
Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Mice
2.3. Cationic Lipid Nanoparticle
2.4. In Vivo Tumor Models
2.5. Single-Cell RNA Sequencing Dataset Analysis
2.6. DNA-Methylation Analysis
2.7. Patient-Dataset Analysis
2.8. Flow Cytometry
2.9. Cell-Death Analysis
2.10. Western Blotting
2.11. Transfection
2.12. Statistical Analysis
3. Results
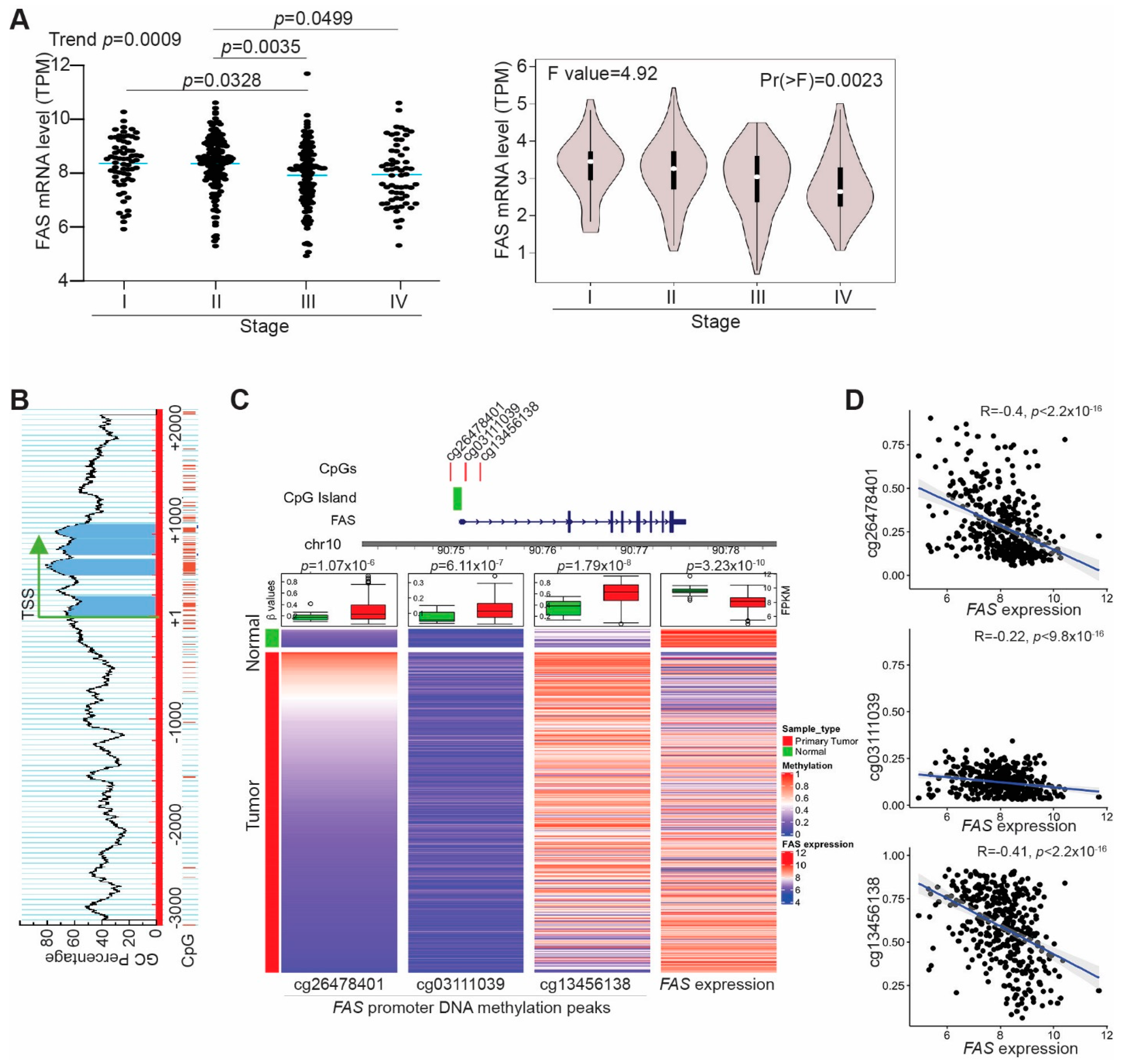
3.1. FAS Is a Suppressor of Human Colorectal-Cancer Metastasis
3.2. FAS Expression Is Repressed by Its Promoter DNA Hypermethylation in Tumor Cells of Human Colorectal Cancer Patients
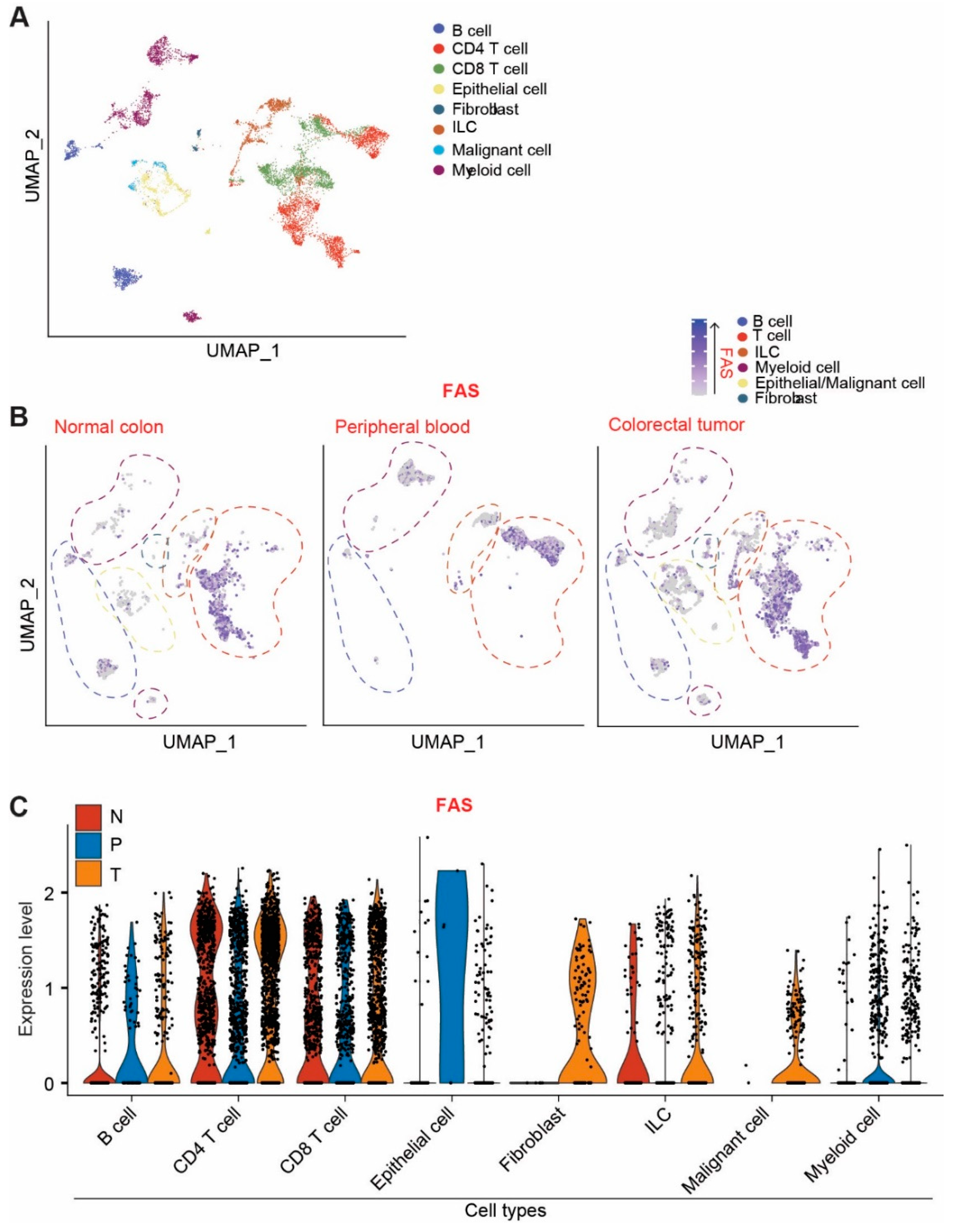
3.3. Single-Cell RNA Sequencing Indicates That FAS Is Highly Expressed in Immune Cells and Normal Epithelial Cells in Human Colorectal-Cancer Patients
3.4. Development of Cationic Lipid Encapsulated Codon-Optimized FAS cDNA-Expressing Plasmid DNA Nanoparticle
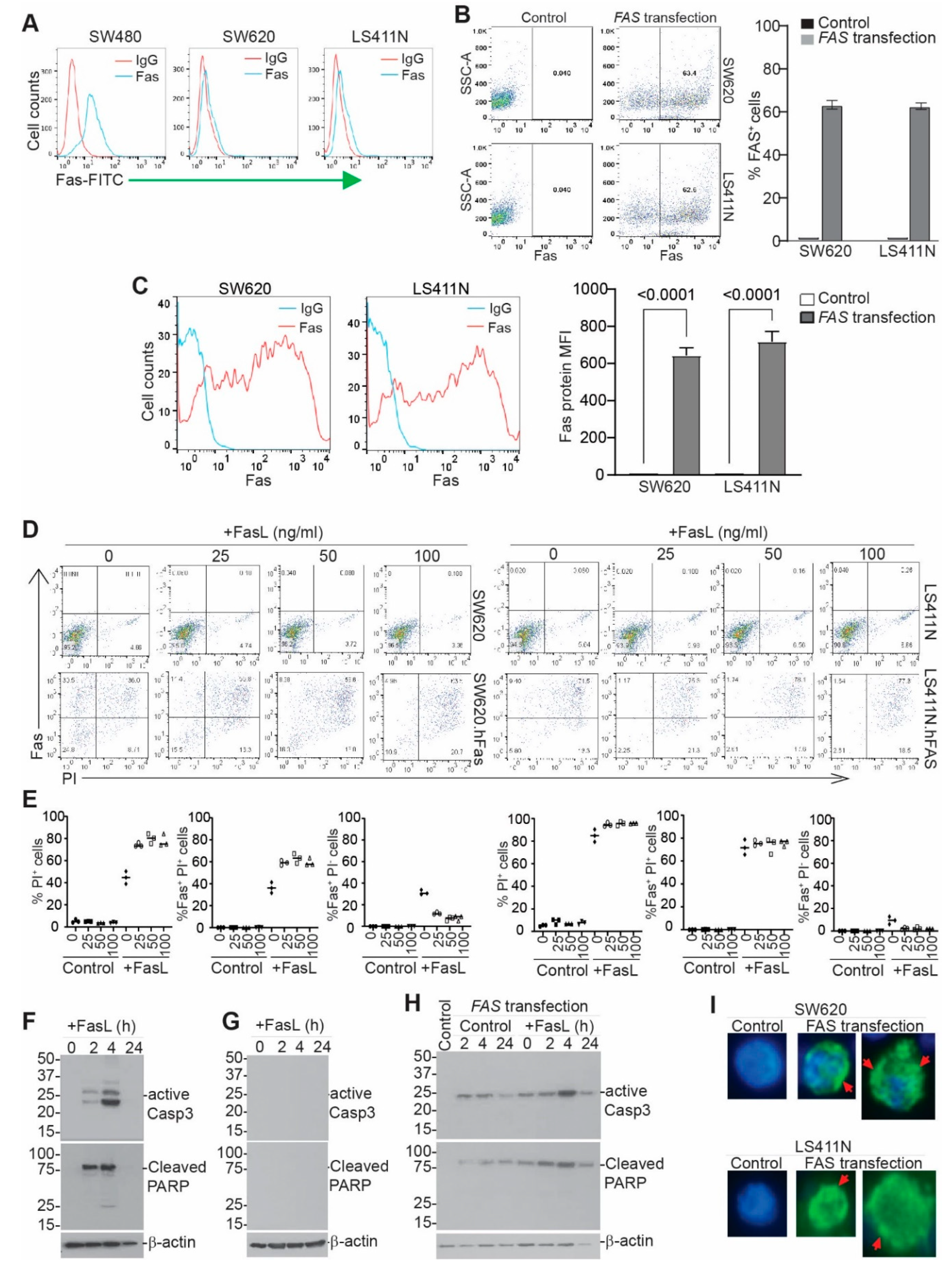
3.5. Restoring FAS Expression Overcomes Colon Tumor Resistance to FASL-Induced Apoptosis In Vitro
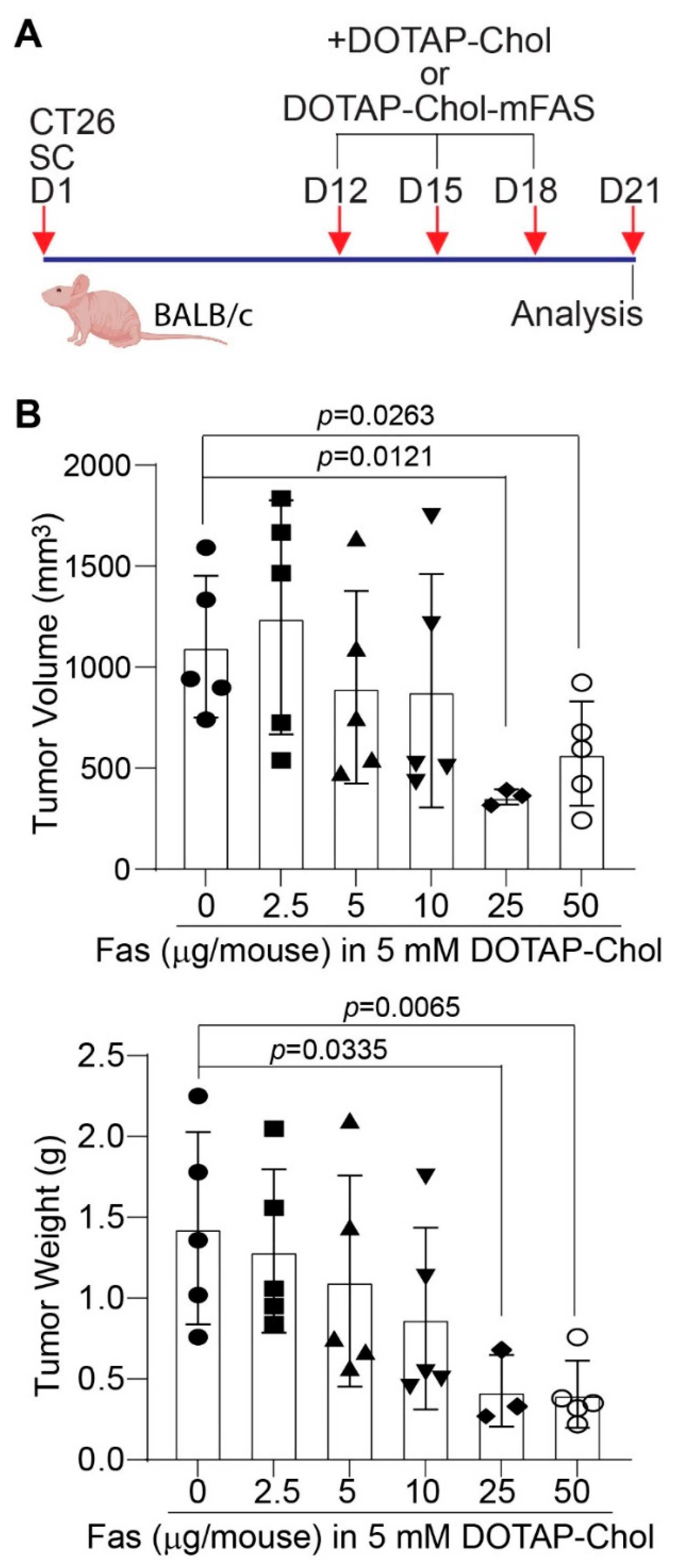
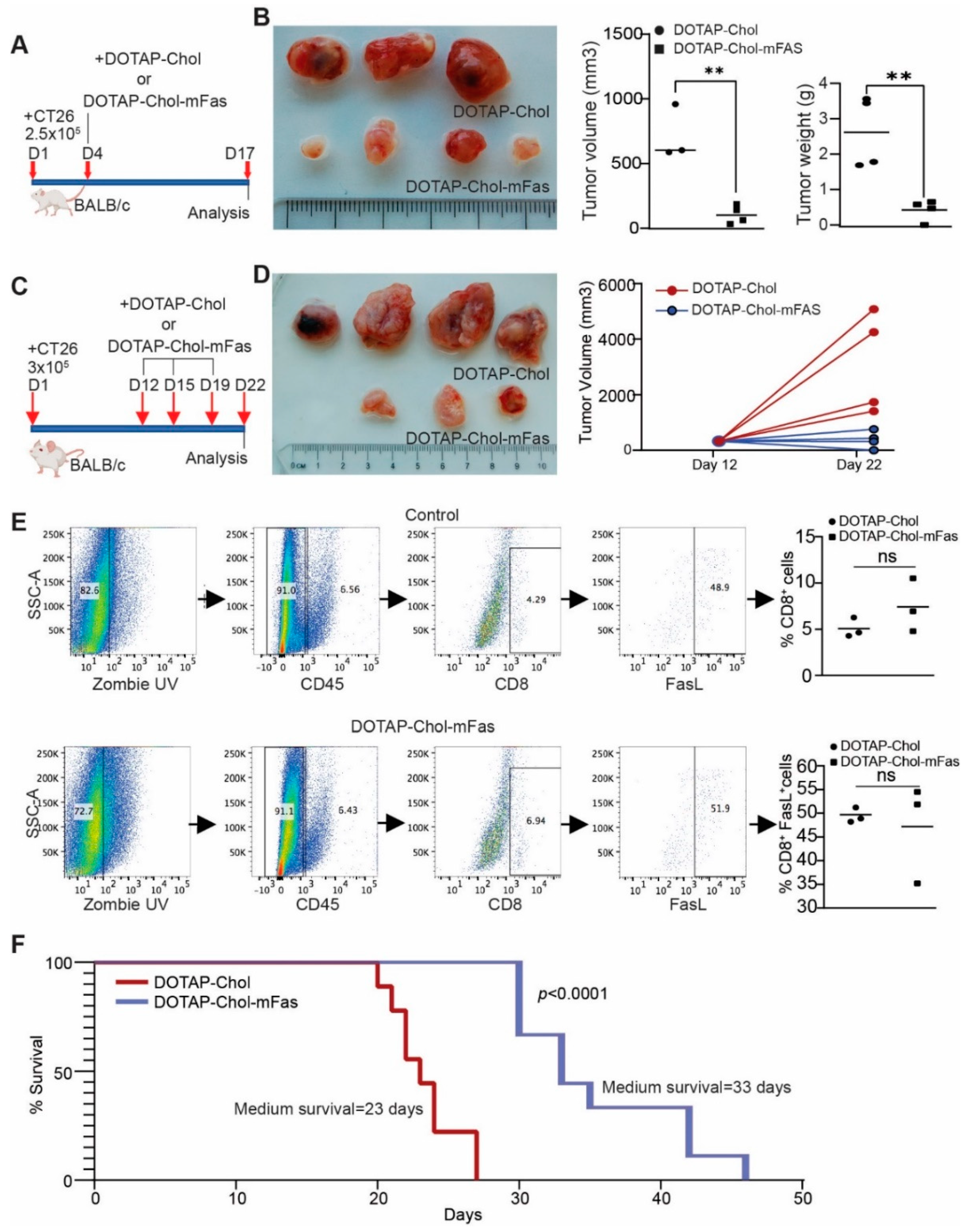
3.6. FAS DNA Nanoparticle Therapy Is Sufficient to Suppress Mouse Colon-Tumor Growth in Immune-Competent Mice
3.7. Exogenous Expression of Codon-Usage-Optimized FAS Induces Metastatic Human Colon-Tumor-Cell FAS Receptor Auto-Oligomerization and Tumor-Cell Auto-Apoptosis In Vitro
