
Transforming Growth Factor-β Activated Kinase 1 (Tak1) Is Activated in Hepatocellular Carcinoma, Mediates Tumor Progression, and Predicts Unfavorable Outcome

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Immunohistochemistry
2.3. Immunoblotting
2.4. Real-Time RT-PCR
2.5. Mouse Experiments
2.6. Plasmids
- TAK1 forward: 5′-CATGGATCCGCCACCATGTCTACAGCCTCTGCCGC-3′
- TAK1 reverse: 5′-CATCTCGAGTGAAGTGCCTTGTCGTTTCT-3′
- TAK1STP reverse: 5′-CATCTCGAGTCATGAAGTGCCTTGTCGTTTC-3′
2.7. Cell Culture
2.8. Migration Assay
2.9. Immunofluorescence Microscopy
2.10. Estimation of Protein Activity in the Cancer Genome Atlas (TCGA) Cohort
2.11. Statistical Analysis
3. Results
3.1. Tak1 Is Induced in Hepatocellular Carcinoma and Further Increased in Distant Metastases
3.2. Tak1 Is Induced in a Chemotoxic HCC Mouse Model and Is Further Induced by a High-Fat Diet
3.3. Tak1, Especially Its Isoform Tak1A, Mediates Increased Tumor Cell Migration
3.4. High Tak1 Expression Is Associated with Unfavorable Prognostic Factors and Reduced Overall Survival
3.5. Combined Analysis of the Cyld-Tak1 Pathway Defines Distinct Prognostic Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- European Association for the Study of the Liver. Electronic address: Easloffice@easloffice.eu; European Association for the Study of the Liver EASL Clinical Practice Guidelines: Management of Hepatocellular Carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Pan, Y.; Liang, Y.; Xu, J.; Cai, X. The Microenvironmental and Metabolic Aspects of Sorafenib Resistance in Hepatocellular Carcinoma. EBioMedicine 2020, 51, 102610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettermann, K.; Vucur, M.; Haybaeck, J.; Koppe, C.; Janssen, J.; Heymann, F.; Weber, A.; Weiskirchen, R.; Liedtke, C.; Gassler, N.; et al. TAK1 Suppresses a NEMO-Dependent but NF-ΚB-Independent Pathway to Liver Cancer. Cancer Cell 2010, 17, 481–496. [Google Scholar] [CrossRef]
- Sakurai, H. Targeting of TAK1 in Inflammatory Disorders and Cancer. Trends Pharmacol. Sci. 2012, 33, 522–530. [Google Scholar] [CrossRef]
- Mihaly, S.R.; Ninomiya-Tsuji, J.; Morioka, S. TAK1 Control of Cell Death. Cell Death Differ. 2014, 21, 1667–1676. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Niu, W.; Peng, C.; Zhang, Z.; Niu, J. The Role of TAK1 Expression in Thyroid Cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 14449–14456. [Google Scholar] [PubMed]
