1. Introduction
Prostate cancer is the second most common cancer in men worldwide, with 1.3 million new cases registered in 2018, and a leading cause of cancer death [
1]. The transformation of normal epithelial cells into malignant tumor cells takes place in the following two fundamental steps: initiation of uncontrolled proliferation and activation of motile spreading of initially healthy, highly differentiated cells. Both processes speed up the mitotic cell cycle and, once the primary tumor has been established, force the motile machinery of the tumor cells to allow spreading into distant organs. A key role in prostate cancer growth activity is played by the insulin growth factor 1 receptor (IGF-1R) with subsequent phosphorylation of the downstream targets, Akt and mechanistic target of rapamycin (mTOR) [
2,
3]. A clinical trial has demonstrated a close association between circulating IGF-1 and prostate cancer development and progression [
2].
Escape of single cells from the primary tumor and resettling as secondary tumors requires interaction with vascular endothelial cells and extracellular matrix substrates such as collagen or fibronectin. These actions are mediated by adhesion receptors of the integrin family [
4,
5], namely heterodimer cell surface molecules, consisting of α- and β-subunits. Following cell–cell- or cell–matrix-binding, integrin-dependent downstream signaling is established via kinases and signaling adaptors, including focal adhesion kinase (FAK), a cytoplasmic non receptor tyrosine kinase [
6,
7]. A multivariable-adjusted Cox regression carried out on 199,698 prostate cancer patients has revealed a close relationship between integrin expression and tumor colonization of the bone microenvironment [
8].
Prostate cancer growth and invasion is complex and tightly coordinated, controlled by intracellular signaling cascades that have controversially been specified. Caromile et al. observed signaling between β1 integrin and IGF-1R [
9]. Akt has been shown to act on apoptosis in concert with integrin α5, although separate activity of either Akt or integrin α5 on apoptosis has been demonstrated as well [
10]. A humanized monoclonal antibody against integrin αV decreased phosphorylation of Akt in prostate cancer cells [
11]. Others, however, have not seen an Akt-αV integrin-interaction in prostate cancer cells [
12].
Using human androgen-independent prostate cancer cell lines (DU-145, PC3) as well as androgen-sensitive LNCaP and VCaP cells, the present study was designed to evaluate how IGF-1 is involved in integrin driven regulation of tumor cell adhesion and migration and, conversely, whether Akt/mTOR signaling activated by IGF-1 is involved in integrin driven regulation of tumor growth.
2. Materials and Methods
2.1. Cell Lines
The human prostate tumor cell lines DU145, PC3, and LNCaP were obtained from DSMZ (Braunschweig, Germany). VCaP cell lines were from the Department of Urology and Pediatric Urology, Saarland University, Homburg/Saar, Germany. DU145, PC3, and LNCaP tumor cell lines were grown in RPMI 1640 medium (Gibco/Invitrogen, Karlsruhe, Germany) supplemented with either 2 or 10% fetal bovine serum (FBS), 2% HEPES (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) buffer (1 M, pH 7.4), 1% GlutaMAX, and 1% penicillin/streptomycin (all: Gibco/Invitrogen) at 37 °C in a humidified incubator with 5% CO2. VCaP were grown in DMEM medium, supplemented with FBS, 1% penicillin/streptomycin, 2% GlutaMAX and 1% sodium pyruvate (all: Gibco/Invitrogen).
2.2. IGF-1R Detection and IGF-1 Stimulation
Surface expression of the insulin-like growth factor 1 receptor (IGF-1R) was evaluated by flow cytometric analysis using the monoclonal antibody phycoerythrin (PE)-conjugated anti Human CD221 (IGF-1R; clone1H7) or the Alexa Fluor 647-labelled phospho-IGFIR (pIGF-1R, IgG1, pY1131, clone K74-218; both BD Pharmingen). Mouse IgG1-PE (κ Isotype, Clone MOPC-31C) and IgG1-Alexa Fluor 647 (κ Isotype, Clone MOPC-31C; all: BD Bioscience) served as the control isotypes. Receptor surface expression was measured using a FACSCanto (BD Biosciences, Heidelberg, Germany; FL-2H or FL-4H (log) channel histogram analysis; 1 × 104 cells per scan) and expressed as mean fluorescence units (MFU). For stimulation studies, tumor cells were incubated with LONG®R3IGF-1 (IGF-1; Sigma Aldrich, München, Germany) at a concentration of 100 ng/mL for different time periods as indicated.
