
Beyond BRCA1/2: Homologous Recombination Repair Genetic Profile in a Large Cohort of Apulian Ovarian Cancers

 , , , , ,
, , , , ,  , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Cohort Characteristics
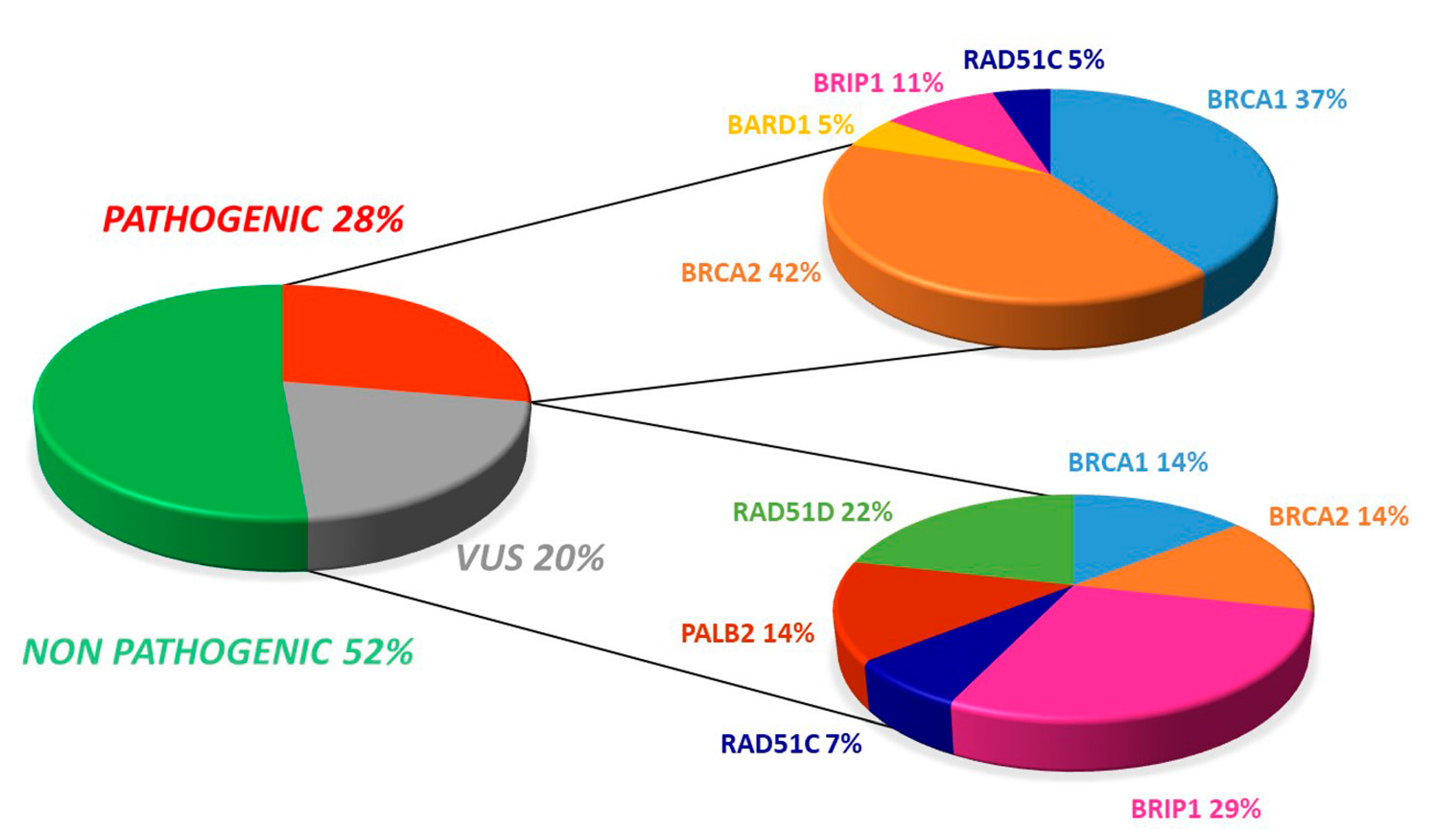
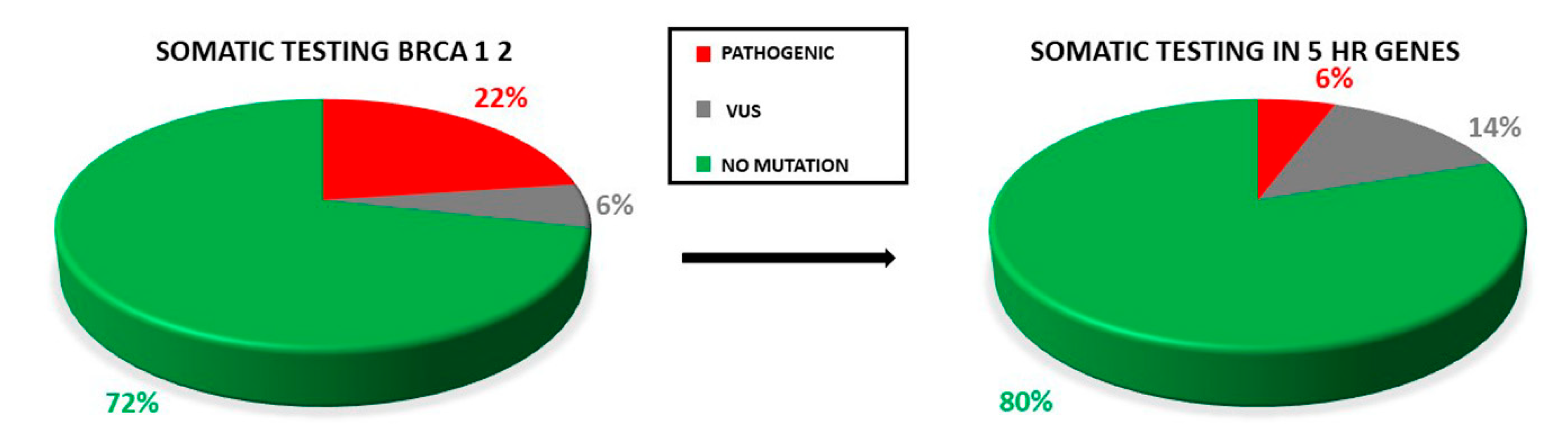
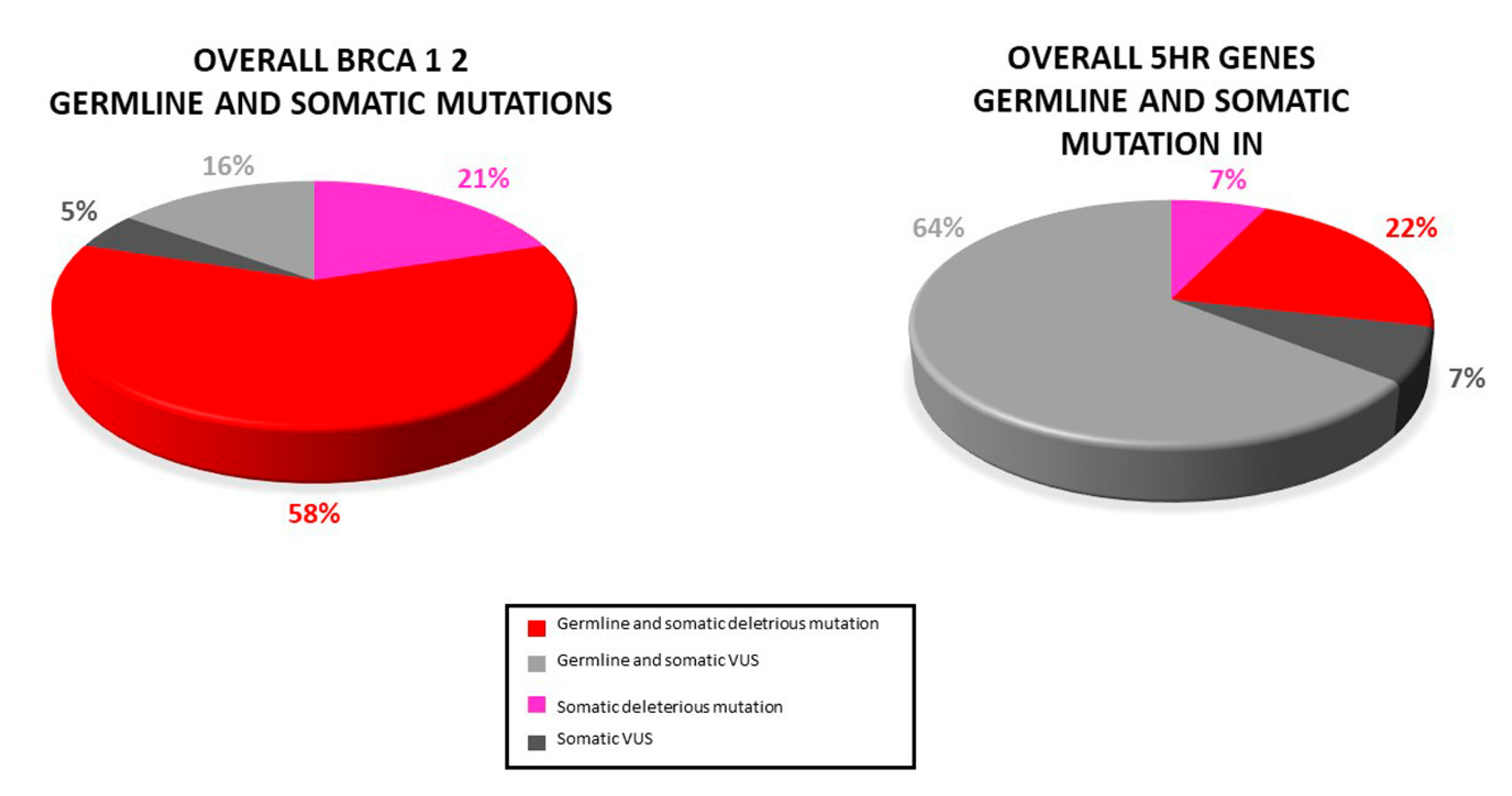
2.2. Overall Mutation Spectrum
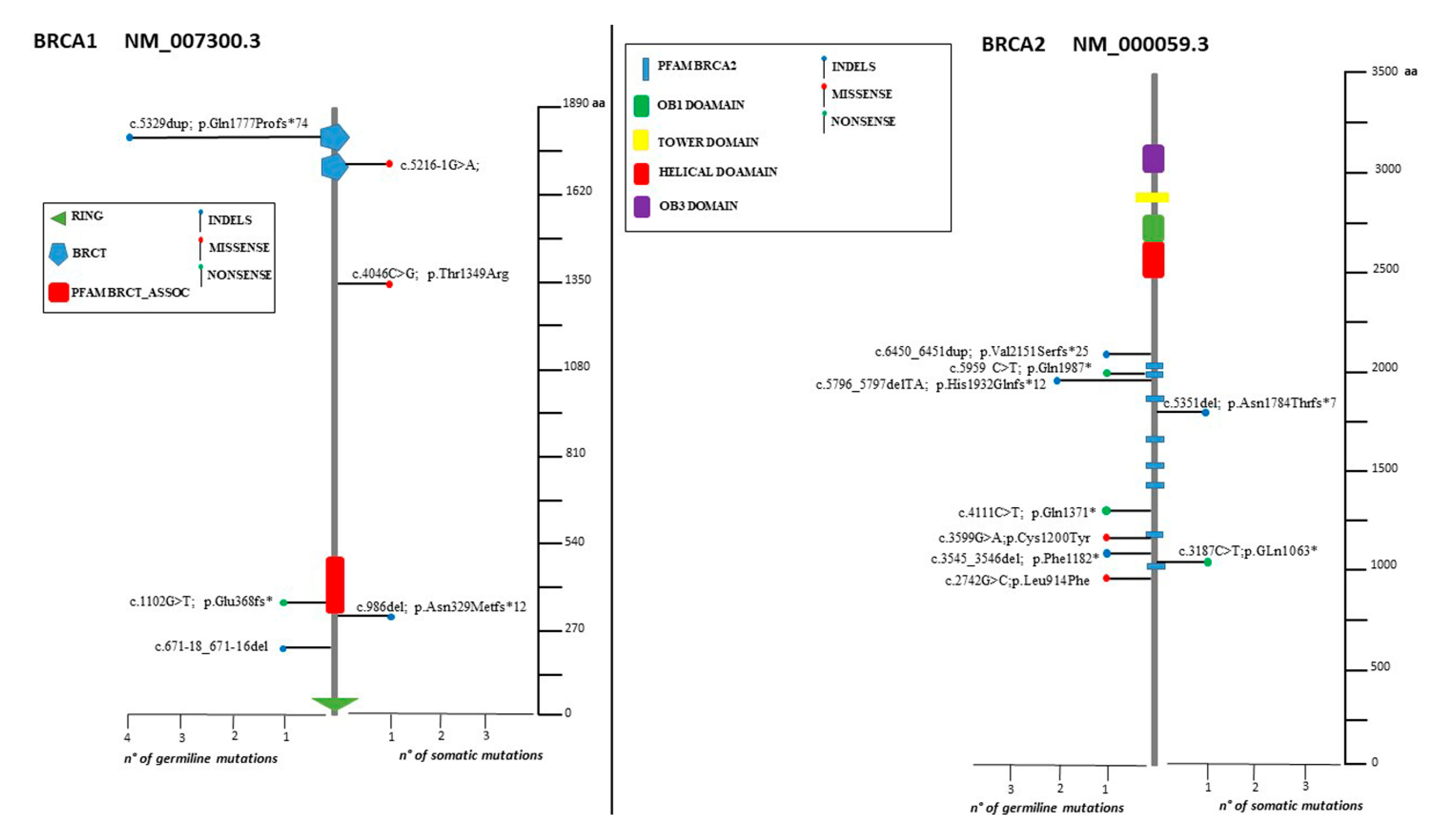
2.3. Distribution of Mutation in BRCA1/2
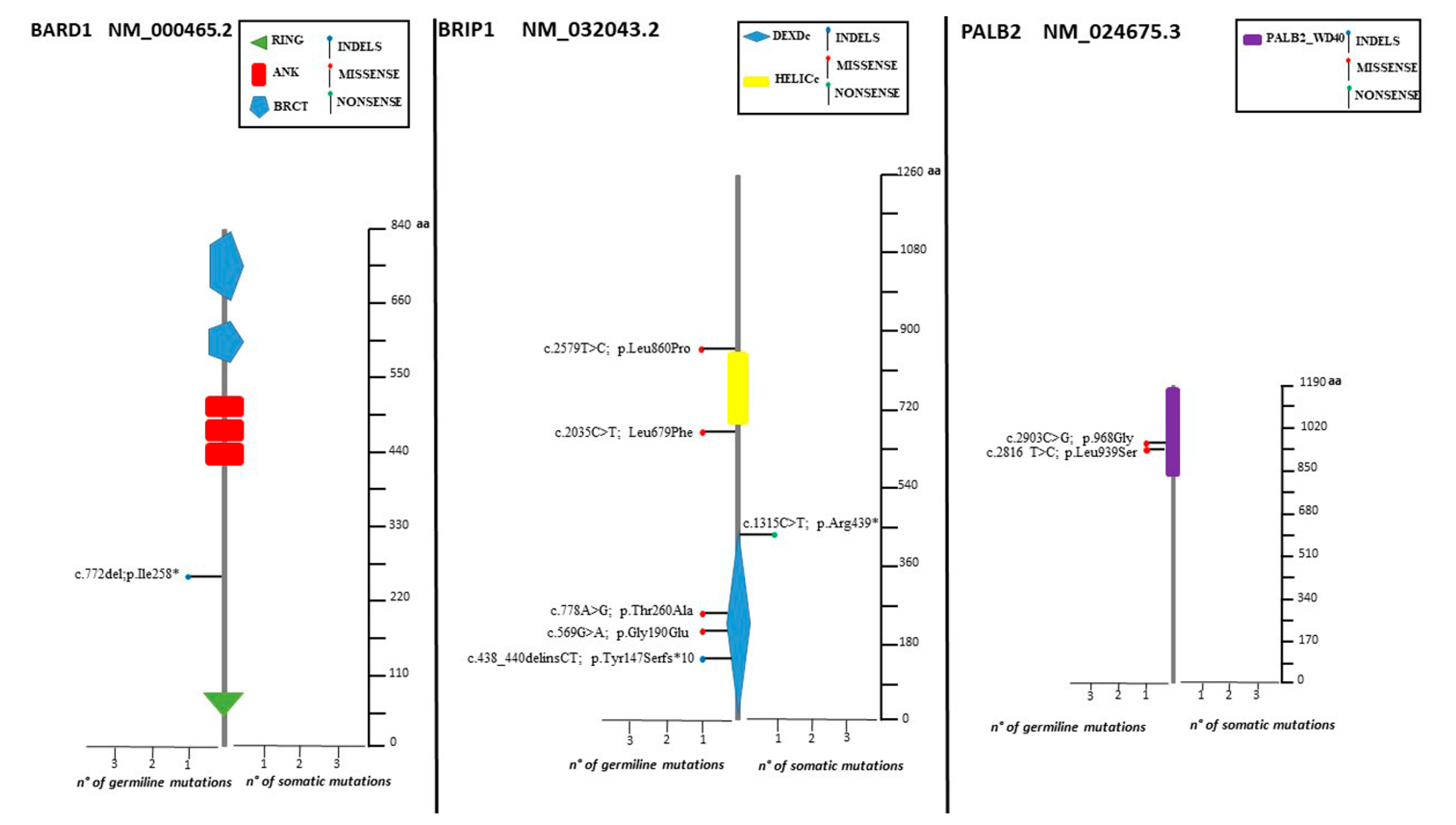
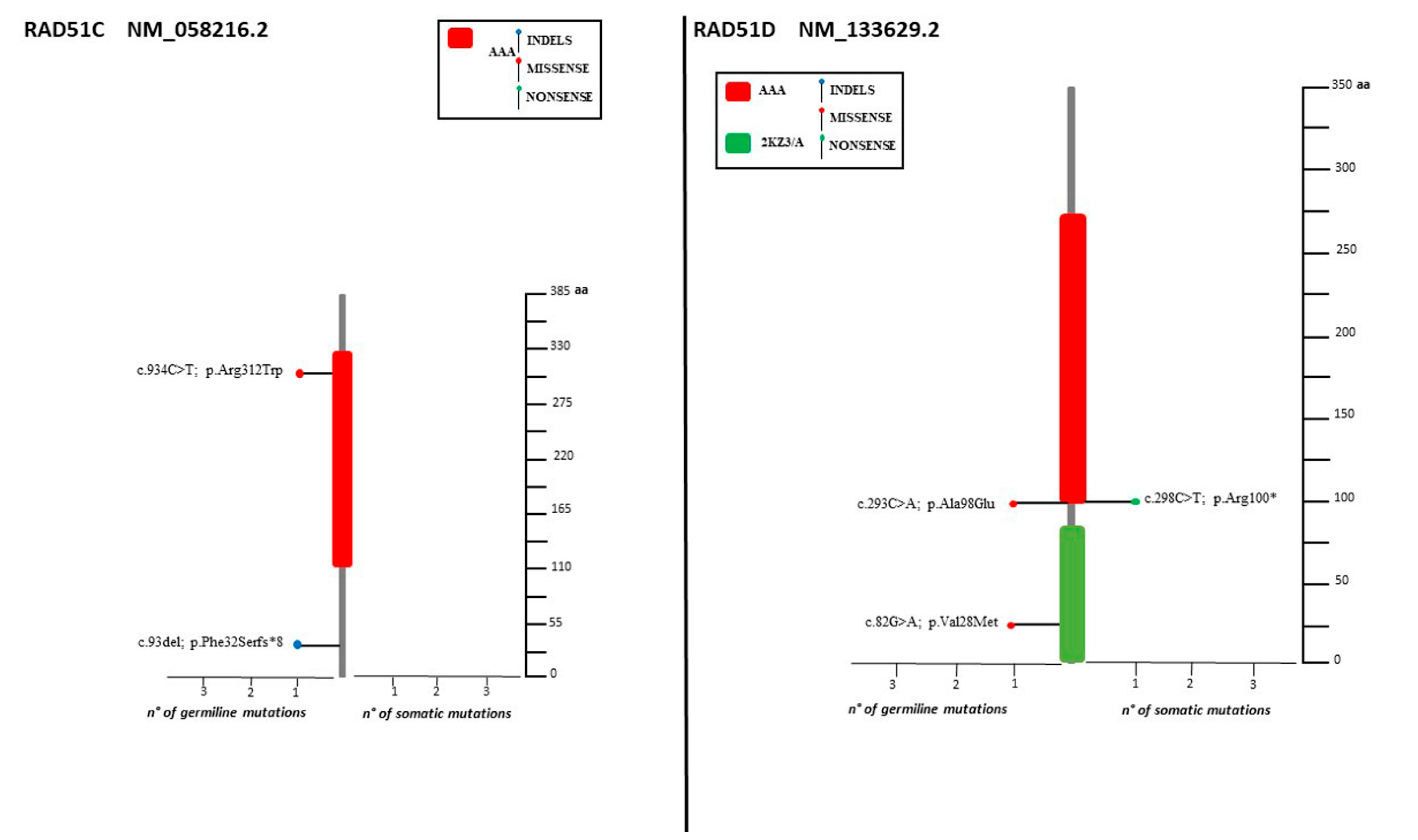
2.4. Distribution of Mutation in Non BRCA Genes
2.5. Germline Mutation Status
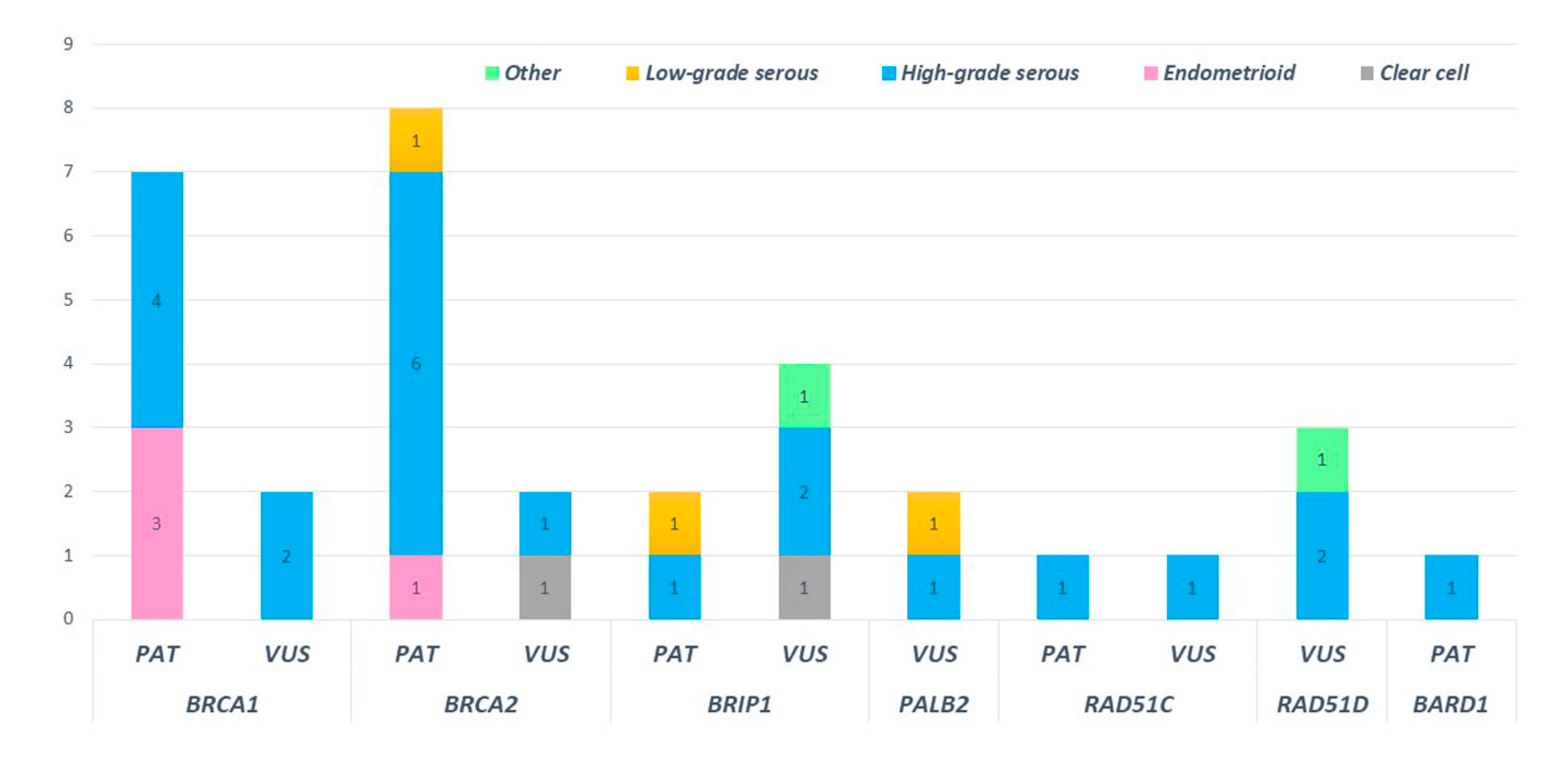
2.6. Mutations and Histology
2.7. BRCA1 and BRCA2 Copy Number Variant
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment
4.2. Next-Generation Sequencing
4.3. Variant Analysis and Data Interpretation
4.4. MLPA
4.5. KRAS and BRAF Mutational Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute Ovarian Cancer-Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/html/ovary.html (accessed on 9 December 2021).
- Prat, J.; D’Angelo, E.; Espinosa, I. Ovarian carcinomas: At least five different diseases with distinct histological features and molecular genetics. Hum. Pathol. 2018, 80, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Mørch, L.S.; Løkkegaard, E.; Andreasen, A.H.; Krüger-Kjær, S.; Lidegaard, Ø. Hormone Therapy and Ovarian Cancer. JAMA 2009, 302, 298–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lheureux, S.; Msc, M.B.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef] [Green Version]