- Yu, M.; Ting, D.T.; Stott, S.L.; Wittner, B.S.; Ozsolak, F.; Paul, S.; Ciciliano, J.C.; Smas, M.E.; Winokur, D.; Gilman, A.J.; et al. RNA Sequencing of Pancreatic Circulating Tumour Cells Implicates WNT Signalling in Metastasis. Nature 2012, 487, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, F.; García de Vinuesa, A.; de Kruijf, E.M.; Mesker, W.E.; Hui, L.; Drabsch, Y.; Li, Y.; Bauer, A.; Rousseau, A.; et al. TRAF4 Promotes TGF-β Receptor Signaling and Drives Breast Cancer Metastasis. Mol. Cell 2013, 51, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Inokuchi, S.; Roh, Y.S.; Song, J.; Loomba, R.; Park, E.J.; Seki, E. Transforming Growth Factor-β Signaling in Hepatocytes Promotes Hepatic Fibrosis and Carcinogenesis in Mice with Hepatocyte-Specific Deletion of TAK1. Gastroenterology 2013, 144, 1042–1054.e4. [Google Scholar] [CrossRef] [Green Version]
- Morioka, S.; Sai, K.; Omori, E.; Ikeda, Y.; Matsumoto, K.; Ninomiya-Tsuji, J. TAK1 Regulates Hepatic Lipid Homeostasis through SREBP. Oncogene 2016, 35, 3829–3838. [Google Scholar] [CrossRef] [Green Version]
- Friemel, J.; Frick, L.; Unger, K.; Egger, M.; Parrotta, R.; Böge, Y.T.; Adili, A.; Karin, M.; Luedde, T.; Heikenwalder, M.; et al. Characterization of HCC Mouse Models: Towards an Etiology-Oriented Subtyping Approach. Mol. Cancer Res. 2019, 17, 1493–1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.-X.; Huang, Z.; Yang, X.; Wang, X.; Zhao, L.-P.; Wang, P.-X.; Zhang, X.-J.; Alves-Bezerra, M.; Cai, L.; Zhang, P.; et al. The Deubiquitinating Enzyme Cylindromatosis Mitigates Nonalcoholic Steatohepatitis. Nat. Med. 2018, 24, 213–223. [Google Scholar] [CrossRef]
- Nikolaou, K.; Tsagaratou, A.; Eftychi, C.; Kollias, G.; Mosialos, G.; Talianidis, I. Inactivation of the Deubiquitinase CYLD in Hepatocytes Causes Apoptosis, Inflammation, Fibrosis, and Cancer. Cancer Cell 2012, 21, 738–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinmann, A.; Koch, S.; Niederle, I.M.; Schulze-Bergkamen, H.; König, J.; Hoppe-Lotichius, M.; Hansen, T.; Pitton, M.B.; Düber, C.; Otto, G.; et al. Trends in Epidemiology, Treatment, and Survival of Hepatocellular Carcinoma Patients between 1998 and 2009: An Analysis of 1066 Cases of a German HCC Registry. J. Clin. Gastroenterol. 2014, 48, 279–289. [Google Scholar] [CrossRef]
- Ridder, D.A.; Schindeldecker, M.; Weinmann, A.; Berndt, K.; Urbansky, L.; Witzel, H.R.; Heinrich, S.; Roth, W.; Straub, B.K. Key Enzymes in Pyrimidine Synthesis, CAD and CPS1, Predict Prognosis in Hepatocellular Carcinoma. Cancers 2021, 13, 744. [Google Scholar] [CrossRef] [PubMed]
