: Genetic Interactions in Cancer Progression and Treatment. Cell 2011, 145, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Garraway, L.A.; Ashworth, A.; Weber, B. Synthetic Lethality as an Engine for Cancer Drug Target Discovery. Nat. Rev. Drug Discov. 2020, 19, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating Genetic Approaches into the Discovery of Anticancer Drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Al-Ahmadie, H.; Iyer, G.; Hohl, M.; Asthana, S.; Inagaki, A.; Schultz, N.; Hanrahan, A.J.; Scott, S.N.; Brannon, A.R.; McDermott, G.C.; et al. Synthetic Lethality in ATM-Deficient RAD50-Mutant Tumors Underlie Outlier Response to Cancer Therapy. Cancer Discov. 2014, 4, 1014. [Google Scholar] [CrossRef]
- Tangutoori, S.; Baldwin, P.; Sridhar, S. PARP Inhibitors: A New Era of Targeted Therapy. Maturitas 2015, 81, 5–9. [Google Scholar] [CrossRef]
- Gunderson, C.C.; Moore, K.N. Olaparib: An Oral PARP-1 and PARP-2 Inhibitor with Promising Activity in Ovarian Cancer. Futur. Oncol. 2015, 11, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef]
- Faraoni, I.; Graziani, G. Role of BRCA Mutations in Cancer Treatment with Poly(ADP-Ribose) Polymerase (PARP) Inhibitors. Cancers 2018, 10, 487. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef]
- Kumar, R.; Khan, R.; Gupta, N.; Seth, T.; Sharma, A.; Kalaivani, M.; Sharma, A. Identifying the Biomarker Potential of Telomerase Activity and Shelterin Complex Molecule, Telomeric Repeat Binding Factor 2 (TERF2), in Multiple Myeloma. Leuk. Lymphoma 2018, 59, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Mullard, A. Synthetic Lethality Screens Point the Way to New Cancer Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Jerby-Arnon, L.; Pfetzer, N.; Waldman, Y.Y.; McGarry, L.; James, D.; Shanks, E.; Seashore-Ludlow, B.; Weinstock, A.; Geiger, T.; Clemons, P.A.; et al. Predicting Cancer-Specific Vulnerability via Data-Driven Detection of Synthetic Lethality. Cell 2014, 158, 1199–1209. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarthy-Leo, C.; Darwiche, F.; Tainsky, M.A. DNA Repair Mechanisms, Protein Interactions and Therapeutic Targeting of the MRN Complex. Cancers 2022, 14, 5278. https://doi.org/10.3390/cancers14215278
McCarthy-Leo C, Darwiche F, Tainsky MA. DNA Repair Mechanisms, Protein Interactions and Therapeutic Targeting of the MRN Complex. Cancers. 2022; 14(21):5278. https://doi.org/10.3390/cancers14215278
Chicago/Turabian StyleMcCarthy-Leo, Claire, Fatima Darwiche, and Michael A. Tainsky. 2022. "DNA Repair Mechanisms, Protein Interactions and Therapeutic Targeting of the MRN Complex" Cancers 14, no. 21: 5278. https://doi.org/10.3390/cancers14215278
APA StyleMcCarthy-Leo, C., Darwiche, F., & Tainsky, M. A. (2022). DNA Repair Mechanisms, Protein Interactions and Therapeutic Targeting of the MRN Complex. Cancers, 14(21), 5278. https://doi.org/10.3390/cancers14215278