- Manchana, T.; Phoolcharoen, N.; Tantbirojn, P. BRCA mutation in high grade epithelial ovarian cancers. Gynecol. Oncol. Rep. 2019, 29, 102–105. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [Green Version]
- Malander, S.; Rambech, E.; Kristoffersson, U.; Halvarsson, B.; Ridderheim, M.; Borg, A.; Nilbert, M. The contribution of the hereditary nonpolyposis colorectal cancer syndrome to the development of ovarian cancer. Gynecol. Oncol. 2006, 101, 238–243. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-Panel Sequencing and the Prediction of Breast-Cancer Risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [Green Version]
- Creeden, J.F.; Nanavaty, N.S.; Einloth, K.R.; Gillman, C.E.; Stanbery, L.; Hamouda, D.M.; Dworkin, L.; Nemunaitis, J. Homologous recombination proficiency in ovarian and breast cancer patients. BMC Cancer 2021, 21, 1154. [Google Scholar] [CrossRef]
- Toh, M.R.; Ngeow, J. Homologous Recombination Deficiency: Cancer Predispositions and Treatment Implications. Oncologist 2021, 26, 1526–1537. [Google Scholar] [CrossRef]
- Haunschild, C.E.; Tewari, K.S. The current landscape of molecular profiling in the treatment of epithelial ovarian cancer. Gynecol. Oncol. 2021, 160, 333–345. [Google Scholar] [CrossRef]