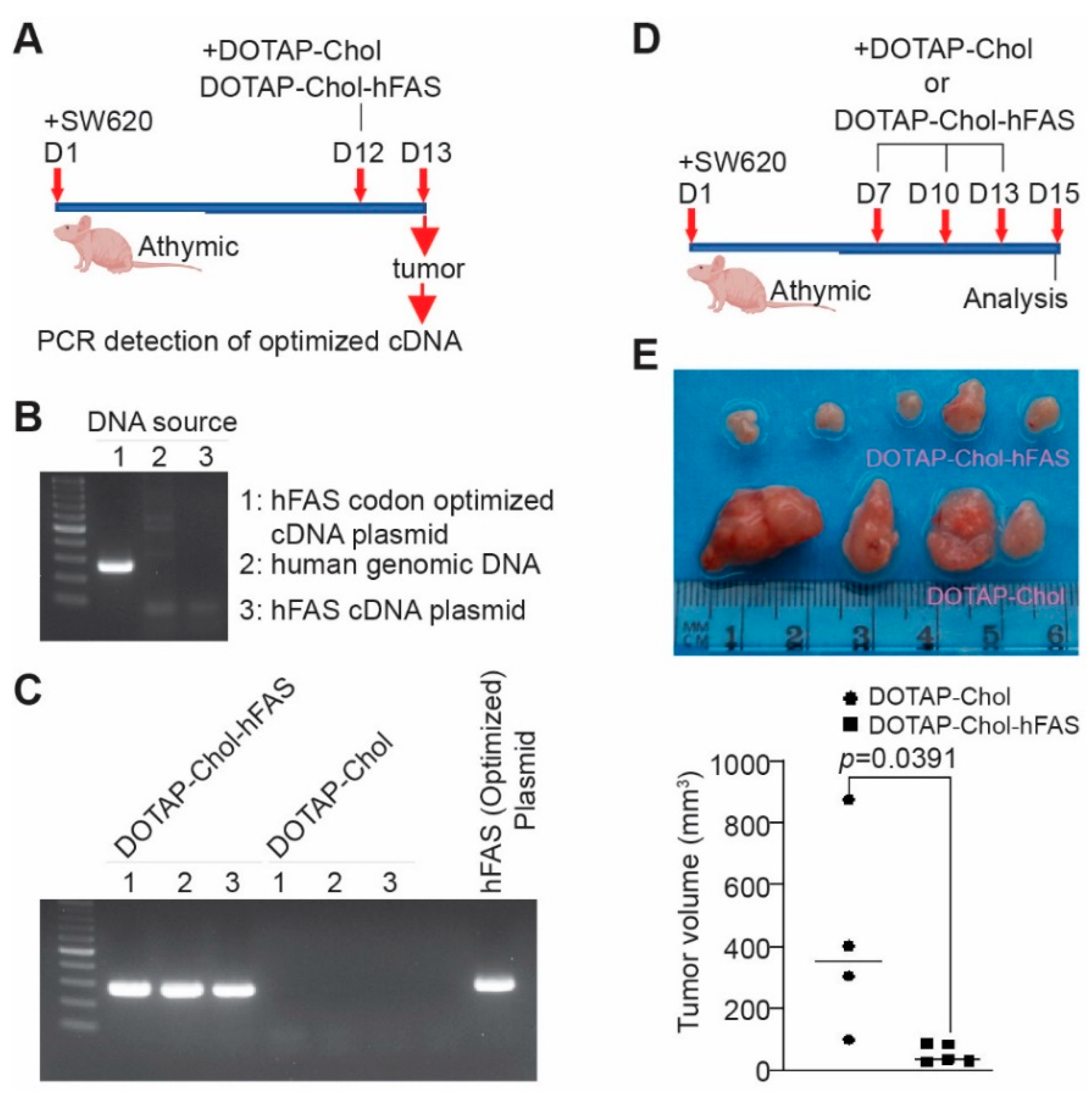
3.8. Restoring FAS Expression Is Sufficient to Suppress Metastatic Human Colon-Tumor Xenograft Growth In Vivo
3.9. FAS DNA Nanoparticle Therapy Has No Significant Toxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Chang, G.J.; Overman, M.J.; Eng, C.; Sargent, D.J.; Larson, D.W.; Grothey, A.; Vauthey, J.N.; Nagorney, D.M.; McWilliams, R.R. Improved survival in metastatic colorectal cancer is associated with adoption of hepatic resection and improved chemotherapy. J. Clin. Oncol. 2009, 27, 3677–3683. [Google Scholar] [CrossRef]
- Brandi, G.; De Lorenzo, S.; Nannini, M.; Curti, S.; Ottone, M.; Dall’Olio, F.G.; Barbera, M.A.; Pantaleo, M.A.; Biasco, G. Adjuvant chemotherapy for resected colorectal cancer metastases: Literature review and meta-analysis. World J. Gastroenterol. 2016, 22, 519–533. [Google Scholar] [CrossRef] [PubMed]
- Moller, P.; Koretz, K.; Leithauser, F.; Bruderlein, S.; Henne, C.; Quentmeier, A.; Krammer, P.H. Expression of APO-1 (CD95), a member of the NGF/TNF receptor superfamily, in normal and neoplastic colon epithelium. Int. J. Cancer 1994, 57, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Liu, Y.; Jiang, C.; Sun, L.; Zhou, H. Identification and clinical validation of metastasis-associated biomarkers based on large-scale samples in colon-adenocarcinoma. Pharm. Res. 2020, 160, 105087. [Google Scholar] [CrossRef]
