
Hepatocellular Carcinoma Is a Natural Target for Adeno-Associated Virus (AAV) 2 Vectors

 , , , , ,
, , , , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. AAV Vector Production
2.2. Animal Studies
2.3. In Vivo and In Situ Imaging
2.4. Human Precision-Cut Liver Slice (PCLS) Transduction
2.5. DNA Isolation and Relative AAV Vector DNA Quantification
2.6. Luciferase Assay
2.7. Immunohistochemical Staining and Analysis
2.8. Uncoating Assay from Tissue
2.9. LDH Measurements
2.10. Statistical Analysis
3. Results
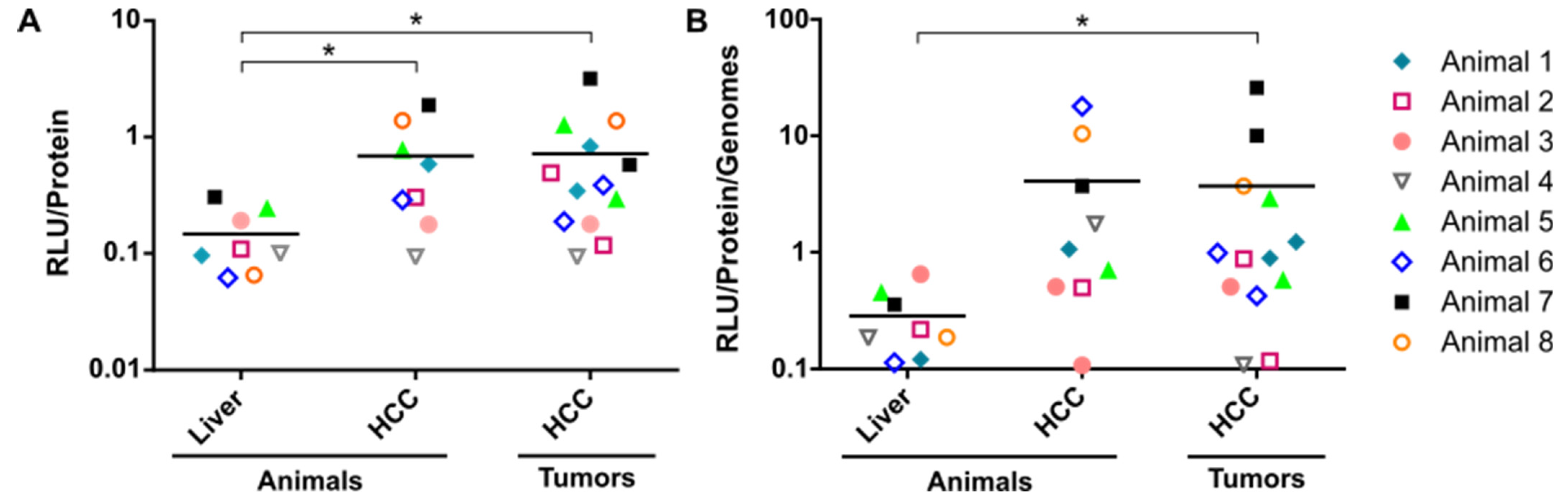
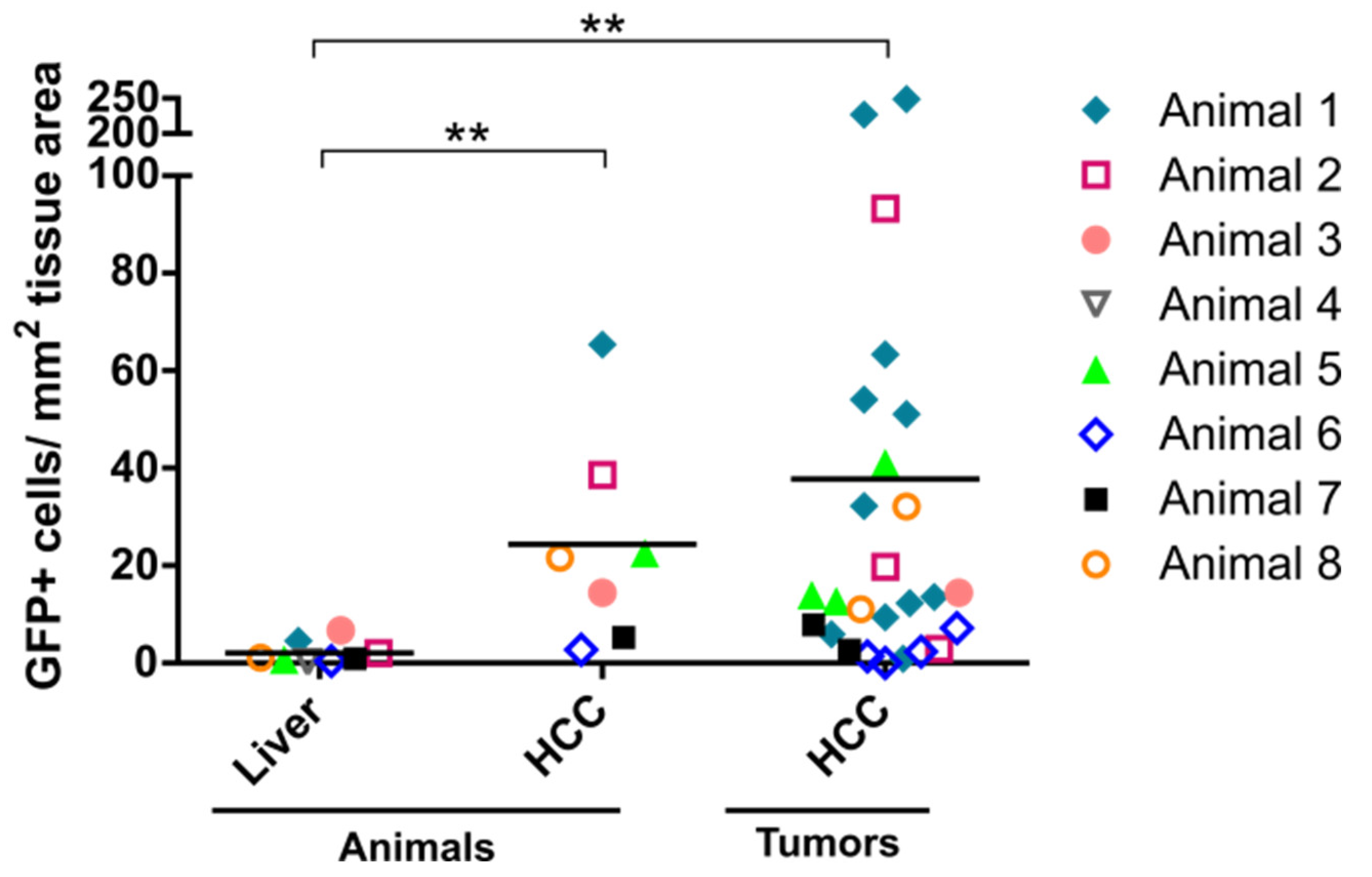
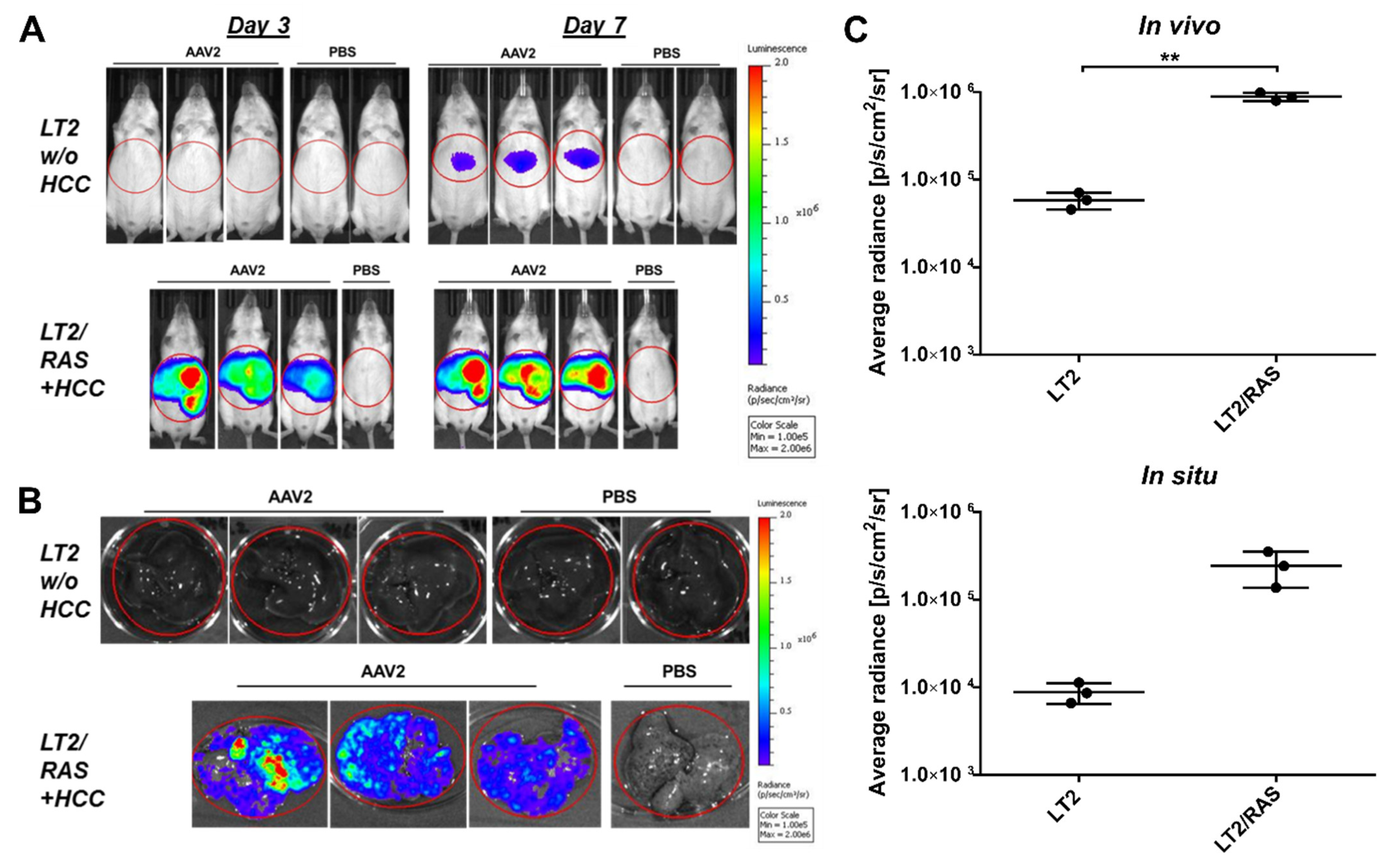
3.1. Enhanced Transgene Expression in HCC
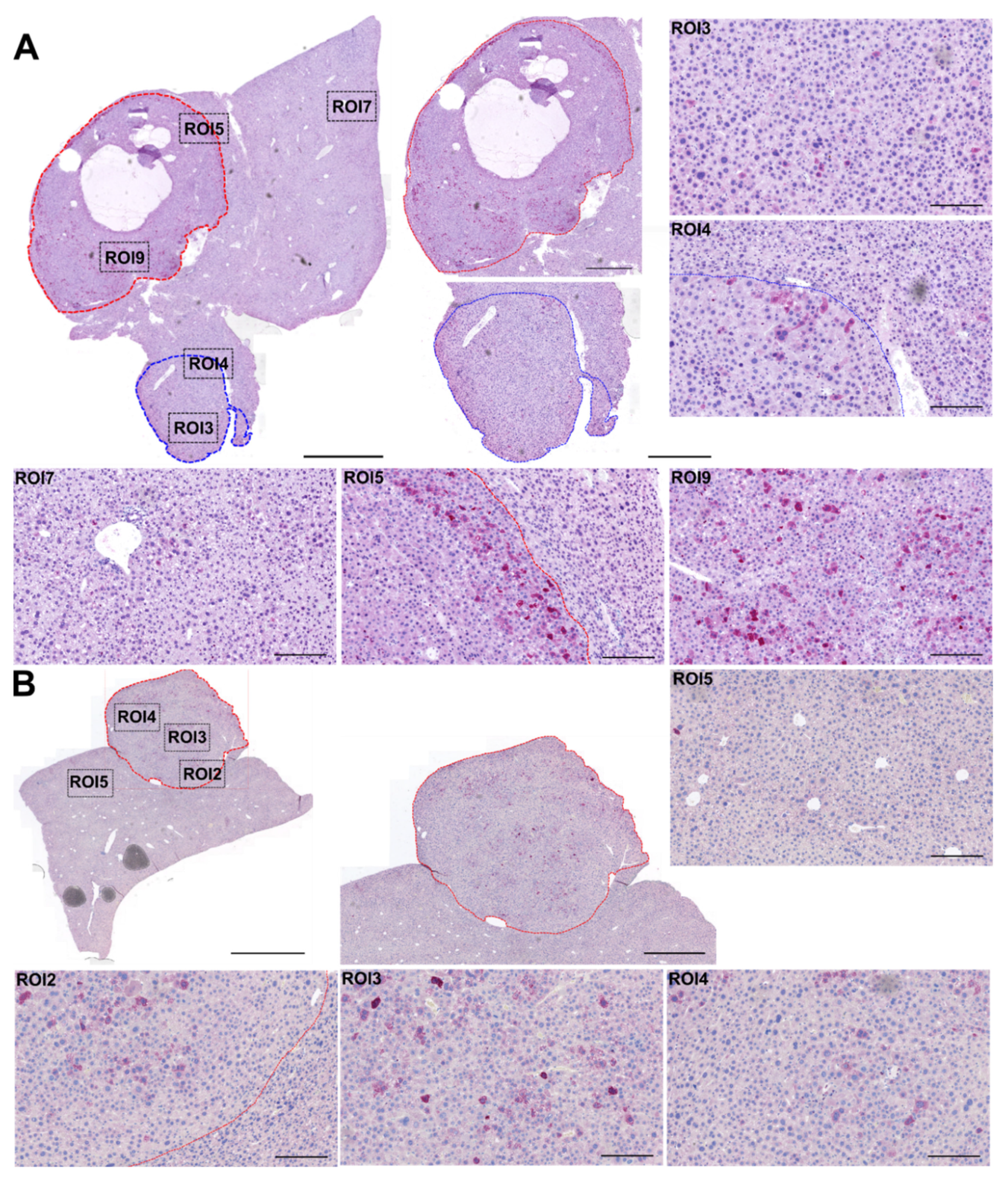
3.2. AAV2 Vector Transduces All Layers, including the Core of Tumor Lesions
3.3. Confirmation of HCC Tropism in Transgenic LT2/RAS HCC Mice
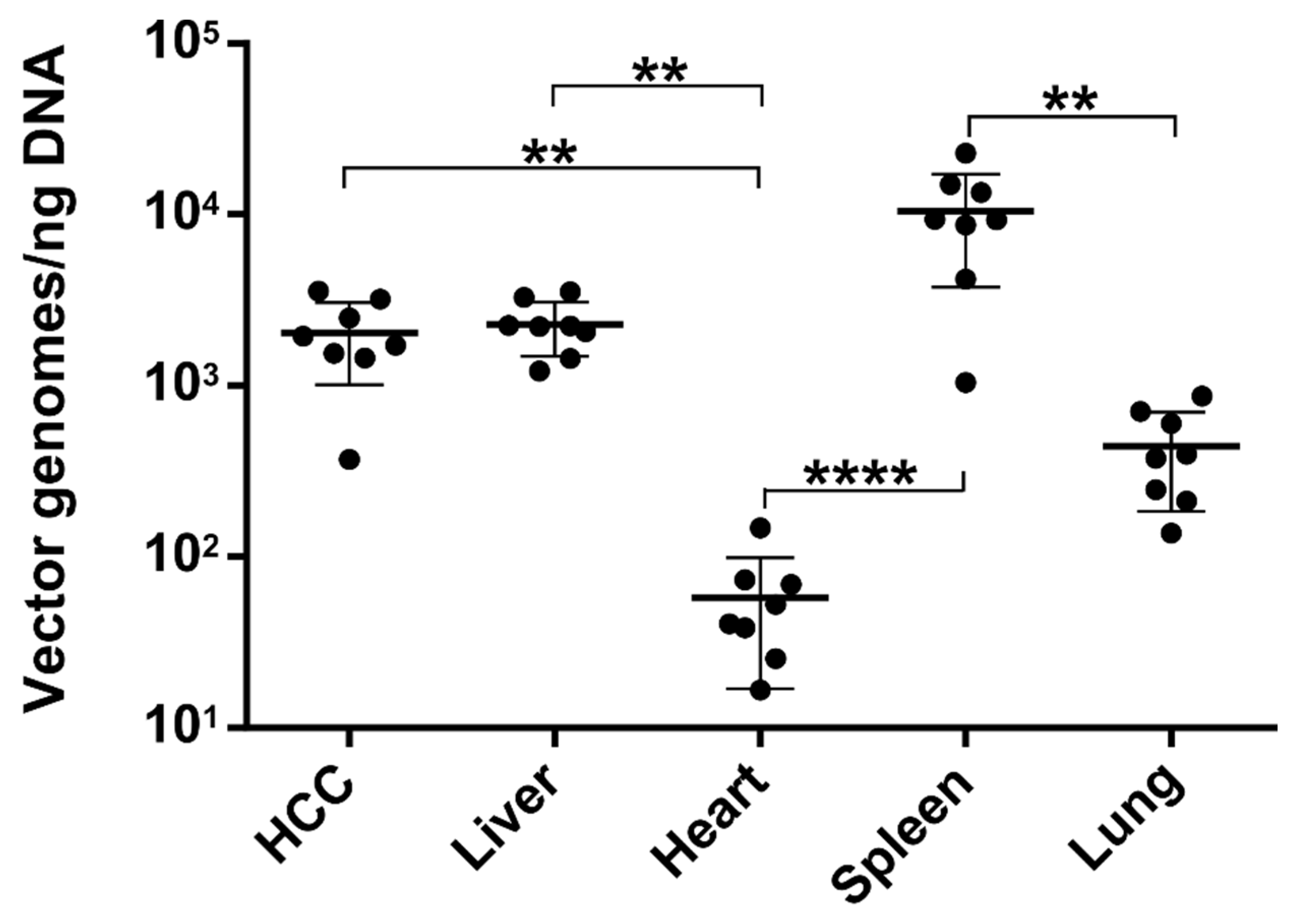
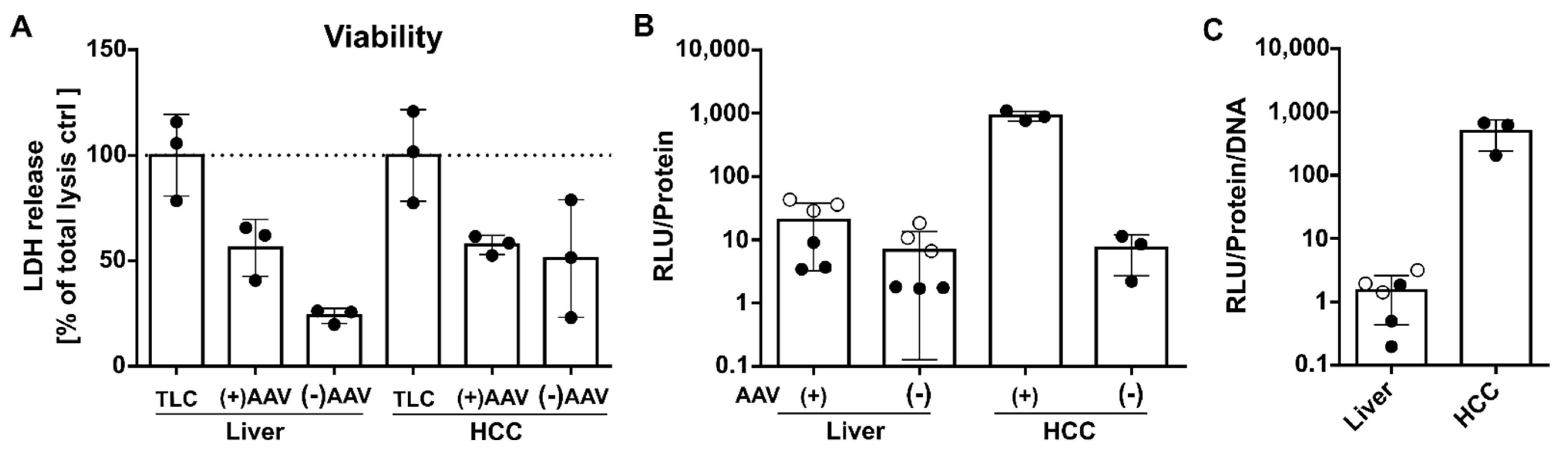
3.4. The Number of Episomal Vector Genomes Differs in HCC and the Liver
3.5. AAV2 Vector Preference for HCC Tissue Observed in Human Precision-Cut Liver Slices
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Valery, P.C.; Laversanne, M.; Clark, P.; Petrick, J.L.; McGlynn, K.A.; Bray, F. Projections of primary liver cancer to 2030 in 30 countries worldwide. Hepatology 2018, 67, 600–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Llovet, J.; Brú, C.; Bruix, J. Prognosis of Hepatocellular Carcinoma: The BCLC Staging Classification. Semin. Liver Dis. 1999, 19, 329–338. [Google Scholar] [CrossRef]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Reghupaty, S.C.; Sarkar, D. Current Status of Gene Therapy in Hepatocellular Carcinoma. Cancers 2019, 11, 1265. [Google Scholar] [CrossRef] [Green Version]
- Hacker, U.T.; Bentler, M.; Kaniowska, D.; Morgan, M.; Büning, H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers 2020, 12, 1889. [Google Scholar] [CrossRef] [PubMed]
- ASGCT. Gene, Cell, and RNA Therapy Landscape. Q2 2021 Quarterly Data Report. 2021. Available online: https://asgct.org/research/news/july-2021/gene-cell-rna-therapy-landscape-q2-2021 (accessed on 18 November 2021).