- Rein, H.L.; Bernstein, K.A.; Baldock, R.A. RAD51 paralog function in replicative DNA damage and tolerance. Curr. Opin. Genet. Dev. 2021, 71, 86–91. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961, Erratum in Lancet 2017, 390, 1948. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Wallbillich, J.J.; Morris, R.T.; Ali-Fehmi, R. Comparing mutation frequencies for homologous recombination genes in uterine serous and high-grade serous ovarian carcinomas: A case for homologous recombination deficiency testing in uterine serous carcinoma. Gynecol. Oncol. 2020, 159, 381–386. [Google Scholar] [CrossRef]
- Yadav, V.; Yadav, G.; Vashisht, M.; Shyam, R. Molecular biomarkers for early detection and prevention of ovarian cancer—A gateway for good prognosis: A narrative review. Int. J. Prev. Med. 2020, 11, 135. [Google Scholar] [CrossRef]
- Amin, N.; Chaabouni, N.; George, A. Genetic testing for epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2020, 65, 125–138. [Google Scholar] [CrossRef]
- Lerner-Ellis, J.; Sopik, V.; Wong, A.; Lázaro, C.; Narod, S.A.; Charames, G.S. Retesting of women who are negative for a BRCA1 and BRCA2 mutation using a 20-gene panel. J. Med. Genet. 2019, 57, 380–384. [Google Scholar] [CrossRef]
- Alenezi, W.M.; Fierheller, C.T.; Recio, N.; Tonin, P.N. Literature Review of BARD1 as a Cancer Predisposing Gene with a Focus on Breast and Ovarian Cancers. Genes 2020, 11, 856. [Google Scholar] [CrossRef]