- Remmele, W.; Stegner, H.E. Recommendation for uniform definition of an immunoreactive score (IRS) for immunohistochemical estrogen receptor detection (ER-ICA) in breast cancer tissue. Pathologe 1987, 8, 138–140. [Google Scholar]
- Tripathi, V.; Shin, J.-H.; Stuelten, C.H.; Zhang, Y.E. TGF-β-Induced Alternative Splicing of TAK1 Promotes EMT and Drug Resistance. Oncogene 2019, 38, 3185–3200. [Google Scholar] [CrossRef] [PubMed]
- Elßner, C.; Goeppert, B.; Longerich, T.; Scherr, A.-L.; Stindt, J.; Nanduri, L.K.; Rupp, C.; Kather, J.N.; Schmitt, N.; Kautz, N.; et al. Nuclear Translocation of RELB Is Increased in Diseased Human Liver and Promotes Ductular Reaction and Biliary Fibrosis in Mice. Gastroenterology 2019, 156, 1190–1205.e14. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Magnuson, M.A. DNA Excision in Liver by an Albumin-Cre Transgene Occurs Progressively with Age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Hövelmeyer, N.; Wunderlich, F.T.; Massoumi, R.; Jakobsen, C.G.; Song, J.; Wörns, M.A.; Merkwirth, C.; Kovalenko, A.; Aumailley, M.; Strand, D.; et al. Regulation of B Cell Homeostasis and Activation by the Tumor Suppressor Gene CYLD. J. Exp. Med. 2007, 204, 2615–2627. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Ramos, M.; Geistlinger, L.; Oh, S.; Schiffer, L.; Azhar, R.; Kodali, H.; de Bruijn, I.; Gao, J.; Carey, V.J.; Morgan, M.; et al. Multiomic Integration of Public Oncology Databases in Bioconductor. JCO Clin. Cancer Inform. 2020, 1, 958–971. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address: Wheeler@bcm.edu; Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Douglass, E.F.; Sonabend, A.M.; Mela, A.; Bose, S.; Gonzalez, C.; Canoll, P.D.; Sims, P.A.; Alvarez, M.J.; Califano, A. Quantitative Assessment of Protein Activity in Orphan Tissues and Single Cells Using the MetaVIPER Algorithm. Nat. Commun. 2018, 9, 1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional Characterization of Somatic Mutations in Cancer Using Network-Based Inference of Protein Activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: https://Www.R-Project.Org/ (accessed on 24 August 2021).
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A Comprehensive and Straightforward Web Application Enabling Rapid Biomarker Cutoff Optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef] [Green Version]
- Venables, W.N.; Ripley, B.D. (Eds.) Modern Applied Statistics with S; Statistics and Computing; Springer: New York, NY, USA, 2002; pp. 301–330. ISBN 978-0-387-21706-2. [Google Scholar]
- Hadian, K.; Krappmann, D. Signals from the Nucleus: Activation of NF-KappaB by Cytosolic ATM in the DNA Damage Response. Sci. Signal 2011, 4, pe2. [Google Scholar] [CrossRef] [PubMed]
