
EGFR Exon 20 Insertion Mutations in Sinonasal Squamous Cell Carcinoma



Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of the Epidermal Growth Factor Receptor (EGFR)
3. EGFR Mutations in Cancer
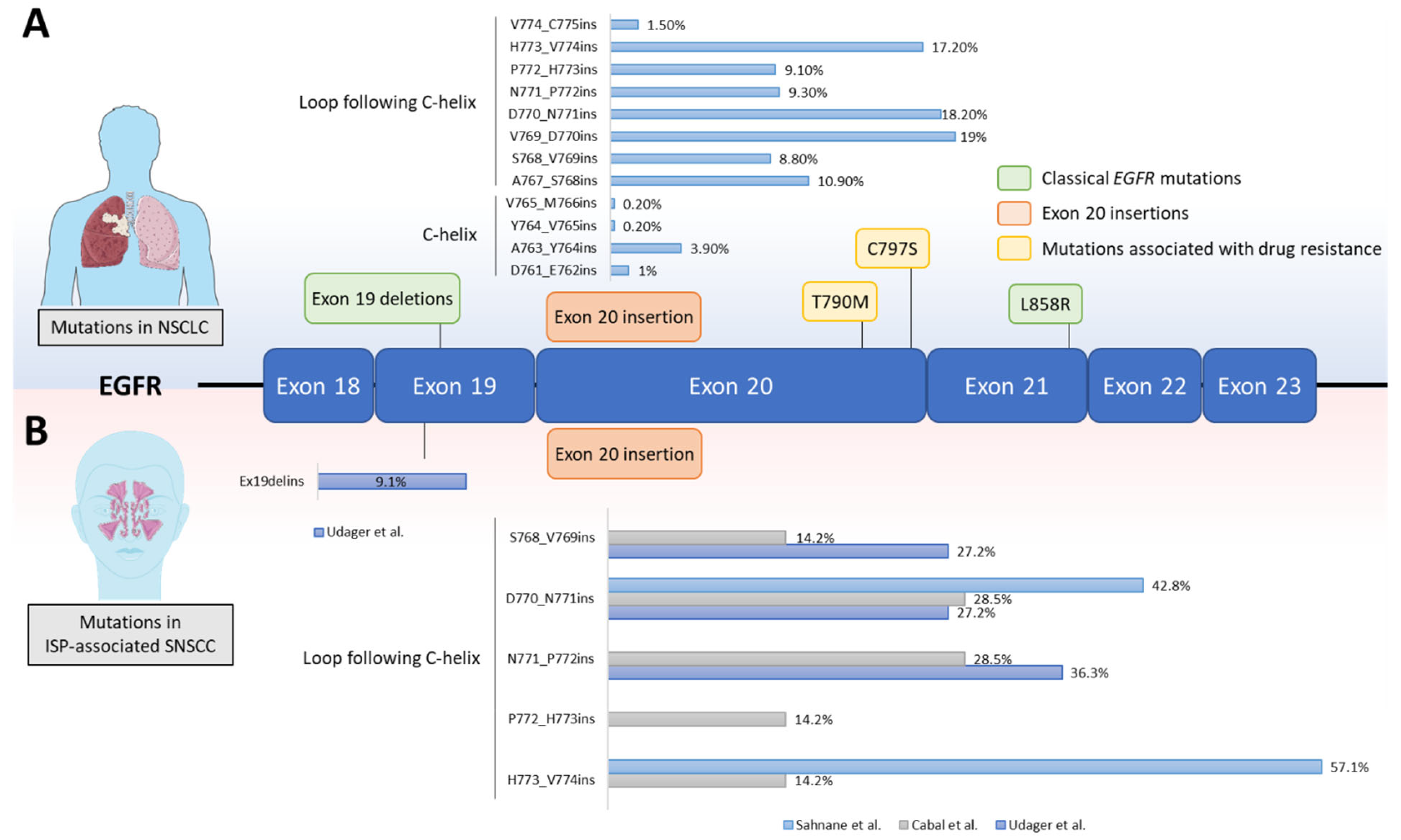
4. EGFR Mutations in SNSCC
5. EGFR Ex20ins Targeted Therapies
6. Lessons from Lung Cancer That Could Be Applied to the Treatment of SNSCC
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Franchi, A. Pathology of Sinonasal Tumors and Tumor-like Lesions, 1st ed.; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Ansa, B.; Goodman, M.; Ward, K.; Kono, S.A.; Owonikoko, T.K.; Higgins, K.; Beitler, J.J.; Grist, W.; Wadsworth, T.; El-Deiry, M.; et al. Paranasal sinus squamous cell carcinoma incidence and survival based on Surveillance, Epidemiology, and End Results data, 1973 to 2009. Cancer 2013, 119, 2602–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghvi, S.; Khan, M.N.; Patel, N.R.; Bs, S.Y.; Baredes, S.; Eloy, J.A. Epidemiology of sinonasal squamous cell carcinoma: A comprehensive analysis of 4994 patients. Laryngoscope 2013, 124, 76–83. [Google Scholar] [CrossRef] [PubMed]
- EI-Naggar, A.K.; Chan, J.K.C.; Grandis, J.R.; Takata, T.; Slootweg, P.J. WHO Classification of Head and Neck Tumours. In WHO Classification of Head and Neck Tumors 2017; International Agency for Research on Cancer (IARC): Lyon, France, 2017. [Google Scholar]
- Llorente, J.L.; López, F.; Suárez, C.; Hermsen, M.A. Sinonasal carcinoma: Clinical, pathological, genetic and therapeutic advances. Nat. Rev. Clin. Oncol. 2014, 11, 460–472. [Google Scholar] [CrossRef]
- Vazquez, A.; Khan, M.N.; Blake, D.M.; Patel, T.D.; Baredes, S.; Eloy, J.A. Sinonasal squamous cell carcinoma and the prognostic implications of its histologic variants: A population-based study. Int. Forum Allergy Rhinol. 2014, 5, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; Ba, P.M.D.; Svider, P.F.; Liu, J.K.; Baredes, S.; Eloy, J.A. Sinonasal malignancies: A population-based analysis of site-specific incidence and survival. Laryngoscope 2015, 125, 2491–2497. [Google Scholar] [CrossRef]
- Haerle, S.K.; Gullane, P.J.; Witterick, I.J.; Zweifel, C.; Gentili, F. Sinonasal carcinomas: Epidemiology, pathology, and management. Neurosurg. Clin. N. Am. 2013, 24, 39–49. [Google Scholar] [CrossRef]
- Cantu, G.; Solero, C.L.; Miceli, R.; Mattana, F.; Riccio, S.; Colombo, S.; Pompilio, M.; Lombardo, G.; Formillo, P.; Quattrone, P. Anterior craniofacial resection for malignant paranasal tumors: A monoinstitutional experience of 366 cases. Head Neck 2011, 34, 78–87. [Google Scholar] [CrossRef]
- Kim, S.A.; Chung, Y.-S.; Lee, B.J. Recurrence patterns of sinonasal cancers after a 5-year disease-free period. Laryngoscope 2019, 129, 2451–2457. [Google Scholar] [CrossRef]
- Hayes, R.; Kardaun, J.; De Bruyn, A. Tobacco use and sinonasal cancer: A case-control study. Br. J. Cancer 1987, 56, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, J.A.; Guo, T.W.; Smith, D.F.; Wang, H.; Ogawa, T.; Pai, S.I.; Westra, W.H. Human Papillomavirus-related Carcinomas of the Sinonasal Tract. Am. J. Surg. Pathol. 2013, 37, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Elgart, K.; Faden, D.L. Sinonasal Squamous Cell Carcinoma: Etiology, Pathogenesis, and the Role of Human Papilloma Virus. Curr. Otorhinolaryngol. Rep. 2020, 8, 111–119. [Google Scholar] [CrossRef]