- Keeler, A.M.; Flotte, T.R. Recombinant Adeno-Associated Virus Gene Therapy in Light of Luxturna (and Zolgensma and Glybera): Where Are We, and How Did We Get Here? Annu. Rev. Virol. 2019, 6, 601–621. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Márquez, E.; Meumann, N.; Büning, H. Adeno-associated virus (AAV) capsid engineering in liver-directed gene therapy. Expert Opin. Biol. Ther. 2021, 21, 749–766. [Google Scholar] [CrossRef]
- Su, H.; Lu, R.; Chang, J.C.; Kan, Y.W. Tissue-specific expression of herpes simplex virus thymidine kinase gene delivered by adeno-associated virus inhibits the growth of human hepatocellular carcinoma in athymic mice. Proc. Natl. Acad. Sci. USA 1997, 94, 13891–13896. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, Y.; Liu, S.; Xu, R.; Zheng, D. Oral adeno-associated virus-sTRAIL gene therapy suppresses human hepatocellular carcinoma growth in mice. Hepatology 2005, 42, 1355–1363. [Google Scholar] [CrossRef]
- Liang, C.-M.; Zhong, C.-P.; Sun, R.-X.; Liu, B.-B.; Huang, C.; Qin, J.; Zhou, S.; Shan, J.; Liu, Y.-K.; Ye, S.-L. Local Expression of Secondary Lymphoid Tissue Chemokine Delivered by Adeno-Associated Virus within the Tumor Bed Stimulates Strong Anti-Liver Tumor Immunity. J. Virol. 2007, 81, 9502–9511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Yang, Z.F.; Gao, Y.; Li, J.C.; Chen, H.X.; Liu, C.C.; Poon, R.T.; Fan, S.T.; Luk, J.; Sze, K.H.; et al. The Kringle 1 Domain of Hepatocyte Growth Factor Has Antiangiogenic and Antitumor Cell Effects on Hepatocellular Carcinoma. Cancer Res. 2008, 68, 404–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Ma, H.; Zhang, J.; Liu, S.; Liu, Y.; Zheng, D. AAV-mediated TRAIL gene expression driven by hTERT promoter suppressed human hepatocellular carcinoma growth in mice. Life Sci. 2008, 82, 1154–1161. [Google Scholar] [CrossRef]
- Tse, L.Y.; Sun, X.; Jiang, H.; Dong, X.; Fung, P.W.; Farzaneh, F.; Xu, R. Adeno-associated virus-mediated expression of kallistatin suppresses local and remote hepatocellular carcinomas. J. Gene Med. 2008, 10, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-H.; Chang, C.-M.; Tang, S.-W.; Pan, W.-Y.; Fang, C.-C.; Chen, Y.; Wu, P.-Y.; Chen, K.-Y.; Ma, H.-I.; Xiao, X.; et al. Differential antitumor effect of interleukin-12 family cytokines on orthotopic hepatocellular carcinoma. J. Gene Med. 2010, 12, 423–434. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, F.; Cai, H.; Wu, Y.; He, G.; Tan, W.-S. The efficacy of combination therapy using adeno-associated virus-TRAIL targeting to telomerase activity and cisplatin in a mice model of hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1827–1837. [Google Scholar] [CrossRef]
- Chang, C.-M.; Lo, C.-H.; Shih, Y.-M.; Chen, Y.; Wu, P.-Y.; Tsuneyama, K.; Roffler, S.R.; Tao, M.-H. Treatment of Hepatocellular Carcinoma with Adeno-Associated Virus Encoding Interleukin-15 Superagonist. Hum. Gene Ther. 2010, 21, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Vanrell, L.; Di Scala, M.; Blanco, L.; Otano, I.; Gil-Farina, I.; Baldim, V.; Paneda, A.; Berraondo, P.; Beattie, S.G.; Chtarto, A.; et al. Development of a Liver-specific Tet-On Inducible System for AAV Vectors and Its Application in the Treatment of Liver Cancer. Mol. Ther. 2011, 19, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Ling, C.; Dai, Y.; Lu, Y.; Glushakova, L.G.; Gee, S.W.Y.; McGoogan, K.; Aslanidi, G.V.; Park, M.; Stacpoole, P.W.; et al. Development of optimized AAV3 serotype vectors: Mechanism of high-efficiency transduction of human liver cancer cells. Gene Ther. 2012, 19, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, S.-H.; Wang, B.; Kota, J.; Yu, J.; Costinean, S.; Kutay, H.; Yu, L.; Bai, S.; La Perle, K.; Chivukula, R.R.; et al. Essential metabolic, anti-inflammatory, and anti-tumorigenic functions of miR-122 in liver. J. Clin. Invest. 2012, 122, 2871–2883. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Wang, Y.; Zhang, Y.; Ejjigani, A.; Yin, Z.; Lu, Y.; Wang, L.; Wang, M.; Li, J.; Hu, Z.; et al. Selective In Vivo Targeting of Human Liver Tumors by Optimized AAV3 Vectors in a Murine Xenograft Model. Hum. Gene Ther. 2014, 25, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.-J.; Fang, C.-C.; Yen, C.-H.; Hsu, S.-M.; Wang, C.-K.; Huang, S.-F.; Liang, Y.-C.; Lin, Y.-Y.; Chu, Y.-T.; Chen, Y.-M.A. Niemann-Pick type C2 protein regulates liver cancer progressionviamodulating ERK1/2 pathway: Clinicopathological correlations and therapeutical implications. Int. J. Cancer 2015, 137, 1341–1351. [Google Scholar] [CrossRef]