2.3. Tumor Cell Growth
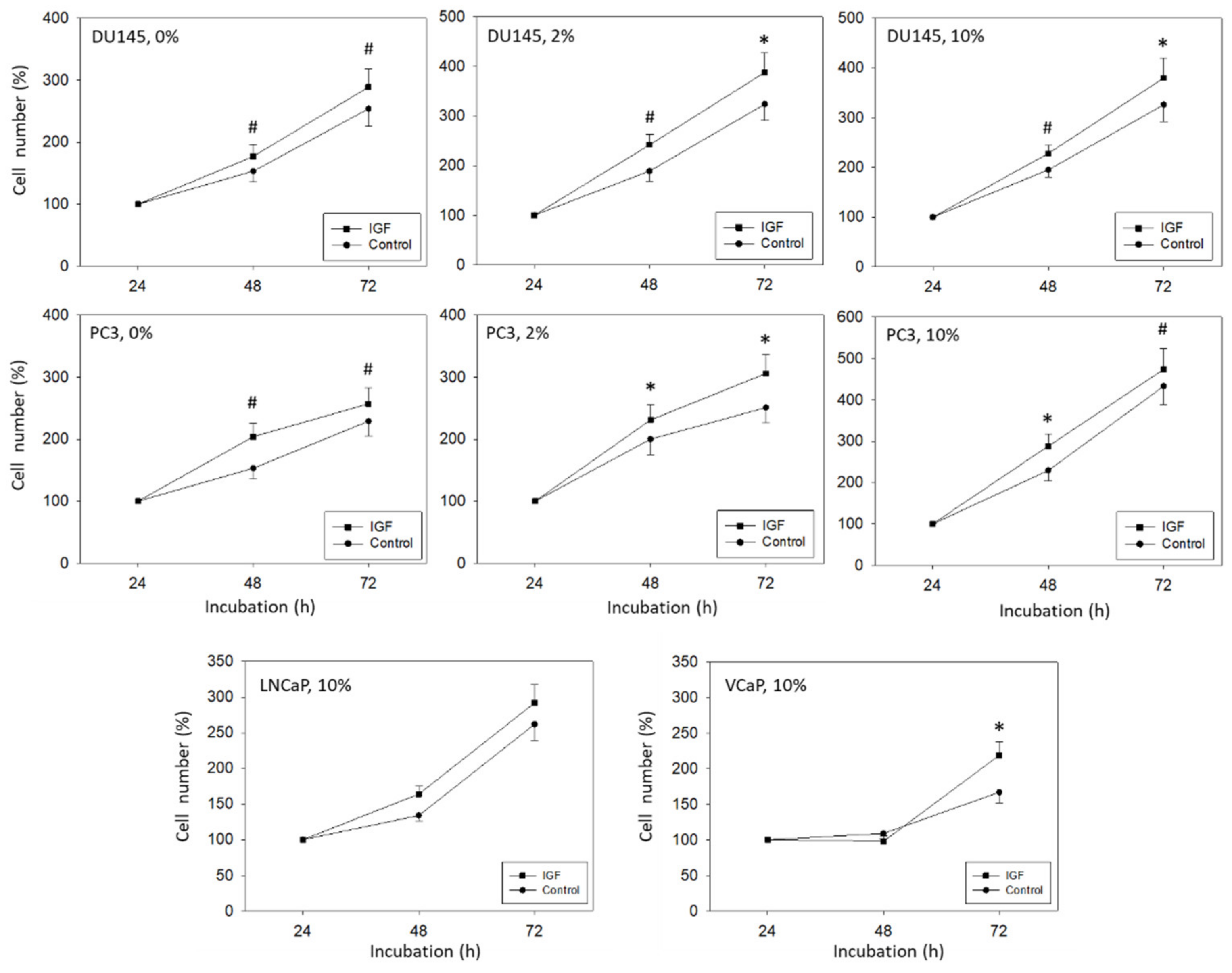
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay (Roche Diagnostics, Penzberg, Germany) was used to evaluate cell growth. For each cell line, 5000 cells were pipetted in triplicate to 96-well plates with medium enriched with soluble IGF-1. Control cells received cell culture medium without IGF-1 and remained untreated. After 24, 48, and 72 h, 10 µL MTT (0.5 mg/mL) was added for an additional 4 h. Cells were then lysed overnight in solubilization buffer (10% SDS in 0.01 M HCl) at 37 °C, 5% CO2. Absorbance at 550 nm was assessed with a microplate enzyme-linked immunosorbent assay (ELISA) reader. To correlate absorbance with cell number, a defined number of cells ranging from 2500 to 160,000 cells/well was added to the microtiter plates (in triplicate). After subtracting background absorbance (cell culture medium alone), results were expressed as mean cell number.
2.4. Tumor Cell Adhesion and Chemotaxis
Tumor cells were incubated with 100 ng/mL IGF-1 for 4 or 24 h (versus unstimulated controls) and were then subjected to adhesion or chemotaxis assays. To analyze tumor cell adhesion, 6-well plates (Falcon Primaria; Corning, Wiesbaden, Germany) were coated with collagen G (matrix mixture consisting of 90% collagen type I and 10% collagen type III; Biochrom, Berlin, Germany) or fibronectin (derived from human plasma; BD Biosciences, Heidelberg, Germany) overnight. Plastic wells were used as background controls. Nonspecific cell adhesion was prevented by washing the plates with PBS and incubating with 1% bovine serum albumin (BSA) in PBS for 60 min. An amount of 2.5 × 105 tumor cells/mL (0.5 mL/well) were then added to each well for 1 h at 37 °C. Following 1 h incubation, non-adherent tumor cells were removed by repeated washing with PBS with Ca2+ and Mg2+. The remaining adherent cells were fixed using 1% glutaraldehyde (Sigma, München, Germany). The mean cellular adhesion rate was calculated microscopically by counting five different fields (each 0.25 mm2) with a raster ocular at 200-fold magnification.
Chemotactic movement of tumor cells was examined by a Boyden double chamber system with 8 µm pore filters (6-well chamber system; Greiner Bio-One, Frickenhausen, Germany). Tumor cells pre-exposed to 100 ng/mL IGF-1 for 4 or 24 h (controls did not receive IGF-1) were detached from the culture flasks and placed in the upper chamber with serum-free medium (2.5 × 105 cells/mL; 2 mL cell suspension/chamber). The lower chamber contained 10% FCS as the chemoattractant. After 24 h incubation time, migrated cells under the membrane were fixed with 1% glutaraldehyde and stained with haematoxylin (Sigma). Non-migrated cells were cleaned from the upper surface of the membrane with cotton-wool tips. The migrated cells were counted microscopically in five different observation fields under a microscope at 200-fold magnification (5 × 0.25 mm2).