- Maeda, Y.; Levy, R.B.; Reddy, P.; Liu, C.; Clouthier, S.G.; Teshima, T.; Ferrara, J.L. Both perforin and FAS ligand are required for the regulation of alloreactive CD8+ T cells during acute graft-versus-host disease. Blood 2005, 105, 2023–2027. [Google Scholar] [CrossRef]
- Golstein, P.; Griffiths, G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018, 18, 527–535. [Google Scholar] [CrossRef]
- Morales-Kastresana, A.; Catalan, E.; Hervas-Stubbs, S.; Palazon, A.; Azpilikueta, A.; Bolanos, E.; Anel, A.; Pardo, J.; Melero, I. Essential complicity of perforin-granzyme and FAS-L mechanisms to achieve tumor rejection following treatment with anti-CD137 mAb. J. Immunother. Cancer 2013, 1, 3. [Google Scholar] [CrossRef]
- Chen, L.; Park, S.M.; Tumanov, A.V.; Hau, A.; Sawada, K.; Feig, C.; Turner, J.R.; Fu, Y.X.; Romero, I.L.; Lengyel, E.; et al. CD95 promotes tumour growth. Nature 2010, 465, 492–496. [Google Scholar] [CrossRef]
- Hong, L.K.; Chen, Y.; Smith, C.C.; Montgomery, S.A.; Vincent, B.G.; Dotti, G.; Savoldo, B. CD30-Redirected Chimeric Antigen Receptor T Cells Target CD30(+) and CD30(-) Embryonal Carcinoma via Antigen-Dependent and FAS/FASL Interactions. Cancer Immunol. Res. 2018, 6, 1274–1287. [Google Scholar] [CrossRef]