- Khan, N.; Bammidi, S.; Chattopadhyay, S.; Jayandharan, G.R. Combination Suicide Gene Delivery with an Adeno-Associated Virus Vector Encoding Inducible Caspase-9 and a Chemical Inducer of Dimerization Is Effective in a Xenotransplantation Model of Hepatocellular Carcinoma. Bioconjug. Chem. 2019, 30, 1754–1762. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Dane, A.P.; Chu, K.; Zhang, Y.; Cunningham, S.C.; Wilson, E.M.; Nygaard, S.; Grompe, M.; Alexander, I.; Kay, M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature 2014, 506, 382–386. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Lu, Y.; Kalsi, J.K.; Jayandharan, G.R.; Li, B.; Ma, W.; Cheng, B.; Gee, S.W.; McGoogan, K.E.; Govindasamy, L.; et al. Human Hepatocyte Growth Factor Receptor Is a Cellular Coreceptor for Adeno-Associated Virus Serotype 3. Hum. Gene Ther. 2010, 21, 1741–1747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.-Q.; Wu, M.-C.; Cong, W.-M. Gene expression of hepatocyte growth factor and its receptor in HCC and nontumorous liver tissues. World J. Gastroenterol. 1999, 5, 119–121. [Google Scholar] [CrossRef]
- Li, S.; Ling, C.; Zhong, L.; Li, M.; Su, Q.; He, R.; Tang, Q.; Greiner, D.L.; Shultz, L.D.; Brehm, M.; et al. Efficient and Targeted Transduction of Nonhuman Primate Liver With Systemically Delivered Optimized AAV3B Vectors. Mol. Ther. 2015, 23, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Bell, P.; Somanathan, S.; Wang, Q.; He, Z.; Yu, H.; McMenamin, D.; Goode, T.; Calcedo, R.; Wilson, J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015, 23, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Vercauteren, K.; Hoffman, B.; Zolotukhin, I.; Keeler, G.D.; Xiao, J.W.; Basner-Tschakarjan, E.; High, K.; Ertl, H.C.; Rice, C.M.; Srivastava, A.; et al. Superior In vivo Transduction of Human Hepatocytes Using Engineered AAV3 Capsid. Mol. Ther. 2016, 24, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Li, J.; Samulski, R.J. Production of High-Titer Recombinant Adeno-Associated Virus Vectors in the Absence of Helper Adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar] [CrossRef] [Green Version]
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef] [Green Version]
- Murakami, H.; Sanderson, N.D.; Nagy, P.; Marino, P.; Merlino, G.; Thorgeirsson, S.S. Transgenic mouse model for synergistic effects of nuclear oncogenes and growth factors in tumorigenesis: Interaction of c-myc and transforming growth factor alpha in hepatic oncogenesis. Cancer Res. 1993, 53, 1719–1723. [Google Scholar] [PubMed]
- Haupenthal, J.; Bihrer, V.; Korkusuz, H.; Kollmar, O.; Schmithals, C.; Kriener, S.; Engels, K.; Pleli, T.; Benz, A.; Canamero, M.; et al. Reduced Efficacy of the Plk1 Inhibitor BI 2536 on the Progression of Hepatocellular Carcinoma due to Low Intratumoral Drug Levels. Neoplasia 2012, 14, IN6–IN10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmithals, C.; Köberle, V.; Korkusuz, H.; Pleli, T.; Kakoschky, B.; Augusto, E.A.; Ibrahim, A.A.; Arencibia, J.M.; Vafaizadeh, V.; Groner, B.; et al. Improving Drug Penetrability with iRGD Leverages the Therapeutic Response to Sorafenib and Doxorubicin in Hepatocellular Carcinoma. Cancer Res. 2015, 75, 3147–3154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, L.; Balakrishnan, A.; Huskey, N.; Jones, K.D.; Jodari, M.; Ng, R.; Song, G.; Riordan, J.; Anderton, B.; Cheung, S.-T.; et al. MicroRNA-494 within an oncogenic microRNA megacluster regulates G 1 /S transition in liver tumorigenesis through suppression of mutated in colorectal cancer. Hepatology 2014, 59, 202–215. [Google Scholar] [CrossRef] [Green Version]
- Komoll, R.-M.; Hu, Q.; Olarewaju, O.; von Döhlen, L.; Yuan, Q.; Xie, Y.; Tsay, H.-C.; Daon, J.; Qin, R.; Manns, M.P.; et al. MicroRNA-342-3p is a potent tumour suppressor in hepatocellular carcinoma. J. Hepatol. 2021, 74, 122–134. [Google Scholar] [CrossRef]
- Granitzny, A.; Knebel, J.; Schaudien, D.; Braun, A.; Steinberg, P.; Dasenbrock, C.; Hansen, T. Maintenance of high quality rat precision cut liver slices during culture to study hepatotoxic responses: Acetaminophen as a model compound. Toxicol. Vitr. 2017, 42, 200–213. [Google Scholar] [CrossRef]
- Manthorpe, M.; Cornefert-Jensen, F.; Hartikka, J.; Felgner, J.; Rundell, A.; Margalith, M.; Dwarki, V. Gene Therapy by Intramuscular Injection of Plasmid DNA: Studies on Firefly Luciferase Gene Expression in Mice. Hum. Gene Ther. 1993, 4, 419–431. [Google Scholar] [CrossRef]