2.5. Scratch Wound Assay
The scratch wound assay was used to examine the horizontal migration of the cancer cells in the presence of IGF-1 (versus controls). Tumor cells were incubated with 100 ng/mL IGF-1 at 37 °C, 5% CO2 for 4 or 24 h and then seeded onto 96-well ImageLock plates (Sartorius, Goettingen, Germany) previously coated with 400 µg/mL collagen at 4 °C for 48 h (100 µL cell suspension, 5 × 105 cells/mL). At 24 h after plating out the cells, a defined scratch of about 700 µm was made with an IncuCyte® WoundMaker (Sartorius). Detached cells were removed by washing with PBS with Ca2+ and Mg2+. Cell culture medium was then renewed with 200 µL medium with 2 or 10% FCS with 100 ng/mL IGF-1. Controls received cell culture medium without IGF-1. Plates were incubated in Incucyte® Zoom (Sartorius) at 37 °C, 5% CO2 and photographed every 2 h for 48 h. Each experiment was conducted in triplicate. Relative wound density was calculated by WimScratch software (Onimagin Technologies SCA, Córdoba, Spain).
2.6. Integrin Surface Expression
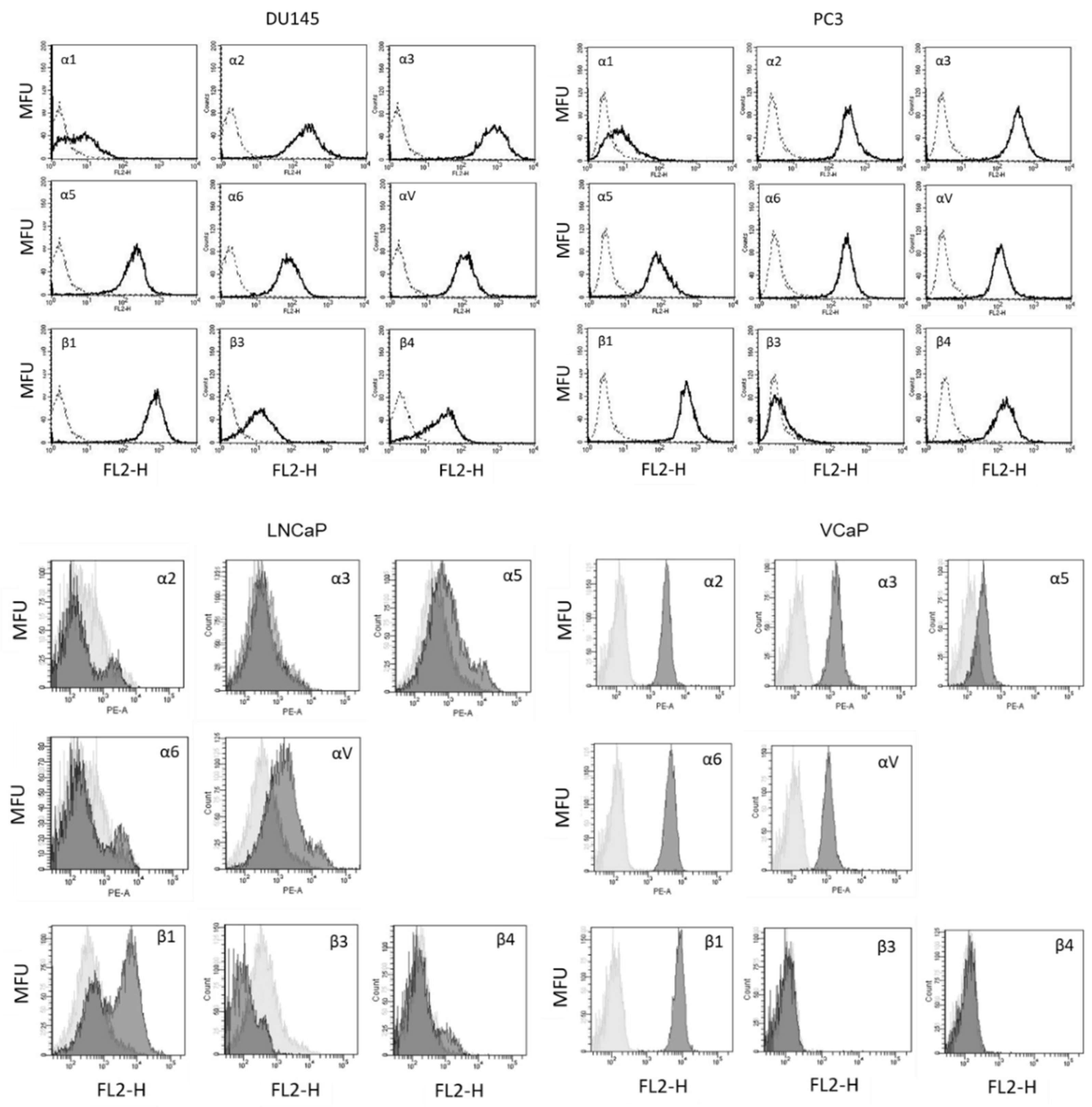
Cancer cells stimulated with IGF-1 for 2, 4, or 24 h were detached from the culture flasks using Accutase® (PAA Laboratories GmbH, Pasching, Austria) and washed with blocking solution (PBS, 0.5% BSA). Subsequently, they were incubated for 1 h at 4 °C with phycoerythrin (PE) conjugated monoclonal antibodies (each 20 µL) directed against the following integrin subtypes: anti-α1 (IgG1; clone SR84, dilution 1:1000), anti-α2 (IgG2a; clone 12F1-H6, dilution 1:250), anti-α3 (IgG1; clone C3II.1, dilution 1:1000), anti-α5 (IgG1; clone IIA1, dilution 1:5000), anti-α6 (IgG2a; clone GoH3, dilution 1:200), anti-β1 (IgG1; clone MAR4, dilution 1:2500), anti-β3 (IgG1; clone VI-PL2, dilution 1:2500), anti-β4 (IgG2a; clone 439-9B, dilution 1:250; all: BD Biosciences), or anti-αV (IgG1; clone 13C2, Southern Biotech, Birmingham, AL, USA). The integrin expression of the tumor cells was then measured and analyzed using a FACSCalibur (BD Biosciences; FL2-H (log) channel histogram analysis; 1 × 104 cells per scan) and expressed as mean fluorescence units. Mouse IgG1-PE (MOPC-21), mouse IgG2a-PE (G155-178) or rat IgG2b-PE (R35-38; all: BD Biosciences) were used as isotype controls.