- Lord, C.; Ashworth, A. BRCAness revisited. Nat. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nishimura, M.; Onoe, T.; Sakai, H.; Urakawa, Y.; Onda, T.; Yaegashi, N. PARP inhibitors for BRCA wild type ovarian cancer; gene alterations, homologous recombination deficiency and combination therapy. Jpn. J. Clin. Oncol. 2019, 49, 703–707. [Google Scholar] [CrossRef]
- Li, H.; Liu, Z.-Y.; Wu, N.; Chen, Y.-C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 1–16. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of Type and Location of BRCA1 and BRCA2 Mutations with Risk of Breast and Ovarian Cancer. JAMA J. Am. Med. Assoc. 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Daly, M.B.; Pal, T.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Goggins, M.; Htton, M.L.L.; et al. Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. NCCN Guidel. 2021, 19, 77–102. [Google Scholar] [CrossRef] [PubMed]
- Figlioli, G.; De Nicolo, A.; Catucci, I.; Manoukian, S.; Peissel, B.; Azzollini, J.; Beltrami, B.; Bonanni, B.; Calvello, M.; Bondavalli, D.; et al. Analysis of Italian BRCA1/2 Pathogenic Variants Identifies a Private Spectrum in the Population from the Bergamo Province in Northern Italy. Cancers 2021, 13, 532. [Google Scholar] [CrossRef] [PubMed]
- Coppa, A.; Buffone, A.; Capalbo, C.; Nicolussi, A.; D’Inzeo, S.; Belardinilli, F.; Colicchia, V.; Petroni, M.; Granato, T.; Midulla, C.; et al. Novel and recurrent BRCA2 mutations in Italian breast/ovarian cancer families widen the ovarian cancer cluster region boundaries to exons 13 and 14. Breast Cancer Res. Treat. 2014, 148, 629–635. [Google Scholar] [CrossRef]