- Urbanik, T.; Boger, R.J.; Longerich, T.; Becker, K.; Ehrenberg, K.R.; Hövelmeyer, N.; Hahn, M.; Schuchmann, M.; Jäger, D.; Waisman, A.; et al. Liver Specific Deletion of CYLDexon7/8 Induces Severe Biliary Damage, Fibrosis and Increases Hepatocarcinogenesis in Mice. J. Hepatol. 2012, 57, 995–1003. [Google Scholar] [CrossRef]
- Welte, S.; Urbanik, T.; Elßner, C.; Kautz, N.; Koehler, B.C.; Waldburger, N.; Bermejo, J.L.; Pinna, F.; Weiss, K.-H.; Schemmer, P.; et al. Nuclear Expression of the Deubiquitinase CYLD Is Associated with Improved Survival in Human Hepatocellular Carcinoma. PLoS ONE 2014, 9, e110591. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Benhamouche, S.; Godard, C.; Boyault, S.; Grimber, G.; Balabaud, C.; Cunha, A.S.; Bioulac-Sage, P.; Perret, C. Differential Effects of Inactivated Axin1 and Activated Beta-Catenin Mutations in Human Hepatocellular Carcinomas. Oncogene 2007, 26, 774–780. [Google Scholar] [CrossRef] [Green Version]
- Renne, S.L.; Woo, H.Y.; Allegra, S.; Rudini, N.; Yano, H.; Donadon, M.; Viganò, L.; Akiba, J.; Lee, H.S.; Rhee, H.; et al. Vessels Encapsulating Tumor Clusters (VETC) Is a Powerful Predictor of Aggressive Hepatocellular Carcinoma. Hepatology 2020, 71, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Lokuhetty, D.; Organisation Mondiale de la Santé; Centre international de Recherche sur le Cancer. WHO Classification of Tumours; WHO: Geneva, Switzerland, 2019; ISBN 978-92-832-4499-8. [Google Scholar]
- Cheng, J.-S.; Tsai, W.-L.; Liu, P.-F.; Goan, Y.-G.; Lin, C.-W.; Tseng, H.-H.; Lee, C.-H.; Shu, C.-W. The MAP3K7-MTOR Axis Promotes the Proliferation and Malignancy of Hepatocellular Carcinoma Cells. Front. Oncol. 2019, 9, 474. [Google Scholar] [CrossRef]
- Cai, P.C.H.; Shi, L.; Liu, V.W.S.; Tang, H.W.M.; Liu, I.J.; Leung, T.H.Y.; Chan, K.K.L.; Yam, J.W.P.; Yao, K.-M.; Ngan, H.Y.S.; et al. Elevated TAK1 Augments Tumor Growth and Metastatic Capacities of Ovarian Cancer Cells through Activation of NF-ΚB Signaling. Oncotarget 2014, 5, 7549–7562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ear, T.; Fortin, C.F.; Simard, F.A.; McDonald, P.P. Constitutive Association of TGF-β–Activated Kinase 1 with the IκB Kinase Complex in the Nucleus and Cytoplasm of Human Neutrophils and Its Impact on Downstream Processes. J. Immunol. 2010, 184, 3897–3906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillo, A.R.; Scarpa, M.; D’Incà, R.; Brun, P.; Scarpa, M.; Porzionato, A.; De Caro, R.; Martines, D.; Buda, A.; Angriman, I.; et al. TAK1 Is a Key Modulator of the Profibrogenic Phenotype of Human Ileal Myofibroblasts in Crohn’s Disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G443–G454. [Google Scholar] [CrossRef] [Green Version]