- Upadhyay, R.; Boiarsky, J.A.; Pantsulaia, G.; Svensson-Arvelund, J.; Lin, M.J.; Wroblewska, A.; Bhalla, S.; Scholler, N.; Bot, A.; Rossi, J.M.; et al. A critical role for FAS-mediated off-target tumor killing in T cell immunotherapy. Cancer Discov. 2021, 11, 599–613. [Google Scholar] [CrossRef]
- Wang, J.; Cao, Z.; Wang, P.; Zhang, X.; Tang, J.; He, Y.; Huang, Z.; Mao, X.; Shi, S.; Kou, X. Apoptotic Extracellular Vesicles Ameliorate Multiple Myeloma by Restoring FAS-Mediated Apoptosis. ACS Nano 2021, 15, 14360–14372. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Lemoine, J.; Ruella, M.; Houot, R. Overcoming Intrinsic Resistance of Cancer Cells to CAR T-Cell Killing. Clin. Cancer Res. 2021, 27, 6298–6306. [Google Scholar] [CrossRef]
- Afshar-Sterle, S.; Zotos, D.; Bernard, N.J.; Scherger, A.K.; Rodling, L.; Alsop, A.E.; Walker, J.; Masson, F.; Belz, G.T.; Corcoran, L.M.; et al. FAS ligand-mediated immune surveillance by T cells is essential for the control of spontaneous B cell lymphomas. Nat. Med. 2014, 20, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.A.; Ryan, M.H.; McDuffie, E.; Abrams, S.I. The FAS/FAS ligand pathway is important for optimal tumor regression in a mouse model of CTL adoptive immunotherapy of experimental CMS4 lung metastases. J. Immunol. 