2.7. Western Blot Analysis
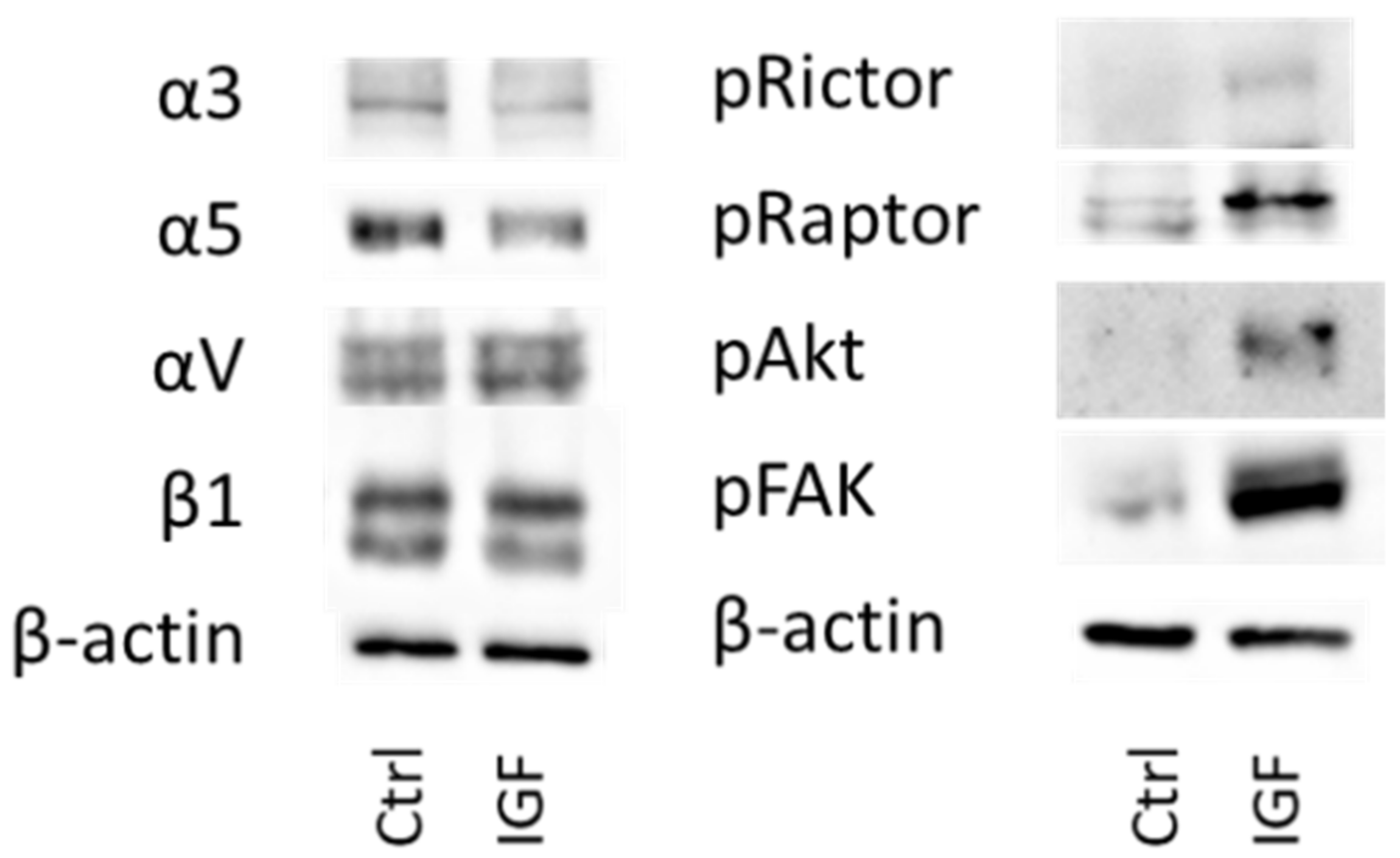
To explore integrin protein expression as well as proteins involved in the Akt-mTOR-pathway under IGF-1 stimulation, tumor cell lysates were applied to a 7–12% polyacrylamide gel, electrophoresed for 90 min at 100 V and transferred to nitrocellulose membranes for 60 min at 100 V. After blocking with non-fat dry milk for 60 min, membranes were incubated overnight with the following unconjugated monoclonal antibodies, directed against integrin proteins and integrin related signaling, as follows: anti-α3 (polyclonal, dilution 1:500), anti-α5 (clone 1/CD49e, dilution 1:5000), anti-β1 (clone 18/CD29, dilution 1:2500; all from BD Biosciences), and anti-αV (Clone 21/CD51, dilution 1:250; Southern Biotech). Anti-FAK (clone 77, dilution 1:1000), anti-pFAK (pY397; clone 18, dilution 1:1000), and anti-ILK (clone 3, dilution 1:1000) antibodies were also from BD Biosciences. The mTOR pathway was investigated through the following proteins: Anti-Rictor (clone D16H9, dilution 1:1000), anti-pRictor (Thr1135, clone D30A3, dilution 1:1000), anti-Raptor (clone 24C12, dilution 1:1000), anti-pRaptor (Ser792, dilution 1:1000; all from Cell Signaling, Cambridge, UK), PKBα/Akt (IgG1, clone 55), and pAkt (IgG1, pS472/pS473, clone 104A282; both from BD Biosciences).