- Kanei-Ishii, C.; Ninomiya-Tsuji, J.; Tanikawa, J.; Nomura, T.; Ishitani, T.; Kishida, S.; Kokura, K.; Kurahashi, T.; Ichikawa-Iwata, E.; Kim, Y.; et al. Wnt-1 Signal Induces Phosphorylation and Degradation of c-Myb Protein via TAK1, HIPK2, and NLK. Genes Dev. 2004, 18, 816–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Fang, W.; Cui, Y.; Shi, H.; Chen, J.; Li, L.; Zhang, L.; Zhang, X. Neddylation Promotes Protein Translocation between the Cytoplasm and Nucleus. Biochem. Biophys. Res. Commun. 2020, 529, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Liang, X.; Liu, Y.; Zheng, M. Neddylation: A Versatile Pathway Takes on Chronic Liver Diseases. Front. Med. 2020, 7, 628. [Google Scholar] [CrossRef]
- Fabregat, I.; Moreno-Càceres, J.; Sánchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; ten Dijke, P.; IT-LIVER Consortium. TGF-β Signalling and Liver Disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [Green Version]
- Verna, L.; Whysner, J.; Williams, G.M. N-Nitrosodiethylamine Mechanistic Data and Risk Assessment: Bioactivation, DNA-Adduct Formation, Mutagenicity, and Tumor Initiation. Pharmacol. Ther. 1996, 71, 57–81. [Google Scholar] [CrossRef]
- Colomer, C.; Margalef, P.; Villanueva, A.; Vert, A.; Pecharroman, I.; Solé, L.; González-Farré, M.; Alonso, J.; Montagut, C.; Martinez-Iniesta, M.; et al. IKKα Kinase Regulates the DNA Damage Response and Drives Chemo-Resistance in Cancer. Mol. Cell 2019, 75, 669–682.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, D.M.; Myers, P.H.; Painter, J.T.; Hoenerhoff, M.J.; Olden, K.; Roberts, J.D. Inhibition of Transforming Growth Factor-β-Activated Kinase-1 Blocks Cancer Cell Adhesion, Invasion, and Metastasis. Br. J. Cancer 2012, 107, 129–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Roderburg, C.; Luedde, T. The Role of the Gut Microbiome in the Development and Progression of Liver Cirrhosis and Hepatocellular Carcinoma. Gut Microbes 2014, 5, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The Immunology of Hepatocellular Carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C. CYLD: A Tumor Suppressor Deubiquitinase Regulating NF-ΚB Activation and Diverse Biological Processes. Cell Death Differ. 