- Santonocito, C.; Rizza, R.; Paris, I.; De Marchis, L.; Paolillo, C.; Tiberi, G.; Scambia, G.; Capoluongo, E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers 2020, 12, 1286. [Google Scholar] [CrossRef]
- Kluz, T.; Jasiewicz, A.; Marczyk, E.; Jach, R.; Jakubowska, A.; Lubiński, J.; Narod, S.A.; Gronwald, J. Frequency of BRCA1 and BRCA2 causative founder variants in ovarian cancer patients in South-East Poland. Hered. Cancer Clin. Pract. 2018, 16, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Cotrim, D.P.; Ribeiro, A.R.G.; Paixão, D.; Soares, D.C.D.Q.; Jbili, R.; Pandolfi, N.C.; Cezana, C.; Mauro, C.D.C.; Mantoan, H.; Bovolim, G.; et al. Prevalence of BRCA1 and BRCA2 pathogenic and likely pathogenic variants in non-selected ovarian carcinoma patients in Brazil. BMC Cancer 2019, 19, 4. [Google Scholar] [CrossRef] [Green Version]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not OnlyBRCA1 and 2 Genes. BioMed Res. Int. 2015, 2015, 341723. [Google Scholar] [CrossRef] [Green Version]
- Moschetta, M.; George, A.; Kaye, S.B.; Banerjee, S. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann. Oncol. 2016, 27, 1449–1455. [Google Scholar] [CrossRef]