Horseradish peroxidase (HRP)-conjugated goat-anti-mouse or goat-anti-rat-IgG (Cell Signaling Technology, Cambridge, UK; dilution 1:3000) served as the secondary antibodies. To visualize proteins, membranes were incubated with enhanced chemiluminescence (ECL) detection reagent (ECLTM, Amersham/GE Healthcare, München, Germany). After incubation they were analyzed by the Fusion FX7 system (Peqlab, Erlangen, Germany). β-actin (1:1000; Sigma, Taufenkirchen, Germany) served as internal control. Pixel density analysis of the protein bands was performed with Gimp 2.8.20 software,
www.gimp.org) before calculating the ratio of protein intensity/β-actin intensity.
2.8. Blocking Studies and siRNA Knock Down
Tumor cells were incubated for 60 min with 10 μg/mL function-blocking anti-integrin α3, α5, αV or anti-integrin β1 mouse mAb (all from Merck Millipore, Darmstadt, Germany) and then subjected to the MTT-assay and the chemotaxis assay. In addition, DU145 cells were treated with 10 µM of the FAK-inhibitor defactinib (Biozol, Eching, Germany) for 24 and 72 h. The mTOR related signaling proteins, Rictor and Raptor, both total and phosphorylated, were then evaluated with Western blotting. Transfection with small interfering RNA (siRNA) was carried out directed against integrin α3 (gene ID: 3675; target Sequence: 5′-CACCATCAACATGGAGAACAA-3′, α5 (gene ID: 3678, GeneGlobe ID-SI02654841, NM_002205) αV (gene ID: 3685; target sequence: 5′-TAGCATGATGTTACAGGAATA-3′) or anti-integrin β1 (gene ID: 891, target sequence: 5′-AATGTAGTCATGGTAAATCAA-3′) as well. Further studies were completed with siRNA directed against FAK (gene ID: 2185; target sequence: AAGCTGATCGGCATCATTGAA) or Akt (gene ID: 207; target sequence: AATCACACCACCTGACCAAGA; all: Qiagen, Hilden, Germany). An amount of 3 × 105 cells were pre-incubated for 24 h with a transfection solution of siRNA and transfection reagent (HiPerFect Transfection Reagent; Qiagen) at a ratio of 1:6. Cells treated with cell culture medium alone and cells treated with 5 nM control siRNA (AllStars Negative Control siRNA; Qiagen) served as controls. Protein expression and tumor cell growth and chemotaxis were then analyzed as described above.
2.9. Statistics
The mean +/− SD was calculated. Graphs were prepared using SigmaPlot 11 (SYSTAT Software, San Jose, CA, USA). To exclude coincidence, all experiments were repeated three to five times. Statistical significance was evaluated with the “Student’s t-Test”. p < 0.05 was considered significant.
4. Discussion
Although the relevance of IGF-1 as a prognostic biomarker in prostate cancer patients has been demonstrated in several cohort studies, its mode of action remains unsettled. The present investigation employing prostate cancer cell lines in vitro point to a dual role of IGF-1. Aside from suppressing tumor growth, IGF-1 acted on cell adhesion and motility as well. Treating PC3 or DU145 cells for 24 h with IGF-1 resulted in a significant increase in adhesion to collagen and, to a minor extent, to fibronectin. This is important, since adhesion of tumor cells to collagen and fibronectin is necessary in forming a pre-metastatic niche, whereby elevated collagen production facilitates cancer cell colonization into the bone [
13].