2010, 17, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Licchetta, A.; Memeo, R.; Argentiero, A.; Solimando, A.G.; Longo, V.; Delcuratolo, S.; Brunetti, O. Role of BRAF in Hepatocellular Carcinoma: A Rationale for Future Targeted Cancer Therapies. Medicina 2019, 55, 754. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Liu, G. Targeting the Ras/Raf/MEK/ERK Pathway in Hepatocellular Carcinoma. Oncol. Lett. 2017, 13, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Colombino, M.; Sperlongano, P.; Izzo, F.; Tatangelo, F.; Botti, G.; Lombardi, A.; Accardo, M.; Tarantino, L.; Sordelli, I.; Agresti, M.; et al. BRAF and PIK3CA Genes Are Somatically Mutated in Hepatocellular Carcinoma among Patients from South Italy. Cell Death Dis. 2012, 3, e259. [Google Scholar] [CrossRef]
- Xia, S.; Ji, L.; Tao, L.; Pan, Y.; Lin, Z.; Wan, Z.; Pan, H.; Zhao, J.; Cai, L.; Xu, J.; et al. TAK1 Is a Novel Target in Hepatocellular Carcinoma and Contributes to Sorafenib Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1121–1143. [Google Scholar] [CrossRef] [PubMed]
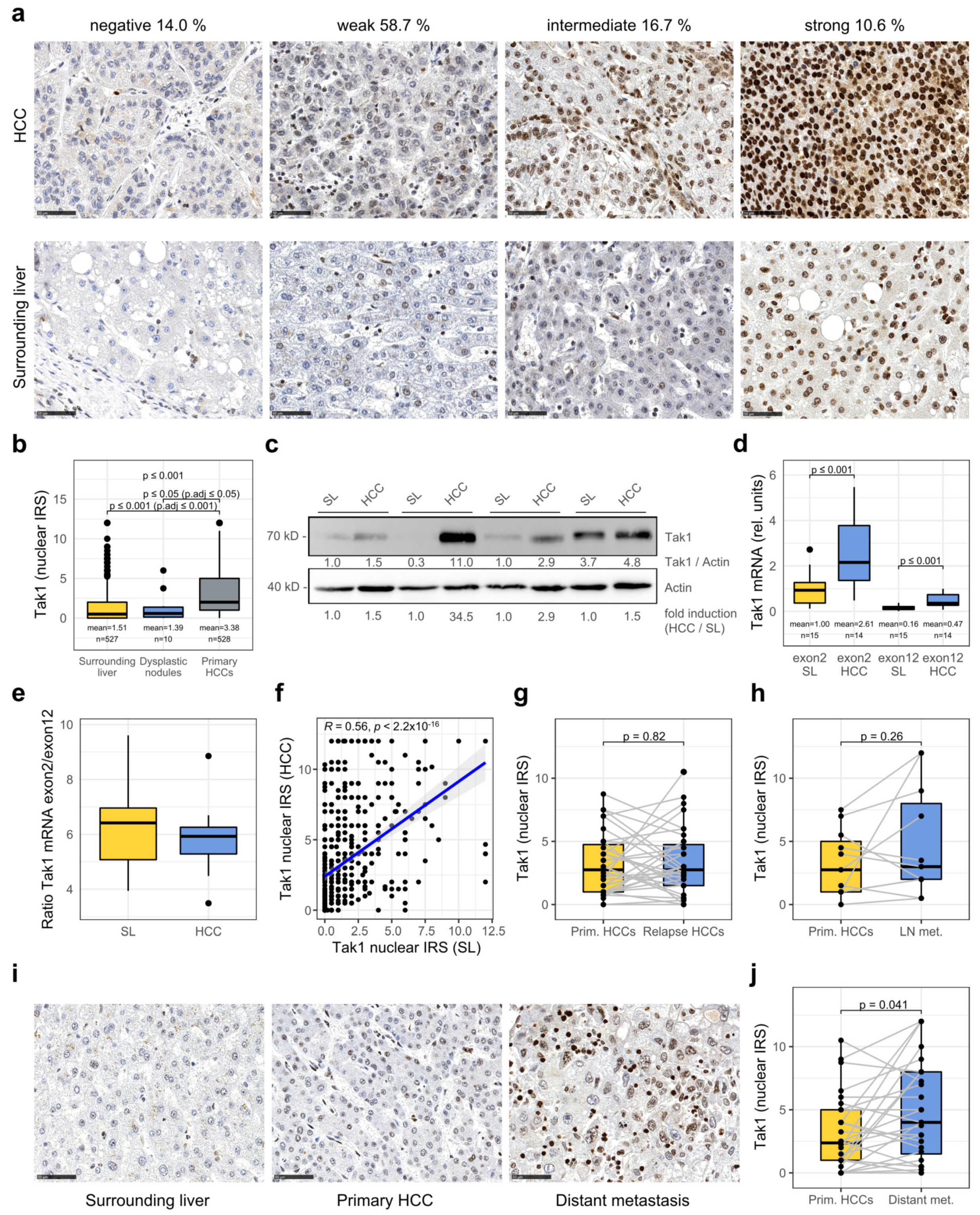
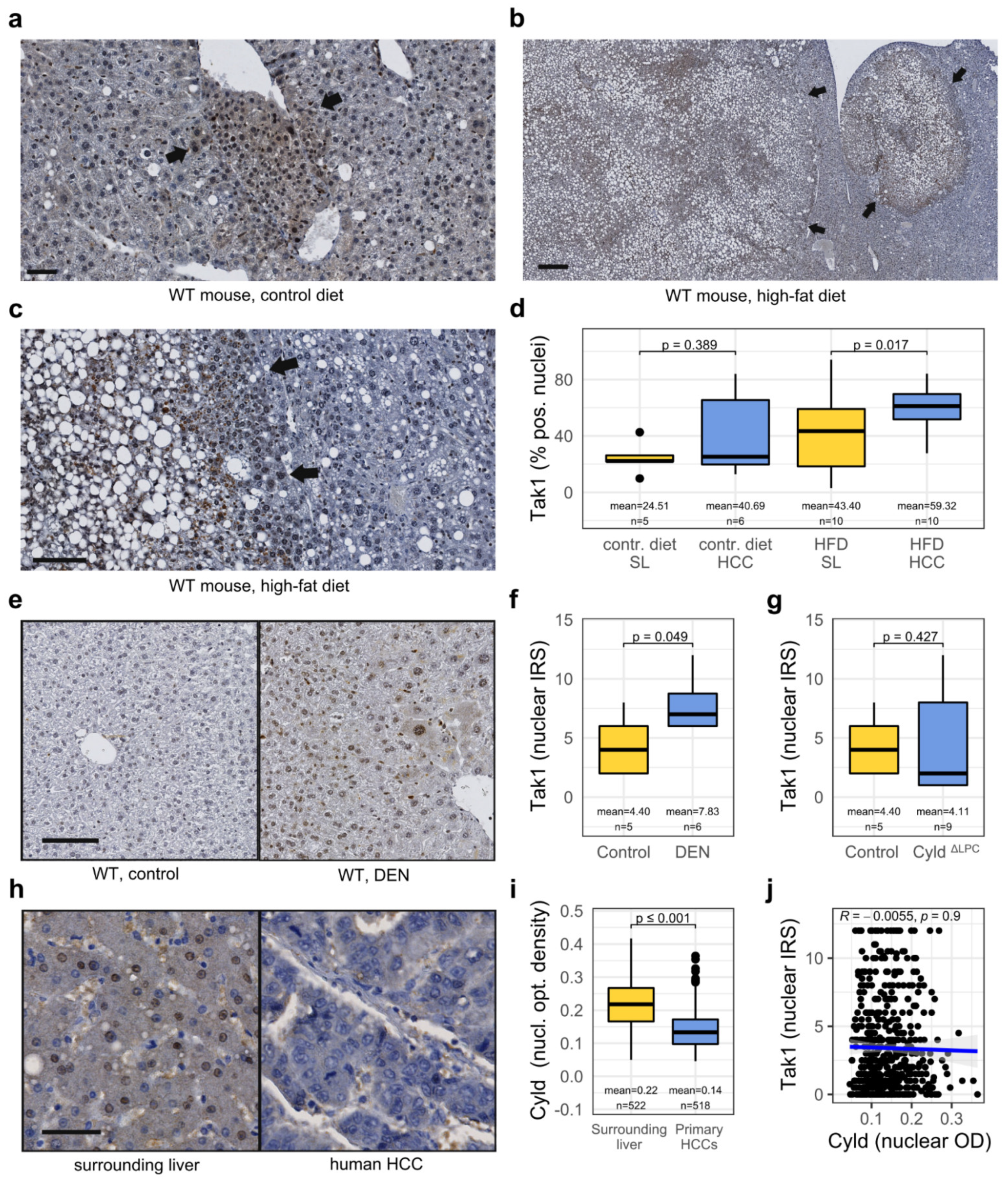
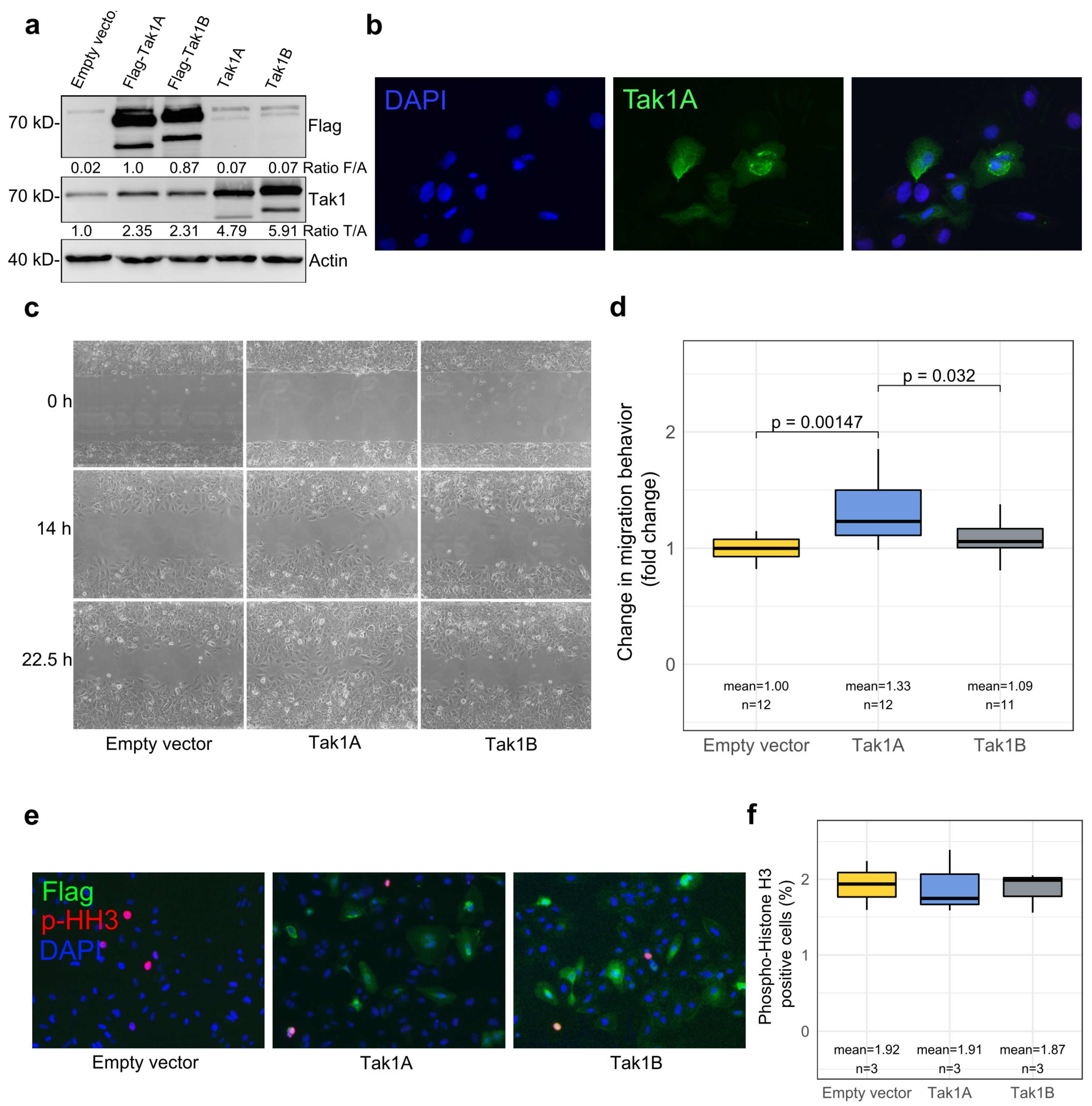

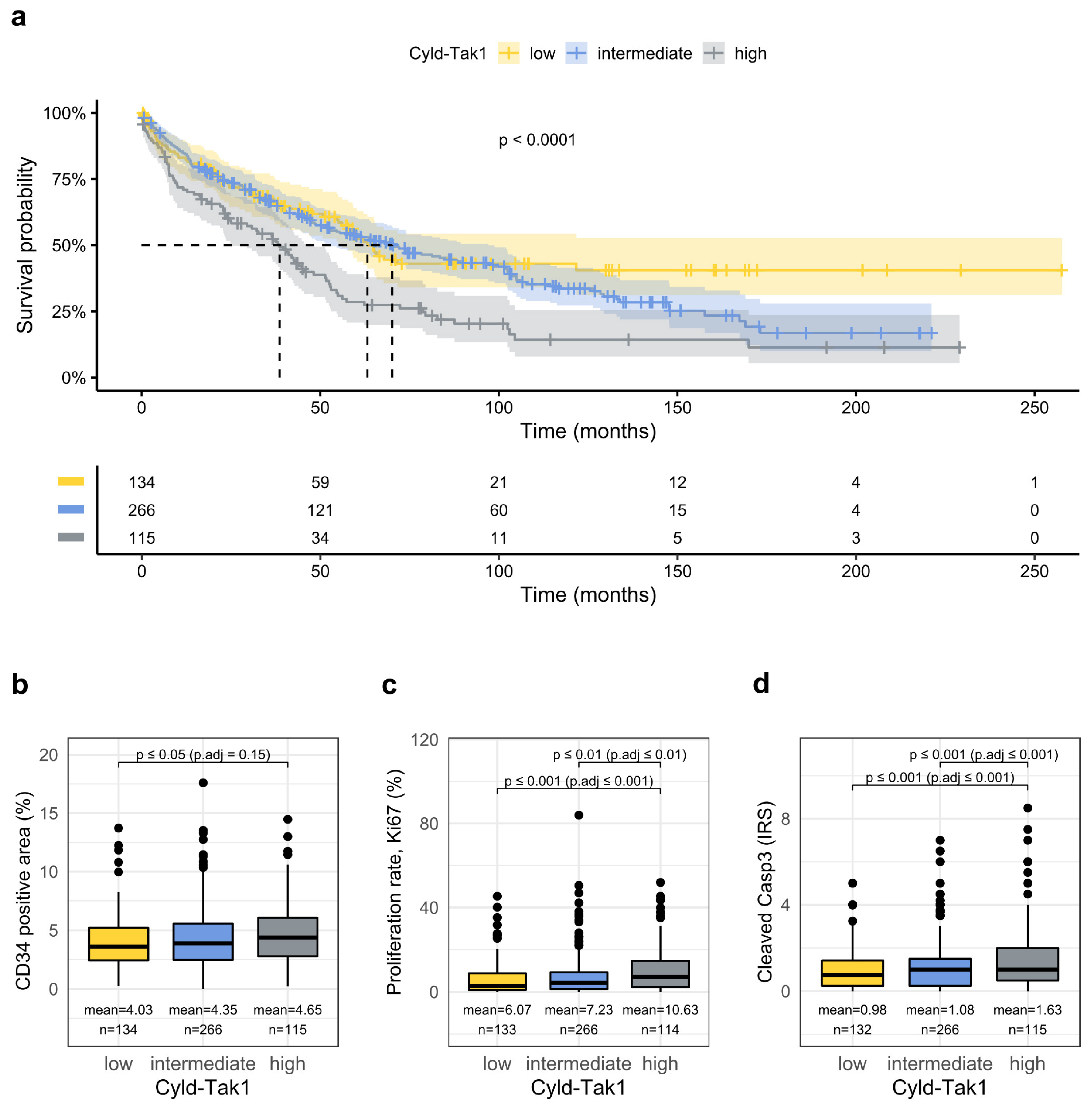

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Nuclear Tak1 Low | Nuclear Tak1 High | p-Value 2 | Cytoplasmic Tak1 Low | Cytoplasmic Tak1 High | p-Value 2 |
|---|---|---|---|---|---|---|
| n = 336 (64%) 1 | n = 192 (36%) 1 | n = 455 (86%) 1 | n = 73 (14%) 1 | |||
| Median age in years | 62.6 (55.7, 69.9) | 66.3 (59.2, 73.2) | <0.001 | 65.2 (56.6, 71.4) | 60.4 (55.5, 66.7) | 0.007 |
| Max. diameter [mm] | 36 (24, 68) | 50 (30, 98) | <0.001 | 41 (25, 80) | 38 (26, 92) | >0.9 |
| Gender | 0.12 | 0.1 | ||||
| Male | 257 (76%) | 158 (82%) | 363 (80%) | 52 (71%) | ||
| Female | 79 (24%) | 34 (18%) | 92 (20%) | 21 (29%) | ||
| Alcohol abuse | 99 (29%) | 56 (29%) | >0.9 | 134 (29%) | 21 (29%) | >0.9 |
| HCV | 78 (23%) | 28 (15%) | 0.017 | 92 (20%) | 14 (19%) | 0.8 |
| HBV | 67 (20%) | 31 (16%) | 0.3 | 84 (18%) | 14 (19%) | 0.9 |
| NASH | 27 (8.0%) | 15 (7.8%) | >0.9 | 34 (7.5%) | 8 (11%) | 0.3 |
| Hemochromatosis | 20 (6.0%) | 4 (2.1%) | 0.04 | 23 (5.1%) | 1 (1.4%) | 0.2 |
| BCLC | <0.001 | 0.8 | ||||
| A | 74 (22%) | 20 (10%) | 79 (17%) | 15 (21%) | ||
| B | 201 (60%) | 138 (72%) | 292 (64%) | 47 (64%) | ||
| C | 34 (10%) | 27 (14%) | 53 (12%) | 8 (11%) | ||
| D | 27 (8.0%) | 7 (3.6%) | 31 (6.8%) | 3 (4.1%) | ||
| ECOG PST | 0.7 | >0.9 | ||||
| 0–1 | 320 (96%) | 185 (97%) | 436 (96%) | 69 (97%) | ||
| 2 | 7 (2.1%) | 2 (1.1%) | 8 (1.8%) | 1 (1.4%) | ||
| 3 | 3 (0.9%) | 2 (1.1%) | 5 (1.1%) | 0 (0%) | ||