2003, 171, 2402–2412. [Google Scholar] [CrossRef] [PubMed]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. FAS triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- LA, O.; Tai, L.; Lee, L.; Kruse, E.A.; Grabow, S.; Fairlie, W.D.; Haynes, N.M.; Tarlinton, D.M.; Zhang, J.G.; Belz, G.T.; et al. Membrane-bound FAS ligand only is essential for FAS-induced apoptosis. Nature 2009, 461, 659–663. [Google Scholar]
- Cremesti, A.; Paris, F.; Grassme, H.; Holler, N.; Tschopp, J.; Fuks, Z.; Gulbins, E.; Kolesnick, R. Ceramide enables FAS to cap and kill. J. Biol. Chem. 2001, 276, 23954–23961. [Google Scholar] [CrossRef]
- Maecker, H.L.; Yun, Z.; Maecker, H.T.; Giaccia, A.J. Epigenetic changes in tumor FAS levels determine immune escape and response to therapy. Cancer Cell 2002, 2, 139–148. [Google Scholar] [CrossRef][Green Version]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The FAS-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Strasser, A.; Jost, P.J. FAS death receptor signalling: Roles of Bid and XIAP. Cell Death Differ. 2012, 19, 42–50. [Google Scholar] [CrossRef]
- Guillen-Ahlers, H.; Suckow, M.A.; Castellino, F.J.; Ploplis, V.A. FAS/CD95 deficiency in ApcMin/+ mice increases intestinal tumor burden. PLoS ONE 2010, 5, e9070. [Google Scholar] [CrossRef]