- Tschanz, S.; Burri, P.; Weibel, E. A simple tool for stereological assessment of digital images: The STEPanizer. J. Microsc. 2011, 243, 47–59. [Google Scholar] [CrossRef]
- Rossi, A.; Dupaty, L.; Aillot, L.; Zhang, L.; Gallien, C.; Hallek, M.; Odenthal, M.; Adriouch, S.; Salvetti, A.; Büning, H. Vector uncoating limits adeno-associated viral vector-mediated transduction of human dendritic cells and vector immunogenicity. Sci. Rep. 2019, 9, 3631. [Google Scholar] [CrossRef]
- Perabo, L.; Goldnau, D.; White, K.; Endell, J.; Boucas, J.; Humme, S.; Work, L.M.; Janicki, H.; Hallek, M.; Baker, A.H.; et al. Heparan Sulfate Proteoglycan Binding Properties of Adeno-Associated Virus Retargeting Mutants and Consequences for Their In Vivo Tropism. J. Virol. 2006, 80, 7265–7269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, E.; Samulski, R.J. Adeno-Associated Virus at 50: A Golden Anniversary of Discovery, Research, and Gene Therapy Success—A Personal Perspective. Hum. Gene Ther. 2015, 26, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, G.-P.; Alvira, M.R.; Wang, L.; Calcedo, R.; Johnston, J.; Wilson, J.M. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 11854–11859. [Google Scholar] [CrossRef] [Green Version]
- Grimm, D.; Zhou, S.; Nakai, H.; Thomas, C.E.; Storm, T.A.; Fuess, S.; Matsushita, T.; Allen, J.; Surosky, R.; Lochrie, M.; et al. Preclinical in vivo evaluation of pseudotyped adeno-associated virus vectors for liver gene therapy. Blood 2003, 102, 2412–2419. [Google Scholar] [CrossRef]
- De, B.P.; Heguy, A.; Hackett, N.R.; Ferris, B.; Leopold, P.L.; Lee, J.; Pierre, L.; Gao, G.; Wilson, J.M.; Crystal, R.G. High Levels of Persistent Expression of α1-Antitrypsin Mediated by the Nonhuman Primate Serotype rh.10 Adeno-associated Virus Despite Preexisting Immunity to Common Human Adeno-associated Viruses. Mol. Ther. 2006, 13, 67–76. [Google Scholar] [CrossRef]
- Summerford, C.; Samulski, R.J. Membrane-Associated Heparan Sulfate Proteoglycan Is a Receptor for Adeno-Associated Virus Type 2 Virions. J. Virol. 1998, 72, 1438–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Large, E.E.; Silveria, M.A.; Zane, G.M.; Weerakoon, O.; Chapman, M.S. Adeno-Associated Virus (AAV) Gene Delivery: Dissecting Molecular Interactions upon Cell Entry. Viruses 2021, 13, 1336. [Google Scholar] [CrossRef]
- Cabanes-Creus, M.; Hallwirth, C.V.; Westhaus, A.; Ng, B.H.; Liao, S.H.; Zhu, E.; Navarro, R.G.; Baltazar, G.; Drouyer, M.; Scott, S.; et al. Restoring the natural tropism of AAV2 vectors for human liver. Sci. Transl. Med. 2020, 12, eaba3312. [Google Scholar] [CrossRef]
- Cabanes-Creus, M.; Westhaus, A.; Navarro, R.G.; Baltazar, G.; Zhu, E.; Amaya, A.K.; Liao, S.H.; Scott, S.; Sallard, E.; Dilworth, K.L.; et al. Attenuation of Heparan Sulfate Proteoglycan Binding Enhances In Vivo Transduction of Human Primary Hepatocytes with AAV2. Mol. Ther.-Methods Clin. Dev. 2020, 17, 1139–1154. [Google Scholar] [CrossRef]
- Geoghegan, J.C.; Keiser, N.W.; Okulist, A.; Martins, I.; Wilson, M.S.; Davidson, B.L. Chondroitin Sulfate is the Primary Receptor for a Peptide-Modified AAV That Targets Brain Vascular Endothelium In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e202. [Google Scholar] [CrossRef]
- Kashiwakura, Y.; Tamayose, K.; Iwabuchi, K.; Hirai, Y.; Shimada, T.; Matsumoto, K.; Nakamura, T.; Watanabe, M.; Oshimi, K.; Daida, H. Hepatocyte Growth Factor Receptor Is a Coreceptor for Adeno-Associated Virus Type 2 Infection. J. Virol. 2005, 79, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Summerford, C.; Samulski, R.J. AAVR: A Multi-Serotype Receptor for AAV. Mol. Ther. 2016, 24, 663–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudek, A.M.; Zabaleta, N.; Zinn, E.; Pillay, S.; Zengel, J.; Porter, C.; Franceschini, J.S.; Estelien, R.; Carette, J.E.; Zhou, G.L.; et al. GPR108 Is a Highly Conserved AAV Entry Factor. Mol. Ther. 2020, 28, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Storm, T.A.; Huang, Z.; Kay, M.A. Rapid Uncoating of Vector Genomes Is the Key toEfficient Liver Transduction with Pseudotyped Adeno-Associated Virus Vectors. J. Virol. 2004, 78, 3110–3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, C.H.; Nakai, H.; Thompson, A.R.; Storm, T.A.; Chiu, W.; Snyder, R.O.; Kay, M.A. Nonrandom Transduction of Recombinant Adeno-Associated Virus Vectors in Mouse Hepatocytes In Vivo: Cell Cycling Does Not Influence Hepatocyte Transduction. J. Virol. 2000, 74, 3793–3803. [Google Scholar] [CrossRef] [Green Version]