- Bishop, J.A.; Andreasen, S.; Hang, J.; Bullock, M.J.; Chen, T.Y.; Franchi, A.; Garcia, J.J.; Gnepp, D.R.; Gomez-Fernandez, C.R.; Ihrler, S.; et al. HPV-related Multiphenotypic Sinonasal Carcinoma: An Expanded Series of 49 Cases of the Tumor Former-ly Known as HPV-related Carcinoma with Adenoid Cystic Carcinoma-like Features. Am. J. Surg. Pathol. 2017, 41, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.A. OSPs and ESPs and ISPs, Oh My! An Update on Sinonasal (Schneiderian) Papillomas. Head Neck Pathol. 2017, 11, 269–277. [Google Scholar] [CrossRef]
- Re, M.; Gioacchini, F.M.; Bajraktari, A.; Tomasetti, M.; Kaleci, S.; Rubini, C.; Bertini, A.; Magliulo, G.; Pasquini, E. Malignant transformation of sinonasal inverted papilloma and related genetic alterations: A systematic review. Eur. Arch. Oto-Rhino-Laryngol. 2017, 274, 2991–3000. [Google Scholar] [CrossRef]
- Nudell, J.; Chiosea, S.; Thompson, L.D.R. Carcinoma ex-Schneiderian papilloma (malignant transformation): A clinico-pathologic and immunophenotypic study of 20 cases combined with a comprehensive review of the literature. Head Neck Pathol. 2014, 8, 269–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maisch, S.; Mueller, S.; Traxdorf, M.; Weyerer, V.; Stoehr, R.; Iro, H.; Hartmann, A.; Agaimy, A. Sinonasal papillomas: A single centre experience on 137 cases with emphasis on malignant transformation and EGFR/KRAS status in “carcinoma ex papilloma”. Ann. Diagn. Pathol. 2020, 46, 151504. [Google Scholar] [CrossRef]
- Udager, A.M.; Rolland, D.C.M.; McHugh, J.B.; Betz, B.L.; Murga-Zamalloa, C.; Carey, T.; Marentette, L.J.; Hermsen, M.A.; DuRoss, K.E.; Lim, M.; et al. High-Frequency Targetable EGFR Mutations in Sinonasal Squamous Cell Carcinomas Arising from Inverted Sinonasal Papilloma. Cancer Res. 2015, 75, 2600–2606. [Google Scholar] [CrossRef] [Green Version]
- Sahnane, N.; Ottini, G.; Turri-Zanoni, M.; Furlan, D.; Battaglia, P.; Karligkiotis, A.; Albeni, C.; Cerutti, R.; Mura, E.; Chiaravalli, A.M.; et al. Comprehensive analysis of HPV infection, EGFR exon 20 mutations and LINE1 hypomethylation as risk factors for malignant transformation of sinonasal-inverted papilloma to squamous cell carcinoma. Int. J. Cancer 2018, 144, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; McHugh, J.B.; Betz, B.L.; Montone, K.T.; Livolsi, V.A.; Seethala, R.R.; Yakirevich, E.; Iwenofu, O.H.; Perez-Ordonez, B.; DuRoss, K.E.; et al. Activating KRAS mutations are characteristic of oncocytic sinonasal papilloma and associated sinonasal squamous cell carcinoma. J. Pathol. 2016, 239, 394–398. [Google Scholar] [CrossRef]
- Purba, E.; Saita, E.; Maruyama, I. Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The ‘Rota-tion Model’. Cells 2017, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J. Receptor Tyrosine Kinases: Legacy of the First Two Decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miettinen, P.J.; Berger, J.E.; Meneses, J.; Phung, Y.; Pedersen, R.A.; Werb, Z.; Derynck, R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 1995, 376, 337–341. [Google Scholar] [CrossRef]
- Sibilia, M.; Wagner, E.F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 1995, 269, 234–238. [Google Scholar] [CrossRef]