- Jorge, S.; McFaddin, A.S.; Doll, K.M.; Pennington, K.P.; Norquist, B.M.; Bennett, R.L.; Pritchard, C.C.; Swisher, E.M. Simultaneous germline and somatic sequencing in ovarian carcinoma: Mutation rate and impact on clinical decision-making. Gynecol. Oncol. 2020, 156, 517–522. [Google Scholar] [CrossRef]
- Lertkhachonsuk, A.-A.; Suprasert, P.; Manchana, T.; Kittisiam, T.; Kantathavorn, N.; Chansoon, T.; Khunamornpong, S.; Pohthipornthawat, N.; Tangjitgamol, S.; Luasiripanthu, T.; et al. Prevalence of Tissue BRCA Gene Mutation in Ovarian, Fallopian Tube, and Primary Peritoneal Cancers: A Multi-Institutional Study. Asian Pac. J. Cancer Prev. 2020, 21, 2381–2388. [Google Scholar] [CrossRef]
- Pavanello, M.; Chan, I.H.Y.; Ariff, A.; Pharoah, P.D.P.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Loizzi, V.; Cicinelli, E.; Santamaria, F.; Murgia, F.; Minicucci, V.; Resta, L.; Resta, N.; Natalicchio, M.I.; Ranieri, G.; Cormio, G. BRCAmut and “founder effect”: A prospective study in a single academic institution. Oncotarget 2018, 9, 22353–22358. [Google Scholar] [CrossRef] [Green Version]
- Moujaber, T.; Balleine, R.L.; Gao, B.; Madsen, I.; Harnett, P.R.; DeFazio, A. New therapeutic opportunities for women with low-grade serous ovarian cancer. Endocr.-Relat. Cancer 2022, 29, R1–R16. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women with Ovarian Cancer. JNCI J. Natl. Cancer Inst. 2015, 107, djv214. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.-K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 1–6. [Google Scholar] [CrossRef]