| 4 | 5 (1.5%) | 1 (0.5%) | 5 (1.1%) | 1 (1.4%) | ||
| Liver cirrhosis | <0.001 | 0.5 | ||||
| Absent | 102 (30%) | 91 (47%) | 164 (36%) | 29 (40%) | ||
| Present | 234 (70%) | 101 (53%) | 291 (64%) | 44 (60%) | ||
| Child–Pugh Score | 0.4 | 0.3 | ||||
| A | 130 (52%) | 58 (53%) | 167 (53%) | 21 (45%) | ||
| B | 94 (37%) | 44 (40%) | 115 (37%) | 23 (49%) | ||
| C | 27 (11%) | 7 (6.4%) | 31 (9.9%) | 3 (6.4%) | ||
| Portal vein thrombosis (PVT) | 34 (10%) | 30 (16%) | 0.062 | 56 (12%) | 8 (11%) | 0.7 |
| Vascular invasion | <0.001 | 0.3 | ||||
| Absent | 237 (71%) | 101 (53%) | 294 (65%) | 44 (60%) | ||
| Micro | 58 (17%) | 51 (27%) | 89 (20%) | 20 (27%) | ||
| Macro | 41 (12%) | 40 (21%) | 72 (16%) | 9 (12%) | ||
| Tumor grading | >0.9 | 0.008 | ||||
| G1 | 59 (20%) | 35 (20%) | 83 (20%) | 11 (17%) | ||
| G2 | 166 (56%) | 100 (56%) | 237 (58%) | 29 (44%) | ||
| G3 | 72 (24%) | 43 (24%) | 89 (22%) | 26 (39%) | ||
| MTM subtype | 0.6 | <0.001 | ||||
| no | 305 (92%) | 174 (93%) | 421 (94%) | 58 (79%) | ||
| yes | 28 (8.4%) | 13 (7.0%) | 26 (5.8%) | 15 (21%) | ||
| VETC | 0.011 | 0.1 | ||||
| negative | 287 (85%) | 147 (77%) | 369 (81%) | 65 (89%) | ||
| positive | 49 (15%) | 45 (23%) | 86 (19%) | 8 (11%) | ||
| GS overexpression | <0.001 | <0.001 | ||||
| no | 286 (86%) | 134 (70%) | 352 (77%) | 68 (96%) | ||
| yes | 48 (14%) | 58 (30%) | 103 (23%) | 3 (4.2%) |
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| Clinical Features | HR | std. Error | p-Value | HR | std. Error | p-Value |
| Age (≥60 vs. <60) | 1.43 | 0.12 | 0.003 | 1.44 | 0.13 | <0.01 |
| Sex (male vs. female) | 1.07 | 0.13 | 0.637 | |||
| Alcohol abuse (true vs. false) | 0.87 | 0.12 | 0.250 | |||
| Hemochromatosis (tr. vs. f.) | 0.70 | 0.27 | 0.186 | 0.66 | 0.28 | 0.140 |
| HBV (pos. vs. neg.) | 0.89 | 0.14 | 0.400 | |||
| HCV (pos. vs. neg.) | 1.07 | 0.14 | 0.608 | |||
| BCLC (B-D vs. A) | 1.83 | 0.16 | <0.001 | 1.58 | 0.18 | 0.010 |
| AFP Serum (> vs. ≤200 ng/mL) | 2.00 | 0.13 | <0.0001 | |||
| Pathologic features | ||||||
| Grading (G3 vs. G1/G2) | 1.50 | 0.13 | 0.003 | 1.29 | 0.14 | 0.075 |
| VI (micro vs. none) | 1.49 | 0.14 | 0.004 | 1.34 | 0.16 | 0.064 |
| VI (macro vs. none) | 3.20 | 0.15 | <0.0001 | 2.94 | 0.16 | <0.0001 |
| Macrotrabecular Subtype | 1.96 | 0.19 | <0.001 | |||
| nuclear Tak1 high | 1.34 | 0.12 | 0.011 | 1.07 | 0.13 | 0.587 |
| Univariate | Multivariate | |||||
|---|---|---|---|---|---|---|
| Clinical Features | HR | std. Error | p-Value | HR | std. Error | p-Value |
| Age (≥60 vs. <60) | 1.43 | 0.12 | 0.003 | 1.42 | 0.13 | <0.01 |