- Huang, P.H.; Xu, A.M.; White, F.M. Oncogenic EGFR Signaling Networks in Glioma. Sci. Signal. 2009, 2, re6. [Google Scholar] [CrossRef] [Green Version]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epi-dermal growth factor receptor. Cell. Signal. 2007, 19, 2013–2023. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Jeuken, J.; Sijben, A.; Alenda, C.; Rijntjes, J.; Dekkers, M.; Boots-Sprenger, S.; McLendon, R.; Wesseling, P. Robust detection of EGFR copy number changes and EGFR variant III: Technical aspects and relevance for glioma diagnostics. Brain Pathol. 2009, 19, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, H.; Zhao, X.; Yuza, Y.; Shimamura, T.; Li, D.; Protopopov, A.; Jung, B.L.; McNamara, K.; Xia, H.; Glatt, K.A.; et al. Epidermal growth factor receptor variant III mutations in lung tumorigenesis and sensitivity to tyrosine kinase in-hibitors. Proc. Natl. Acad. Sci. USA 2006, 103, 7817–7822. [Google Scholar] [CrossRef] [Green Version]
- Chagoya, G.; Kwatra, S.G.; Nanni, C.W.; Roberts, C.; Phillips, S.; Nullmeyergh, S.; Gilmore, S.P.; Spasojevic, I.; Corcoran, D.L.; Young, C.C.; et al. Efficacy of osimertinib against EGFRvIII+ glioblastoma. Oncotarget 2020, 11, 2074–2082. [Google Scholar] [CrossRef]
- Kwatra, M.M. A Rational Approach to Target the Epidermal Growth Factor Receptor in Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 290–296. [Google Scholar] [CrossRef]
- Pakkala, S.; Ramalingam, S.S. Personalized therapy for lung cancer: Striking a moving target. JCI Insight 2018, 3, e120858. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, S.P.; Pietanza, M.C.; Johnson, M.L.; Riely, G.J.; Miller, V.A.; Sima, C.S.; Zakowski, M.F.; Rusch, V.; Ladanyi, M.; Kris, M.G. Incidence of EGFR Exon 19 Deletions and L858R in Tumor Specimens from Men and Cigarette Smokers With Lung Adenocarcinomas. J. Clin. Oncol. 2011, 29, 2066–2070. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, Y.; Mitsudomi, T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: Perspec-tives for individualized treatment strategy. Cancer Sci. 2016, 107, 1179–1186. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Yasuda, H.; Kobayashi, S.; Costa, D. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Arcila, M.E.; Nafa, K.; Chaft, J.; Rekhtman, N.; Lau, C.; Reva, B.; Zakowski, M.F.; Kris, M.; Ladanyi, M. EGFR Exon 20 Insertion Mutations in Lung Adenocarcinomas: Prevalence, Molecular Heterogeneity, and Clinicopathologic Characteristics. Mol. Cancer Ther. 2013, 12, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Lo, P.C.; Nishino, M.; Dahlberg, S.; Lindeman, N.I.; Butaney, M.; Jackman, D.M.; Johnson, B.E.; Jänne, P.A. Natural History and Molecular Characteristics of Lung Cancers Harboring EGFR Exon 20 Insertions. J. Thorac. Oncol. 2013, 8, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Park, E.; Yun, C.-H.; Sng, N.J.; Lucena-Araujo, A.R.; Yeo, W.-L.; Huberman, M.S.; Cohen, D.W.; Nakayama, S.; Ishioka, K.; et al. Structural, Biochemical, and Clinical Characterization of Epidermal Growth Factor Receptor (EGFR) Exon 20 Insertion Mutations in Lung Cancer. Sci. Transl. Med. 2013, 5, 216ra177. [Google Scholar] [CrossRef] [Green Version]