- Azwar, S.; Seow, H.F.; Abdullah, M.; Faisal Jabar, M.; Mohtarrudin, N. Recent Updates on Mechanisms of Resistance to 5-Fluorouracil and Reversal Strategies in Colon Cancer Treatment. Biology 2021, 10, 854. [Google Scholar] [CrossRef] [PubMed]
- Rani, I.; Sharma, B.; Kumar, S.; Kaur, S.; Agnihotri, N. Apoptosis mediated chemosensitization of tumor cells to 5-fluorouracil on supplementation of fish oil in experimental colon carcinoma. Tumor Biol. 2017, 39, 1010428317695019. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.S.; Choi, B.Y.; Keum, Y.S. Sensitization of 5-Fluorouracil-Resistant SNUC5 Colon Cancer Cells to Apoptosis by alpha-Mangostin. Biomol. Ther. 2016, 24, 604–609. [Google Scholar] [CrossRef][Green Version]
- Cervantes-Villagrana, R.D.; Albores-Garcia, D.; Cervantes-Villagrana, A.R.; Garcia-Acevez, S.J. Tumor-induced neurogenesis and immune evasion as targets of innovative anti-cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 99. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Torres, E.; Lopez-Perez, T.V.; Morales-Martinez, M.; Huerta-Yepez, S. Yin-Yang-1 decreases FAS-induced apoptosis in acute lymphoblastic leukemia under hypoxic conditions: Its implications in immune evasion. Boletín Médico Hosp. Infant. México 2020, 77, 186–194. [Google Scholar] [CrossRef]