Elevated adhesion was accompanied by elevated migration activity, shown by both the Boyden chamber assay evaluating vertical migration towards a chemotactic stimulus and the wound healing assay focusing on horizontal cell movement. Our investigation provides evidence that IGF-1 acts on the expression level of integrin α and β subtypes, notably α3, α5, αV, and β1. Marelli et al. observed complete antagonization of IGF-1-induced migration of DU145 and PC3 cells following αVβ3 function blockade [
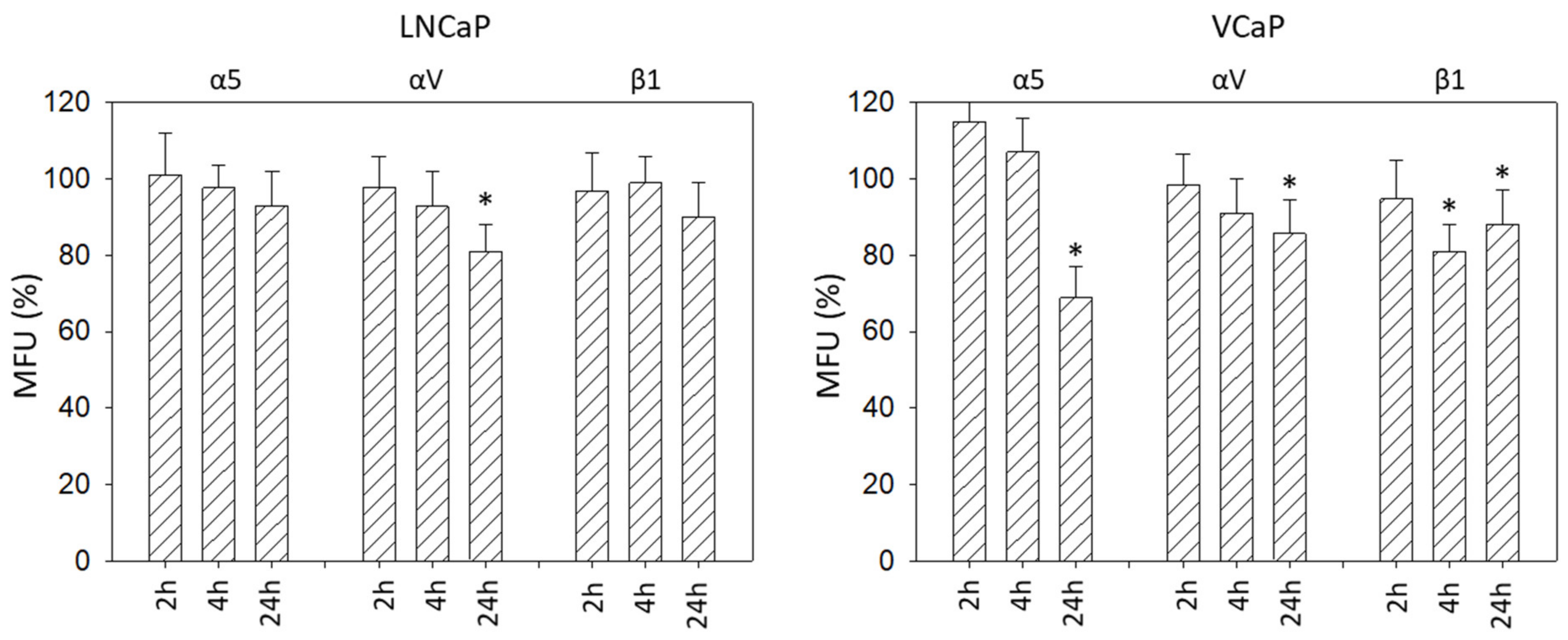
14]. Since β3 integrin was not expressed on PC3 cells, as demonstrated by FACS analysis, we did not further deal with this subtype. In accordance with our experiments, we assume that αV rather than β3 contributed to the IGF-1-induced migration effects observed by Marelli et al. Still, the role of IGF-1 in regard to integrin expression is not straightforward. We have observed that the kind of integrin modulation strongly depends on the IGF-1 incubation time. DU145 surface expression of α3 and αV increased strongly after 4 h but was reduced after 24 h. The β1 subtype was elevated already after 2 h with subsequent down-regulation at 4 and 24 h, in contrast to α5, which was distinctly elevated after 24 h.
Slight differences in integrin modulation have also been noted between DU145 and PC3 cells. Particularly, αV was enhanced on PC3 cells after 4 or 24 h IGF-1 incubation. Hypothetically, this difference could explain why blocking αV surface expression correlated with an increased adhesion of DU145 cells but decreased PC3 adhesion to immobilized collagen.
The protein profile, investigated after 24 h IGF-1 incubation, indicates enhancement of α3 and αV and diminution of α5 in DU145 cells. This could point to IGF-1 causing a translocation of α3 and αV from the cell membrane into the cytoplasm and translocation of integrin α5 from inside the cell to the surface membrane. Integrin β1 was not distinctly modified (only a slight, insignificant reduction was apparent after 24 h IGF-1 treatment). Sayeed et al. have demonstrated that β1 integrin regulation by IGF-1R does not occur at the mRNA level [
15]. This may explain why we did not observe intracellular β1 alterations at the 24 h time point. Based on their and our data, it may be assumed that β1 is shifted from the cancer cell surface only after 24 h. Protein expression at earlier time points has not been investigated. Thus, further modes of IGF-1 action should be considered, including β1-trafficking, as has been documented in an in vivo mouse xenograft model [
16]. Since differences are seen in initial integrin expression levels, with β3 verified on DU145 but not on PC3 cells, integrin trafficking, if it does take place, must also differ. We have shown that integrin protein expression in response to IGF-1 does differ between DU145 and PC3 cells. Differences in the genetic pattern of different tumor types may possibly be involved in modulating particular integrin subtypes since DU145 harbors mutations in CDKN2A, RB1, and TP53, whereas PC3 harbors mutations in PTEN and TP53.
Time-dependent integrin alteration caused by IGF-1 has not been dealt with by others as yet. However, time-dependent expression kinetics of several integrin subtypes (α2, α5, αV, β1, β3) has been observed with the human umbilical vein cell line EA.hy926 and the hepatoblastoma cell line HepG2. The temporal sequence of integrin up- and down-regulation has been interpreted such that coordinated assembly and disassembly of these receptors might be necessary to allow coordinated cell migration [
17,
18]. Interaction between c-Met and β1-integrin receptors has recently been described [
19]. This is notable, since stimulation of PC3 cells with IGF-1 induced a time-dependent phosphorylation of c-Met, reaching a maximum 18–24 h after IGF-1 addition [
20].