- Vyse, S.; Huang, P.H. Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Beau-Faller, M.; Prim, N.; Ruppert, A.M.; Nanni-Metéllus, I.; Lacave, R.; Lacroix, L.; Escande, F.; Lizard, S.; Pretet, J.L.; Rouquette, I.; et al. Rare EGFR exon 18 and exon 20 mutations in non-small-cell lung cancer on 10 117 patients: A multicen-tre observational study by the French ERMETIC-IFCT network. Ann. Oncol. 2014, 25, 126. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Sima, C.S.; Rodriguez, K.; Busby, N.; Nafa, K.; Ladanyi, M.; Riely, G.J.; Kris, M.; Arcila, M.E.; Yu, H.A. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer 2015, 121, 3212–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.C.-H.; Sequist, L.V.; Geater, S.L.; Tsai, C.-M.; Mok, T.; Schuler, M.; Yamamoto, N.; Yu, C.-J.; Ou, S.-H.I.; Zhou, C.; et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: A combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015, 16, 830–838. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Elamin, Y.Y.; Tan, Z.; Carter, B.W.; Zhang, S.; Liu, S.; Li, S.; Chen, T.; Poteete, A.; Estrada-Bernal, A.; et al. Mechanisms and clinical activity of an EGFR and HER2 exon 20–selective kinase inhibitor in non–small cell lung cancer. Nat. Med. 2018, 24, 638–646. [Google Scholar] [CrossRef]
- Barnes, L. Schneiderian Papillomas and Nonsalivary Glandular Neoplasms of the Head and Neck. Mod. Pathol. 2002, 15, 279–297. [Google Scholar] [CrossRef] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The Mutational Landscape of Head and Neck Squamous Cell Carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2016, 45, D777–D783. [Google Scholar] [CrossRef]
- Cabal, V.; Menendez, M.; Vivanco, B.; Potes-Ares, S.; Riobello, C.; Suarez-Fernandez, L.; Garcia-Marin, R.; Blanco-Lorenzo, V.; Lopez, F.; Alvarez-Marcos, C.; et al. EGFR mutation and HPV infection in sinonasal inverted papilloma and squamous cell carcinoma. Rhinol. J. 2020, 58, 368–376. [Google Scholar] [CrossRef]
- Sasaki, E.; Nishikawa, D.; Hanai, N.; Hasegawa, Y.; Yatabe, Y. Sinonasal squamous cell carcinoma and EGFR mutations: A molecular footprint of a benign lesion. Histopathology 2018, 73, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, H.; Hu, L.; Zhou, J.; Zhai, C.; Wang, D.; Sun, X. EGFR and KRAS mutations in Chinese patients with sinonasal inverted papilloma and oncocytic papilloma. Histopathology 2019, 75, 274–281. [Google Scholar] [CrossRef]
- Udager, A.; McHugh, J.; Goudsmit, C.; Weigelin, H.; Lim, M.; Elenitoba-Johnson, K.; Betz, B.; Carey, T.; Brown, N. Human papillomavirus (HPV) and somatic EGFR mutations are essential, mutually exclusive oncogenic mechanisms for inverted sinonasal papillomas and associated sinonasal squamous cell carcinomas. Ann. Oncol. 2017, 29, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Hongo, T.; Yamamoto, H.; Jiromaru, R.; Nozaki, Y.; Yasumatsu, R.; Hashimoto, K.; Yoneda, R.; Sugii, A.; Taguchi, K.; Masuda, M.; et al. Clinicopathologic Significance of EGFR Mutation and HPV Infection in Sinonasal Squamous Cell Carcinoma. Am. J. Surg. Pathol. 2020, 45, 108–118. [Google Scholar] [CrossRef]
- Cho, J.; Kim, S.; Du, J.; Meyerson, M. Autophosphorylation of the carboxyl-terminal domain is not required for oncogenic transformation by lung-cancer derived EGFR mutants. Int. J. Cancer 2018, 143, 679–685. [Google Scholar] [CrossRef]
- Ferrari, M.; Taboni, S.; Carobbio, A.; Emanuelli, E.; Maroldi, R.; Bossi, P.; Nicolai, P. Sinonasal Squamous Cell Carcinoma, a Narrative Reappraisal of the Current Evidence. Cancers 2021, 13, 2835. [Google Scholar] [CrossRef]