- Kaldawy, A.; Segev, Y.; Lavie, O.; Auslender, R.; Sopik, V.; Narod, S.A. Low-grade serous ovarian cancer: A review. Gynecol. Oncol. 2016, 143, 433–438. [Google Scholar] [CrossRef]
- Vineyard, M.A.; Daniels, M.S.; Urbauer, D.L.; Deavers, M.T.; Sun, C.C.; Boerwinkle, E.; Bodurka, D.C.; Gershenson, D.M.; Crawford, J.; Lu, K.H. Is low-grade serous ovarian cancer part of the tumor spectrum of Hereditary Breast and Ovarian Cancer? Gynecol. Oncol. 2011, 120, 229–232. [Google Scholar] [CrossRef]
- Kalachand, R.D.; O’Riain, C.; Toomey, S.; Carr, A.; Timms, K.M.; O’Toole, S.; Madden, S.; Bates, M.; O’Leary, J.J.; Gleeson, N.; et al. Prevalence of tumor BRCA1 and BRCA2 dysfunction in unselected patients with ovarian cancer. Obstet. Gynecol. Sci. 2020, 63, 643–654. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Tan, D.S.P. The role of homologous recombination deficiency testing in ovarian cancer and its clinical implications: Do we need it? ESMO Open 2021, 6, 100144. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristic | |
|---|---|
| Age of Diagnosis | # Patients |
| ≤40 | 2 |
| 40–50 | 13 |
| 50–60 | 19 |
| 60–70 | 16 |
| >70 | 19 |
| Tumor Histology | |
| High-grade serous | 38 |
| Low-grade serous | 7 |
| Clear cell | 5 |
| Endometrioid | 10 |
| Mucinous | 1 |