Therefore, we assume that time-dependent alterations in the integrin subtype expression pattern, along with integrin translocation processes, are pivotal mechanisms accounting for how IGF-1 may force metastatic spread. Integrin alterations might further be coupled to cascaded downstream signaling, as observed in a HepG2 cell migration assay [
21]. Our data point to an elevation of Akt/Rictor signaling under IGF. Since IGF-1 enhanced the levels of integrin α3 and αV while reducing α5, and Akt knock-down diminished α3 and αV but up-regulated integrin α5, cross-communication between these integrin subtypes and Akt seems likely. Interestingly, FAK also became activated by IGF-1, but was not altered following Akt knock-down, showing that Akt may not serve as an upstream modulator of FAK. This does not exclude the possibility that FAK may regulate IGF-1R activation and subsequently Akt, as the downstream effector. We did not investigate this issue, but FAK is known to interact with integrins and IGF-1R in osteogenic cells, which results in up-regulation of Akt phosphorylation [
22].
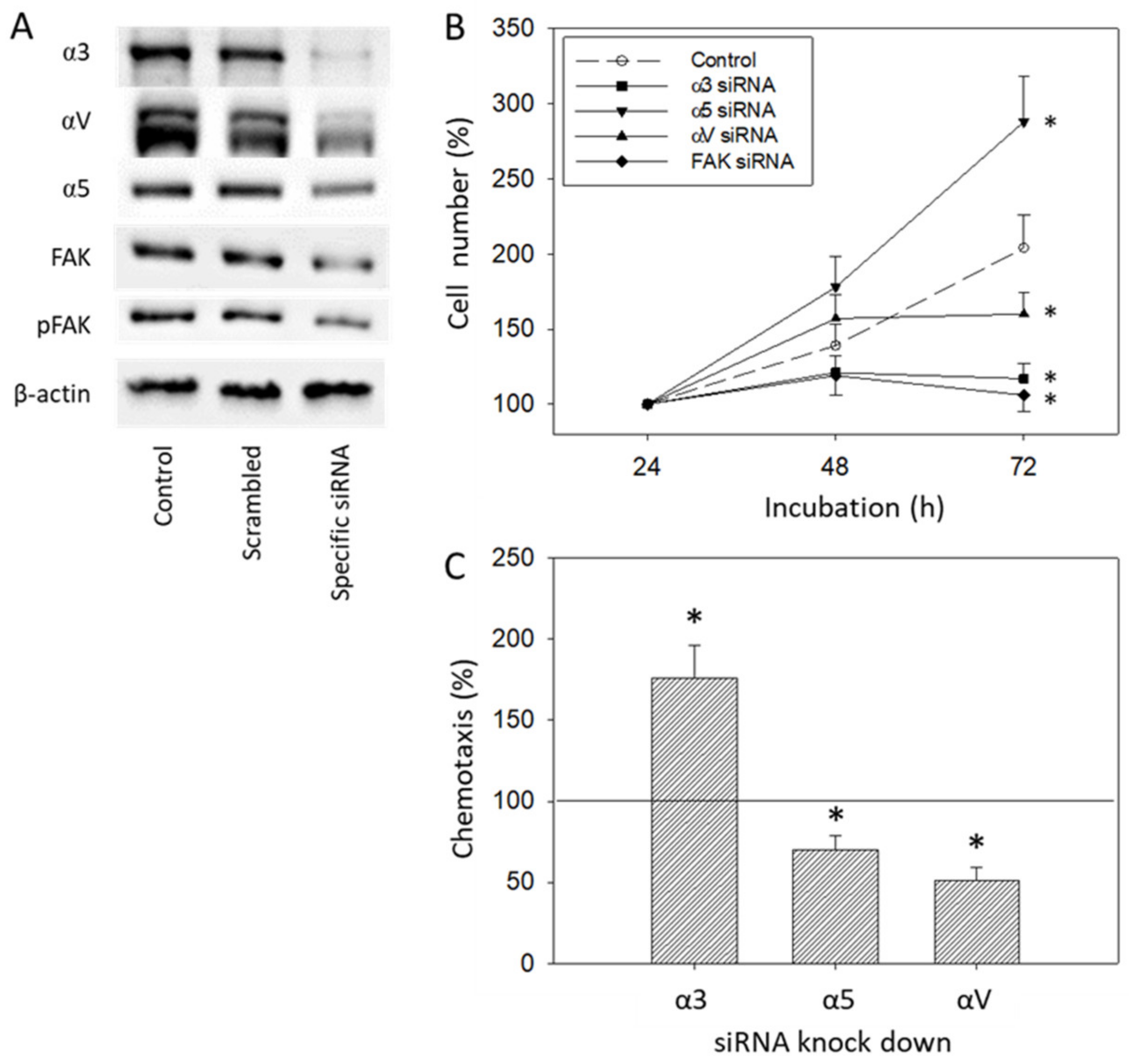
The IGF-1-integrin axis is not only involved in tumor cell invasion but also in the control of tumor cell growth. Depending on the kind of integrin blockade and the type of integrin which was blocked, different modes of action were apparent. Surface blockade of integrin α5 did not cause any effects on cell growth but correlated with increased chemotaxis. In contrast, α5 knock-down was found to increase tumor growth but reduce chemotaxis. Blocking αV with its monoclonal antibody was associated with both diminished growth and migration, whereas siRNA knock-down enhanced tumor growth at the 48 h time point. Loss of the α3 subtype was associated with reduced growth, independent from the kind of integrin modulation. However, chemotaxis was diminished following α3 surface blockade, but elevated following α3 knock-down. We, therefore, conclude that IGF-1 alters integrin subtypes in a concerted and fine-tuned manner, which may finally accelerate both prostate cancer growth and invasion.