- Farrell, N.F.; Mace, J.C.; Detwiller, K.Y.; Li, R.; Andersen, P.E.; Smith, T.L.; Clayburgh, D.R.; Geltzeiler, M. Predictors of survival outcomes in sinonasal squamous cell carcinoma: An analysis of the National Cancer Database. Int. Forum Allergy Rhinol. 2020, 11, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.S. Sinonasal Squamous Cell Carcinoma: A Review with Emphasis on Emerging Histologic Subtypes and the Role of Human Papillomavirus. Head Neck Pathol. 2016, 10, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.H.; Reh, D.D. Incidence and survival in patients with sinonasal cancer: A historical analysis of population-based data. Head Neck 2011, 34, 877–885. [Google Scholar] [CrossRef]
- Lee, J.J.; Peterson, A.M.; Embry, T.W.; Wamkpah, N.S.; Kallogjeri, D.; Doering, M.M.; Schneider, J.S.; Klatt-Cromwell, C.N.; Pipkorn, P. Survival Outcomes of De Novo vs Inverted Papilloma-Associated Sinonasal Squamous Cell Carcinoma: A Sys-tematic Review and Meta-analysis. JAMA Otolaryngol. Head Neck Surg. 2021, 147, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, D.; Sasaki, E.; Suzuki, H.; Beppu, S.; Sawabe, M.; Terada, H.; Sone, M.; Hanai, N. Treatment outcome and pattern of recurrence of sinonasal squamous cell carcinoma with EGFR-mutation and human papillomavirus. J. Cranio-Maxillofacial Surg. 2021, 49, 494–500. [Google Scholar] [CrossRef]
- Robichaux, J.P.; Le, X.; Vijayan, R.S.K.; Hicks, J.K.; Heeke, S.; Elamin, Y.Y.; Lin, H.Y.; Udagawa, H.; Skoulidis, F.; Tran, H.; et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature 2021, 597, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Eaton, B.G.; Zacharchuk, C.; Freyman, A.; Powell, C.; Ananthakrishnan, R.; et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef]
- Kim, T.; Ock, C.-Y.; Kim, M.; Kim, S.; Keam, B.; Kim, Y.; Kim, D.-W.; Lee, J.-S.; Heo, D. Phase II study of osimertinib in NSCLC patients with EGFR exon 20 insertion mutation: A multicenter trial of the Korean Cancer Study Group (LU17-19). Ann. Oncol. 2019, 30, v628. [Google Scholar] [CrossRef]
- Yang, J.C.-H.; Reckamp, K.L.; Kim, Y.C.; Novello, S.; Smit, E.F.; Lee, J.S.; Su, W.C.; Akerley, W.L.; Blakely, C.M.; Groen, H.J.M.; et al. Efficacy and Safety of Rociletinib Versus Chemotherapy in Patients with EGFR-Mutated NSCLC: The Results of TIGER-3, a Phase 3 Randomized Study. JTO Clin. Res. Rep. 2021, 2, 100114. [Google Scholar] [CrossRef]
- Kosaka, T.; Tanizaki, J.; Paranal, R.M.; Endoh, H.; Lydon, C.; Capelletti, M.; Repellin, C.E.; Choi, J.; Ogino, A.; Calles, A.; et al. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 2017, 77, 2712–2721. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Boss, D.S.; Camidge, D.R.; Britten, C.D.; Engelman, J.A.; Garon, E.B.; Guo, F.; Wong, S.; Liang, J.; Letrent, S.; et al. Phase I Dose-Escalation Study of the Pan-HER Inhibitor, PF299804, in Patients with Advanced Malignant Solid Tumors. Clin. Cancer Res. 2011, 17, 1131–1139. [Google Scholar] [CrossRef] [Green Version]
- Janne, P.A.; Neal, J.W.; Camidge, D.R.; Spira, A.I.; Piotrowska, Z.; Horn, L.; Costa, D.B.; Tsao, A.S.; Patel, J.D.; Gadgeel, S.M.; et al. Antitumor activity of TAK-788 in NSCLC with EGFR exon 20 insertions. J. Clin. Oncol. 2019, 37, 9007. [Google Scholar] [CrossRef]
- Doebele, R.C.; Riely, G.J.; Spira, A.I.; Horn, L.; Piotrowska, Z.; Costa, D.B.; Neal, J.W.; Zhang, S.; Reichmann, W.; Kerstein, D.; et al. First report of safety, PK, and preliminary antitumor activity of the oral EGFR/HER2 exon 20 inhibitor TAK-788 (AP32788) in non–small cell lung cancer (NSCLC). J. Clin. Oncol. 2018, 36, 9015. [Google Scholar] [CrossRef]