- Snyder, R.O.; Miao, C.H.; Patijn, G.A.; Spratt, S.K.; Danos, O.; Nagy, D.; Gown, A.M.; Winther, B.; Meuse, L.; Cohen, L.K.; et al. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat. Genet. 1997, 16, 270–276. [Google Scholar] [CrossRef]
- Ponnazhagan, S.; Mukherjee, P.; Yoder, M.C.; Wang, X.-S.; Zhou, S.Z.; Kaplan, J.; Wadsworth, S.; Srivastava, A. Adeno-associated virus 2-mediated gene transfer in vivo: Organ-tropism and expression of transduced sequences in mice. Gene 1997, 190, 203–210. [Google Scholar] [CrossRef]
- Xiao, W.; Berta, S.C.; Lu, M.M.; Moscioni, A.D.; Tazelaar, J.; Wilson, J.M. Adeno-Associated Virus as a Vector for Liver-Directed Gene Therapy. J. Virol. 1998, 72, 10222–10226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, C.H.; Snyder, R.O.; Schowalter, D.B.; Patijn, G.A.; Donahue, B.; Winther, B.; Kay, M.A. The kinetics of rAAV integration in the liver. Nat. Genet. 1998, 19, 13–15. [Google Scholar] [CrossRef]
- Chen, S.J.; Tazelaar, J.; Moscioni, A.D.; Wilson, J.M. In Vivo Selection of Hepatocytes Transduced with Adeno-Associated Viral Vectors. Mol. Ther. 2000, 1, 414–422. [Google Scholar] [CrossRef]
- Nakai, H.; Thomas, C.E.; Storm, T.A.; Fuess, S.; Powell, S.; Wright, J.F.; Kay, M.A. A Limited Number of Transducible Hepatocytes Restricts a Wide-Range Linear Vector Dose Response in Recombinant Adeno-Associated Virus-Mediated Liver Transduction. J. Virol. 2002, 76, 11343–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-L.; Jensen, R.L.; Schnepp, B.C.; Connell, M.J.; Shell, R.; Sferra, T.J.; Bartlett, J.S.; Clark, K.R.; Johnson, P.R. Molecular Characterization of Adeno-Associated Viruses Infecting Children. J. Virol. 2005, 79, 14781–14792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hösel, M.; Huber, A.; Bohlen, S.; Lucifora, J.; Ronzitti, G.; Puzzo, F.; Boisgerault, F.; Hacker, U.T.; Kwanten, W.J.; Klöting, N.; et al. Autophagy determines efficiency of liver-directed gene therapy with adeno-associated viral vectors. Hepatology 2017, 66, 252–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hösel, M.; Lucifora, J.; Michler, T.; Holz, G.; Gruffaz, M.; Stahnke, S.; Zoulim, F.; Durantel, D.; Heikenwalder, M.; Nierhoff, D.; et al. Hepatitis B virus infection enhances susceptibility toward adeno-associated viral vector transductionin vitroandin vivo. Hepatology 2014, 59, 2110–2120. [Google Scholar] [CrossRef]
- Fang, C.-C.; Wu, C.-F.; Liao, Y.-J.; Huang, S.-F.; Chen, M.; Chen, Y.-M.A. AAV serotype 8-mediated liver specific GNMT expression delays progression of hepatocellular carcinoma and prevents carbon tetrachloride-induced liver damage. Sci. Rep. 2018, 8, 13802. [Google Scholar] [CrossRef]
- Lemken, M.-L.; Wybranietz, W.A.; Schmidt, U.; Graepler, F.; Armeanu, S.; Bitzer, M.; Lauer, U.M. Liver-directed gene expression employing synthetic transcriptional control units. World J. Gastroenterol. 2005, 11, 5295–5302. [Google Scholar] [CrossRef]
- Dhungel, B.; Ramlogan-Steel, C.A.; Steel, J.C. Synergistic and independent action of endogenous microRNAs 122a and 199a for post-transcriptional liver detargeting of gene vectors. Sci. Rep. 2018, 8, 15539. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meumann, N.; Schmithals, C.; Elenschneider, L.; Hansen, T.; Balakrishnan, A.; Hu, Q.; Hook, S.; Schmitz, J.; Bräsen, J.H.; Franke, A.-C.; et al. Hepatocellular Carcinoma Is a Natural Target for Adeno-Associated Virus (AAV) 2 Vectors. Cancers 2022, 14, 427. https://doi.org/10.3390/cancers14020427
Meumann N, Schmithals C, Elenschneider L, Hansen T, Balakrishnan A, Hu Q, Hook S, Schmitz J, Bräsen JH, Franke A-C, et al. Hepatocellular Carcinoma Is a Natural Target for Adeno-Associated Virus (AAV) 2 Vectors. Cancers. 2022; 14(2):427. https://doi.org/10.3390/cancers14020427
Chicago/Turabian StyleMeumann, Nadja, Christian Schmithals, Leroy Elenschneider, Tanja Hansen, Asha Balakrishnan, Qingluan Hu, Sebastian Hook, Jessica Schmitz, Jan Hinrich Bräsen, Ann-Christin Franke, and et al. 2022. "Hepatocellular Carcinoma Is a Natural Target for Adeno-Associated Virus (AAV) 2 Vectors" Cancers 14, no. 2: 427. https://doi.org/10.3390/cancers14020427
APA StyleMeumann, N., Schmithals, C., Elenschneider, L., Hansen, T., Balakrishnan, A., Hu, Q., Hook, S., Schmitz, J., Bräsen, J. H., Franke, A.-C., Olarewaju, O., Brandenberger, C., Talbot, S. R., Fangmann, J., Hacker, U. T., Odenthal, M., Ott, M., Piiper, A., & Büning, H. (2022). Hepatocellular Carcinoma Is a Natural Target for Adeno-Associated Virus (AAV) 2 Vectors. Cancers, 14(2), 427. https://doi.org/10.3390/cancers14020427