| Sex (male vs. female) | 1.07 | 0.13 | 0.637 | |||
| Alcohol abuse (true vs. false) | 0.87 | 0.12 | 0.250 | |||
| Hemochromatosis (tr. vs. f.) | 0.70 | 0.27 | 0.186 | 0.69 | 0.28 | 0.183 |
| HBV (pos. vs. neg.) | 0.89 | 0.14 | 0.400 | |||
| HCV (pos. vs. neg.) | 1.07 | 0.14 | 0.608 | |||
| BCLC (B-D vs. A) | 1.83 | 0.16 | <0.001 | 1.50 | 0.18 | 0.024 |
| AFP Serum (> vs. ≤200 ng/mL) | 2.00 | 0.13 | <0.0001 | |||
| Pathologic features | ||||||
| Grading (G3 vs. G1/G2) | 1.50 | 0.13 | 0.003 | 1.30 | 0.14 | 0.073 |
| VI (micro vs. none) | 1.49 | 0.14 | 0.004 | 1.24 | 0.16 | 0.172 |
| VI (macro vs. none) | 3.20 | 0.15 | <0.0001 | 2.74 | 0.16 | <0.0001 |
| Macrotrabecular Subtype | 1.96 | 0.19 | <0.001 | |||
| intermed. comb. score (vs. low) | 1.15 | 0.15 | 0.351 | 1.09 | 0.16 | 0.607 |
| high comb. score (vs. low) | 1.93 | 0.17 | <0.0001 | 1.50 | 0.18 | 0.028 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ridder, D.A.; Urbansky, L.L.; Witzel, H.R.; Schindeldecker, M.; Weinmann, A.; Berndt, K.; Gerber, T.S.; Köhler, B.C.; Nichetti, F.; Ludt, A.; et al. Transforming Growth Factor-β Activated Kinase 1 (Tak1) Is Activated in Hepatocellular Carcinoma, Mediates Tumor Progression, and Predicts Unfavorable Outcome. Cancers 2022, 14, 430. https://doi.org/10.3390/cancers14020430
Ridder DA, Urbansky LL, Witzel HR, Schindeldecker M, Weinmann A, Berndt K, Gerber TS, Köhler BC, Nichetti F, Ludt A, et al. Transforming Growth Factor-β Activated Kinase 1 (Tak1) Is Activated in Hepatocellular Carcinoma, Mediates Tumor Progression, and Predicts Unfavorable Outcome. Cancers. 2022; 14(2):430. https://doi.org/10.3390/cancers14020430
Chicago/Turabian StyleRidder, Dirk Andreas, Lana Louisa Urbansky, Hagen Roland Witzel, Mario Schindeldecker, Arndt Weinmann, Kristina Berndt, Tiemo Sven Gerber, Bruno Christian Köhler, Federico Nichetti, Annekathrin Ludt, and et al. 2022. "Transforming Growth Factor-β Activated Kinase 1 (Tak1) Is Activated in Hepatocellular Carcinoma, Mediates Tumor Progression, and Predicts Unfavorable Outcome" Cancers 14, no. 2: 430. https://doi.org/10.3390/cancers14020430
APA StyleRidder, D. A., Urbansky, L. L., Witzel, H. R., Schindeldecker, M., Weinmann, A., Berndt, K., Gerber, T. S., Köhler, B. C., Nichetti, F., Ludt, A., Gehrke, N., Schattenberg, J. M., Heinrich, S., Roth, W., & Straub, B. K. (2022). Transforming Growth Factor-β Activated Kinase 1 (Tak1) Is Activated in Hepatocellular Carcinoma, Mediates Tumor Progression, and Predicts Unfavorable Outcome. Cancers, 14(2), 430. https://doi.org/10.3390/cancers14020430