- Riely, G.J.; Neal, J.W.; Camidge, D.R.; Spira, A.I.; Piotrowska, Z.; Costa, D.B.; Tsao, A.S.; Patel, J.D.; Gadgeel, S.M.; Bazhenova, L.; et al. Activity and Safety of Mobocertinib (TAK-788) in Previously Treated Non–Small Cell Lung Cancer with EGFR Exon 20 Insertion Mutations from a Phase I/II Trial. Cancer Discov. 2021, 11, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Zhou, C.; Kim, T.M.; Kim, S.-W.; Yang, J.C.-H.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Campelo, M.R.G.; Felip, E.; et al. Mobocertinib (TAK-788) in EGFR exon 20 insertion (ex20ins)+ metastatic NSCLC (mNSCLC): Additional results from platinum-pretreated patients (pts) and EXCLAIM cohort of phase 1/2 study. J. Clin. Oncol. 2021, 39, 9014. [Google Scholar] [CrossRef]
- Zhou, C.; Ramalingam, S.S.; Kim, T.M.; Kim, S.-W.; Yang, J.C.-H.; Riely, G.J.; Mekhail, T.; Nguyen, D.; Campelo, M.R.G.; Felip, E.; et al. Treatment Outcomes and Safety of Mobocertinib in Platinum-Pretreated Patients with EGFR Exon 20 Insertion–Positive Metastatic Non–Small Cell Lung Cancer. JAMA Oncol. 2021, 7, e214761. [Google Scholar] [CrossRef]
- Park, K.; Haura, E.B.; Leighl, N.B.; Mitchell, P.; Shu, C.A.; Girard, N.; Viteri, S.; Han, J.-Y.; Kim, S.-W.; Lee, C.K.; et al. Amivantamab in EGFR Exon 20 Insertion–Mutated Non–Small-Cell Lung Cancer Progressing on Platinum Chemotherapy: Initial Results from the CHRYSALIS Phase I Study. J. Clin. Oncol. 2021, 39, 3391–3402. [Google Scholar] [CrossRef]
- Park, K.; John, T.; Kim, S.-W.; Lee, J.S.; Shu, C.A.; Kim, D.-W.; Ramirez, S.V.; Spira, A.I.; Sabari, J.K.; Han, J.-Y.; et al. Amivantamab (JNJ-61186372), an anti-EGFR-MET bispecific antibody, in patients with EGFR exon 20 insertion (exon20ins)-mutated non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2020, 38, 9512. [Google Scholar] [CrossRef]
- Udagawa, H.; Hasako, S.; Ohashi, A.; Fujioka, R.; Hakozaki, Y.; Shibuya, M.; Abe, N.; Komori, T.; Haruma, T.; Terasaka, M.; et al. TAS6417/CLN-081 is a pan-mutation-selective EGFR tyrosine kinase inhibitor with a broad spectrum of preclinical activity against clinically relevant EGFR mutations. Mol. Cancer Res. 2019, 17, 2233–2243. [Google Scholar] [CrossRef] [Green Version]
- Piotrowska, Z.; Costa, D.; Oxnard, G.; Huberman, M.; Gainor, J.; Lennes, I.; Muzikansky, A.; Shaw, A.; Azzoli, C.; Heist, R.; et al. Activity of the Hsp90 inhibitor luminespib among non-small-cell lung cancers harboring EGFR exon 20 insertions. Ann. Oncol. 2018, 29, 2092–2097. [Google Scholar] [CrossRef]
- Jorge, S.E.; Lucena-Araujo, A.R.; Yasuda, H.; Piotrowska, Z.; Oxnard, G.R.; Rangachari, D.; Huberman, M.S.; Sequist, L.V.; Kobayashi, S.S.; Costa, D.B. EGFR Exon 20 Insertion Mutations Display Sensitivity to Hsp90 Inhibition in Preclinical Models and Lung Adenocarcinomas. Clin. Cancer Res. 2018, 24, 6548–6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piotrowska, Z.; Yu, H.A.; Yang, J.C.-H.; Koczywas, M.; Smit, E.F.; Tan, D.S.-W.; Lee, V.H.-F.; Soo, R.A.; Wrangle, J.M.; Spira, A.I.; et al. Safety and activity of CLN-081 (TAS6417) in NSCLC with EGFR Exon 20 insertion mutations (Ins20). J. Clin. Oncol. 2021, 39, 9077. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, H.; Yasuda, H.; Hamamoto, J.; Masuzawa, K.; Tani, T.; Nukaga, S.; Hirano, T.; Kobayashi, K.; Manabe, T.; Terai, H.; et al. Efficacy of afatinib or osimertinib plus cetuximab combination therapy for non-small-cell lung cancer with EGFR exon 20 insertion mutations. Lung Cancer 2018, 127, 146–152. [Google Scholar] [CrossRef]