Since adhesion and chemotaxis were more strongly influenced by IGF-1 than was tumor cell growth, the action of IGF-1 on integrin subtype expression might be particularly important in forcing metastatic progression. Although this is speculative, we observed the most prominent alterations on the integrin αV subtype being excessively enhanced on the cell surface following 4 h IGF-1 incubation and considerably elevated in the cytoplasm after 24 h. Since αV expression strongly correlated with DU145 chemotaxis, as verified by both blocking and knocking down αV, this hypothesis seems plausible. In good accordance, αV has been found to be aberrantly expressed in bone-metastases of prostate adenocarcinomas and is suggested to play a candidate role in the bone dissemination of aggressive prostate cancer [
8]. Recently, Ibrahim et al. reported that exposing breast cancer cells to IGF-1 may cause a shift from IGF-1 receptor substrate (IRS)-1 phosphorylation to IRS-2 phosphorylation, leading to activated cell migration [
23]. In this context, expression of IRS-1 resulted in IGF-1-stimulated proliferation, but did not affect motility, whereas expression of IRS-2 enhanced IGF-1-stimulated motility but did not stimulate proliferation [
24]. This is notable, since IRS-2 has already been linked to integrin αV as a cell migration regulator [
25].
Our results also point to integrins as tumor growth modulators, being related to prior IGF-1 stimulation. The most prominent influence on tumor cell growth was exerted by the integrin subtype α3. Since α3 accumulates in the cell cytoplasm, at least after a 24 h IGF-1 stimulation, we postulate that this process might be relevant to the IGF-1 induced enhancement of the cell growth rate. Integrin α3 has been shown to activate colorectal cancer cell proliferation [
26], and evaluation of papillary thyroid cancer tissues and cell lines indicate an influence of integrin α3 on cell cycling and autophagy [
27].
The role of integrins in tumor growth has been demonstrated by others, whereby most studies concentrate on integrin β1. β1 activated IGF-1R signaling in DU145 and PC3 cells and promoted cell survival and proliferation [
28,
29]. The role of the β1 integrin as a trigger factor for IGF-1-mediated mitogenic and transforming activities has also been verified in androgen-sensitive LNCaP cells [
30]. Signaling between the β1 integrin and IGF-1R is thought to enable Akt activation and further signaling pathways to start the mitotic cascade [
31]. In our experimental model, the integrin β1 surface level was elevated at a very early time point and rapidly decreased under the influence of IGF-1 in both DU145 and PC3 cells. How far this regulatory mechanism may contribute to tumor cell mitosis is not clear. Nevertheless, all β1-related studies cited here have in common that integrin β1 served as an upstream mediator of IGF-1-triggered IGF-1R activation, which contrasts with our observations. We did not investigate whether β1 and further integrin family members, α3, α5 and αV, may contribute to IGF-1-related signaling, but rather investigated the inverse mechanism. Therefore, different integrin action modes should be considered, depending on their activation order.
IGF-1-stimulation did not alter LNCaP growth and only integrin αV was slightly reduced after 24 h incubation. We, therefore, assume that the IGF-1-integrin-interaction seen in the androgen-resistant DU145 and PC3 cells is not highly relevant in the androgen-sensitive LNCaP cell line. Integrin blockage resulted in no significant difference in cell growth after 72 h. However, in contrast to LNCaP, IGF-1 did activate VCaP tumor cell growth and caused diminishment of the integrins α5, αV, and β1, pointing to differences between the LNCaP and VCaP cell lines. The IGF ligand-neutralizing antibody xentuzumab has been shown to alter the proliferative activity of VCaP but not of PTEN-null LNCaP cells. The authors assumed that PTEN may be involved in IGF-1 triggered cell proliferation [
32]. Considering that PTEN-null PC3 cells responded well to IGF-1 treatment in terms of cell growth and integrin regulation, this postulate may not hold true for our model. However, IGF-1R was strongly expressed on VCaP but not on LNCaP cells, which may at least partially account for the differences observed. Since the response of the androgen-sensitive LNCaP and VCaP cell lines to IGF-1-stimulation differs, androgen-sensitivity per se does not appear to be a characteristic that uniformly influences the response to IGF-1-stimulation.
Overall, an inconsistent picture is presented here. IGF-1 driven cross-communication between Akt/mTOR and FAK-integrin signaling has been demonstrated in the androgen-resistant prostate cancer cell lines DU145 and PC3. Since integrin β1 was altered similarly by IGF-1 and shown to down-regulate growth and invasion of both cell lines, functional blockade of β1 might be an option to treat prostate cancer once castration-resistance has been established. Differences between the androgen-resistant and the androgen-sensitive cells became obvious. LNCaP cells did not respond to IGF-1 in terms of growth activation and integrin expression (excepting a slight loss of αV). VCaP differed in a way that blockade of integrin αV did not diminish cell growth as seen with DU145 and PC3 but was rather associated with an accelerated cell growth. Presumably, IGF-1 driven Akt-integrin cross-talk might (at least partially) depend on androgen related signaling. The initial integrin equipment which varied considerably between DU145/PC3, LNCaP, and VCaP, may also be a crucial factor determining the kind of Akt-integrin communication.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