| others | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turchiano, A.; Loconte, D.C.; De Nola, R.; Arezzo, F.; Chiarello, G.; Pantaleo, A.; Iacoviello, M.; Bagnulo, R.; De Luisi, A.; Perrelli, S.; et al. Beyond BRCA1/2: Homologous Recombination Repair Genetic Profile in a Large Cohort of Apulian Ovarian Cancers. Cancers 2022, 14, 365. https://doi.org/10.3390/cancers14020365
Turchiano A, Loconte DC, De Nola R, Arezzo F, Chiarello G, Pantaleo A, Iacoviello M, Bagnulo R, De Luisi A, Perrelli S, et al. Beyond BRCA1/2: Homologous Recombination Repair Genetic Profile in a Large Cohort of Apulian Ovarian Cancers. Cancers. 2022; 14(2):365. https://doi.org/10.3390/cancers14020365
Chicago/Turabian StyleTurchiano, Antonella, Daria Carmela Loconte, Rosalba De Nola, Francesca Arezzo, Giulia Chiarello, Antonino Pantaleo, Matteo Iacoviello, Rosanna Bagnulo, Annunziata De Luisi, Sonia Perrelli, and et al. 2022. "Beyond BRCA1/2: Homologous Recombination Repair Genetic Profile in a Large Cohort of Apulian Ovarian Cancers" Cancers 14, no. 2: 365. https://doi.org/10.3390/cancers14020365
APA StyleTurchiano, A., Loconte, D. C., De Nola, R., Arezzo, F., Chiarello, G., Pantaleo, A., Iacoviello, M., Bagnulo, R., De Luisi, A., Perrelli, S., Martino, S., Ranieri, C., Garganese, A., Stella, A., Forleo, C., Loizzi, V., Marinaccio, M., Cicinelli, E., Cormio, G., & Resta, N. (2022). Beyond BRCA1/2: Homologous Recombination Repair Genetic Profile in a Large Cohort of Apulian Ovarian Cancers. Cancers, 14(2), 365. https://doi.org/10.3390/cancers14020365