- van Veggel, B.; de Langen, A.J.; Hashemi, S.M.S.; Monkhorst, K.; Heideman, D.A.M.; Thunnissen, E.; Smit, E.F. Afatinib and Cetuximab in Four Patients with EGFR Exon 20 Insertion–Positive Advanced NSCLC. J. Thorac. Oncol. 2018, 13, 1222–1226. [Google Scholar] [CrossRef] [PubMed]
- van Veggel, B.; van der Wekken, A.J.; Paats, M.; Hashemi, S.M.; Hendriks, L.; Sikorska, K.; Broek, D.V.D.; Monkhorst, K.; Smit, E.F.; De Langen, J. Interim results of a phase II single arm trial combining afatinib with cetuximab in patients with EGFRex20ins positive NSCLC. J. Clin. Oncol. 2021, 39, 9112. [Google Scholar] [CrossRef]
- Bagchi, A.; Haidar, J.N.; Eastman, S.W.; Vieth, M.; Topper, M.; Iacolina, M.D.; Walker, J.M.; Forest, A.; Shen, Y.; Novosiadly, R.D.; et al. Molecular Basis for Necitumumab Inhibition of EGFR Variants Associated with Acquired Cetuximab Resistance. Mol. Cancer Ther. 2018, 17, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Riess, J.W.; Groshen, S.G.; Reckamp, K.L.; Wakelee, H.A.; Oxnard, G.R.; Padda, S.K.; Koczywas, M.; Piotrowska, Z.; Sholl, L.M.; Paweletz, C.P.; et al. Osimertinib (Osi) plus necitumumab (Neci) in EGFR-mutant NSCLC: An ETCTN California cancer consortium phase I study. J. Clin. Oncol. 2019, 37, 9057. [Google Scholar] [CrossRef]
- Shu, C.A.; Goto, K.; Cho, B.C.; Griesinger, F.; Yang, J.C.-H.; Felip, E.; Xie, J.; Chen, J.; Mahoney, J.; Thayu, M.; et al. CHRYSALIS-2: A phase 1/1b study of lazertinib as monotherapy and in combination with amivantamab in patients with EGFR-mutant NSCLC. J. Clin. Oncol. 2021, 39, TPS9132. [Google Scholar] [CrossRef]
- Pacini, L.; Jenks, A.D.; Vyse, S.; Wilding, C.P.; Arthur, A.; Huang, P.H. Tackling Drug Resistance in EGFR Exon 20 Insertion Mutant Lung Cancer. Pharm. Pers. Med. 2021, 14, 301–317. [Google Scholar] [CrossRef]
- Elamin, Y.; Robichaux, J.; Carter, B.; Altan, M.; Gibbons, D.; Fossella, F.; Simon, G.; Lam, V.; Blumenschein, G.; Tsao, A.; et al. MA09.03 Identification of Mechanisms of Acquired Resistance to Poziotinib in EGFR Exon 20 Mutant Non-Small Cell Lung Cancer (NSCLC). J. Thorac. Oncol. 2019, 14, S282–S283. [Google Scholar] [CrossRef]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R.; et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouitar, J.; Vincent, S.; Brake, R.; Li, S. P2.13-32 TAK-788 is a Novel and Potent Tyrosine Kinase Inhibitor with Selective Activity Against EGFR/HER2. J. Thorac. Oncol. 2018, 13, S811. [Google Scholar] [CrossRef] [Green Version]
- Qin, Q.; Li, X.; Liang, X.; Zeng, L.; Wang, J.; Sun, L.; Zhong, D. CDK4/6 inhibitor palbociclib overcomes acquired resistance to third-generation EGFR inhibitor osimertinib in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET Amplification Leads to Gefitinib Resistance in Lung Cancer by Activating ERBB3 Signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Brown, N.A.; Plouffe, K.R.; Yilmaz, O.; Weindorf, S.C.; Betz, B.L.; Carey, T.E.; Seethala, R.R.; McHugh, J.B.; Tomlins, S.A.; Udager, A.M. TP53 mutations and CDKN2A mutations/deletions are highly recurrent molecular alterations in the ma-lignant progression of sinonasal papillomas. Mod. Pathol. 2020, 34, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Uchi, R.; Jiromaru, R.; Yasumatsu, R.; Yamamoto, H.; Hongo, T.; Manako, T.; Sato, K.; Hashimoto, K.; Wakasaki, T.; Matsuo, M.; et al. Genomic Sequencing of Cancer-related Genes in Sinonasal Squamous Cell Carcinoma and Coexisting Inverted Papilloma. Anticancer. Res. 2021, 41, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Riess, J.W.; Gandara, D.R.; Frampton, G.M.; Madison, R.; Peled, N.; Bufill, J.A.; Dy, G.K.; Ou, S.H.I.; Stephens, P.J.; McPherson, J.D.; et al. Diverse EGFR Exon 20 Insertions and Co-Occurring Molecular Alterations Identified by Comprehensive Ge-nomic Profiling of Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2018, 13, 1560. [Google Scholar] [CrossRef] [Green Version]
- Shien, K.; Toyooka, S.; Yamamoto, H.; Soh, J.; Jida, M.; Thu, K.L.; Maki, Y.; Ichihara, E.; Asano, H.; Tsukuda, K.; et al. Acquired Resistance to EGFR Inhibitors Is Associated with a Manifestation of Stem cell-like Properties in Cancer Cells. Cancer Res. 2015, 73, 3051–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, C.H.; Chen, L.Y.; Lin, Y.C.; Shih, J.Y.; Lin, Y.C.; Tseng, R.Y.; Chiu, A.C.; Yeh, Y.H.; Liu, C.; Lin, Y.T.; et al. Epithelial-mesenchymal transition (EMT) beyond EGFR mutations per se is a common mechanism for ac-quired resistance to EGFR TKI. Oncogene 2019, 38, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.-E.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.-H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
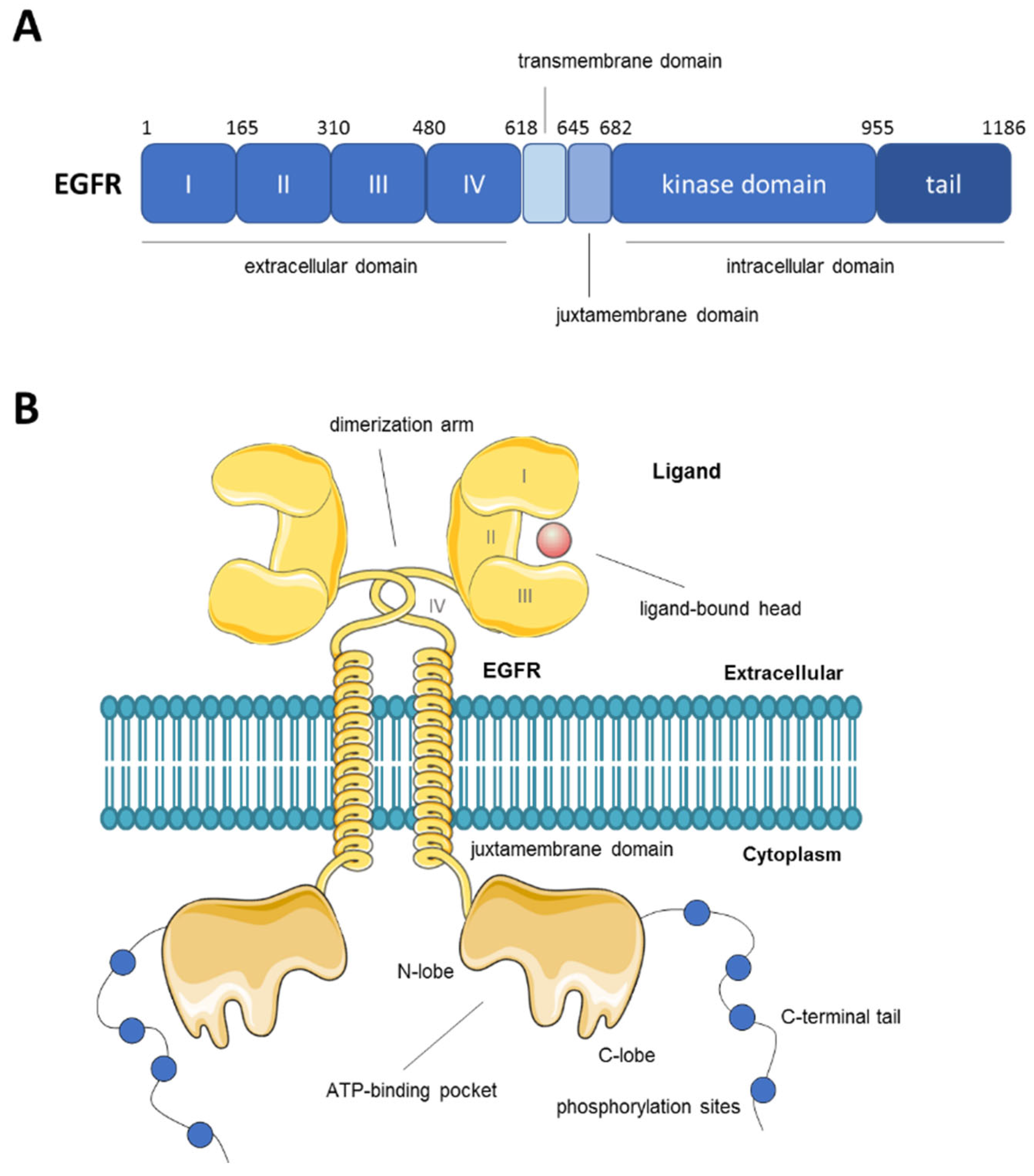

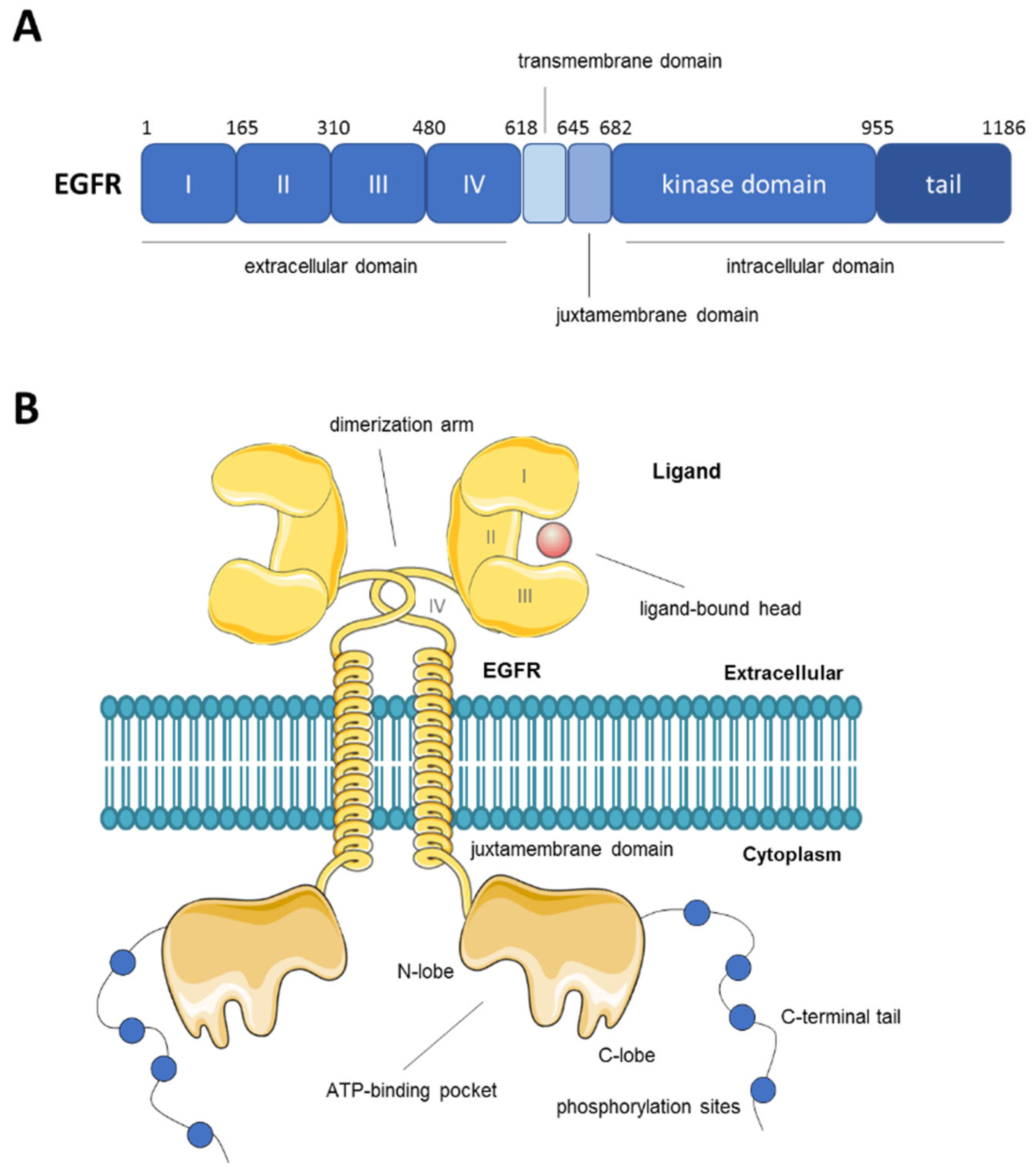{kind=link}
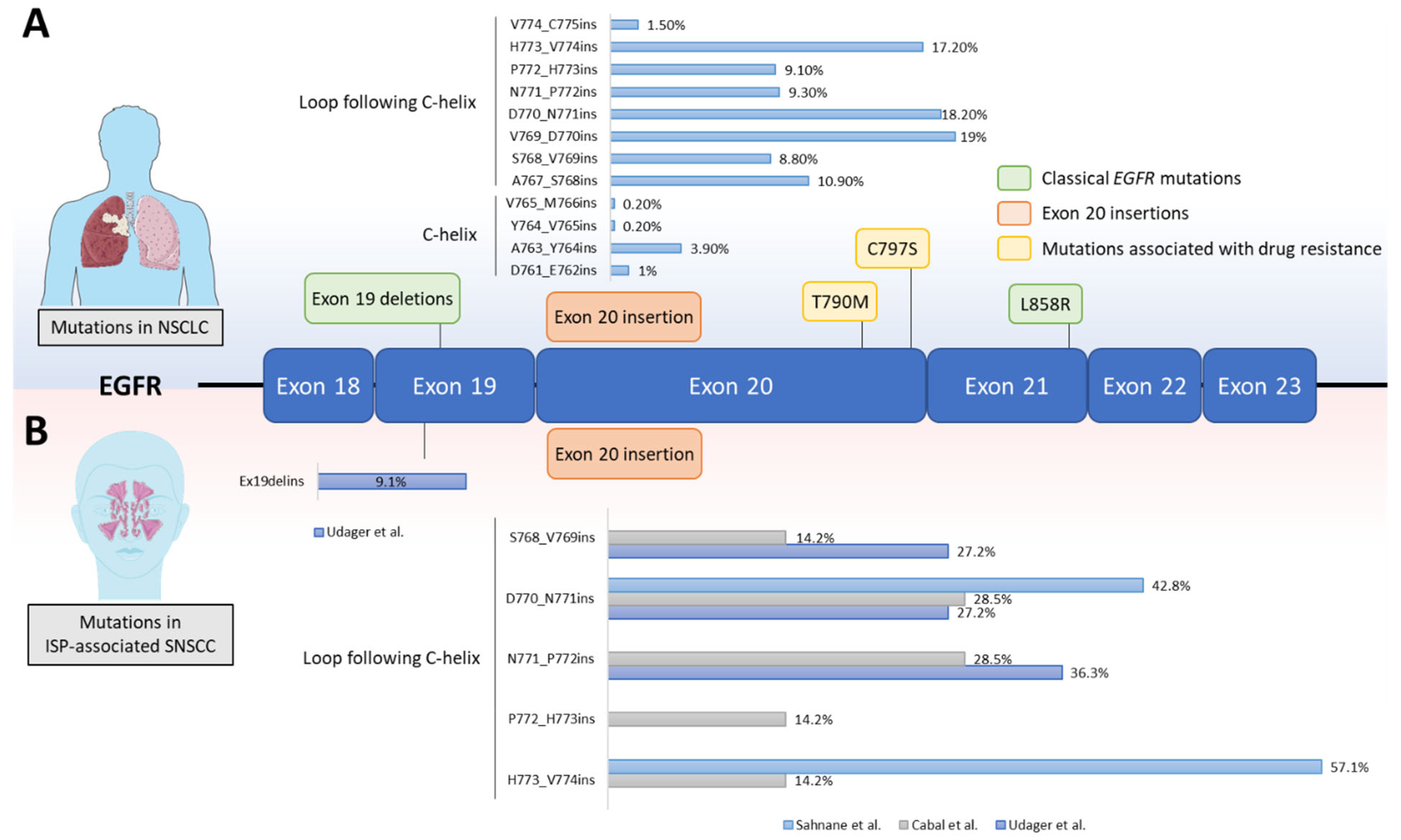{kind=link}
| Mutation | Frequency per Tumour Types | Ref. | ||
|---|---|---|---|---|
| ISP | ISP-Associated SNSCC | De Novo SNSCC | ||
| A767_V769dup | 2% | [19] | ||
| S768_D770dup | 25% | 24% | [19] | |
| V769_D770insGSV | 2% | [19] | ||
| D770_P772dup | 5% | [19] | ||
| D770_N771insGF | 2–17% | [19,58] | ||
| D770_N771insSVD | 5–24% | 1–25% | [20,56,58,60] | |
| D770_N771insGD | 2% | 6% | [19] | |
| D770_N771insSVE | 2% | 6% | [19] | |
| D770_N771insG | 2–6% | 2–7% | 1% | [19,20,56,58,60] |
| D770_N771insGL | 2–9% | 6–7% | [19,56,60] | |
| ]N771delinsGS | 2% | [56] | ||
| N771delinsGF | 9% | 1% | [19,60] | |
| N771delinsGY | 2% | 14% | [19,56] | |
| ]N771delinsSG | 2% | [19] | ||
| N771_P772insV | 2% | 6% | [19,56] | |
| N771_H773dup | 18% | 29% | [19] | |
| N771_P772insPDN | 15% | 1% | [60] | |
| P772_H773dupPDN | 1% | [56] | ||
| P772_H773insDNP | 9% | [58] | ||
| H773_V774insGCRH | 2% | [19] | ||
| H773dup | 11% | [19] | ||
| H773_V774dup | 2% | [19] | ||
| H773_V774insPH | 1% | [20] | ||
| H773_V774insH | 3–4% | 1% | [20,58] | |
| H773_V774insNPH | 3–9% | 2–45% | 1–2% | [20,56,58,60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pacini, L.; Cabal, V.N.; Hermsen, M.A.; Huang, P.H. EGFR Exon 20 Insertion Mutations in Sinonasal Squamous Cell Carcinoma. Cancers 2022, 14, 394. https://doi.org/10.3390/cancers14020394
Pacini L, Cabal VN, Hermsen MA, Huang PH. EGFR Exon 20 Insertion Mutations in Sinonasal Squamous Cell Carcinoma. Cancers. 2022; 14(2):394. https://doi.org/10.3390/cancers14020394
Chicago/Turabian StylePacini, Laura, Virginia N. Cabal, Mario A. Hermsen, and Paul H. Huang. 2022. "EGFR Exon 20 Insertion Mutations in Sinonasal Squamous Cell Carcinoma" Cancers 14, no. 2: 394. https://doi.org/10.3390/cancers14020394
APA StylePacini, L., Cabal, V. N., Hermsen, M. A., & Huang, P. H. (2022). EGFR Exon 20 Insertion Mutations in Sinonasal Squamous Cell Carcinoma. Cancers, 14(2), 394. https://doi.org/10.3390/cancers14020394