- Coe, G.L.; Redd, P.S.; Paschall, A.V.; Lu, C.; Gu, L.; Cai, H.; Albers, T.; Lebedyeva, I.O.; Liu, K. Ceramide mediates FASL-induced caspase 8 activation in colon carcinoma cells to enhance FASL-induced cytotoxicity by tumor-specific cytotoxic T lymphocytes. Sci. Rep. 2016, 6, 30816. [Google Scholar] [CrossRef]
- Pryczynicz, A.; Guzinska-Ustymowicz, K.; Kemona, A. FAS/FASL expression in colorectal cancer. An immunohistochemical study. Folia Histochem. Cytobiol. 2010, 48, 425–429. [Google Scholar] [CrossRef]
- Cacan, E.; Ozmen, Z.C. Regulation of FAS in response to bortezomib and epirubicin in colorectal cancer cells. J. Chemother. 2020, 32, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.J. Apoptosis and colorectal cancer. Gut 2004, 53, 1701–1709. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Alam, K.J.; Kang, I.H.; Park, W.C.; Seo, G.S.; Choi, S.C.; Kim, H.S.; Moon, H.B.; Yun, K.J.; Chae, S.C. MicroRNA 196B regulates FAS-mediated apoptosis in colorectal cancer cells. Oncotarget 2015, 6, 2843–2855. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Lee, K.F.; Tung, S.Y.; Huang, W.S.; Lee, L.Y.; Chen, W.P.; Chen, C.C.; Teng, C.C.; Shen, C.H.; Hsieh, M.C.; et al. Induction Apoptosis of Erinacine A in Human Colorectal Cancer Cells Involving the Expression of TNFR, FAS, and FAS Ligand via the JNK/p300/p50 Signaling Pathway with Histone Acetylation. Front. Pharm. 2019, 10, 1174. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Mitchell, C.; Zhang, G.; Yacoub, A.; Allegood, J.; Haussinger, D.; Reinehr, R.; Larner, A.; Spiegel, S.; Fisher, P.B.; et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res. 2010, 70, 6313–6324. [Google Scholar] [CrossRef]
- Petak, I.; Danam, R.P.; Tillman, D.M.; Vernes, R.; Howell, S.R.; Berczi, L.; Kopper, L.; Brent, T.P.; Houghton, J.A. Hypermethylation of the gene promoter and enhancer region can regulate FAS expression and sensitivity in colon carcinoma. Cell Death Differ. 2003, 10, 211–217. [Google Scholar] [CrossRef]
- Paschall, A.V.; Yang, D.; Lu, C.; Choi, J.H.; Li, X.; Liu, F.; Figueroa, M.; Oberlies, N.H.; Pearce, C.; Bollag, W.B.; et al. H3K9 Trimethylation Silences FAS Expression to Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J. Immunol 2015, 195, 1868–1882. [Google Scholar] [CrossRef]
- Lu, C.; Klement, J.D.; Yang, D.; Albers, T.; Lebedyeva, I.O.; Waller, J.L.; Liu, K. SUV39H1 regulates human colon carcinoma apoptosis and cell cycle to promote tumor growth. Cancer Lett. 2020, 476, 87–96. [Google Scholar] [CrossRef]
- Klymenko, Y.; Nephew, K.P. Epigenetic Crosstalk between the Tumor Microenvironment and Ovarian Cancer Cells: A Therapeutic Road Less Traveled. Cancers 2018, 10, 295. [Google Scholar] [CrossRef]
- Williams, J.A. Vector Design for Improved DNA Vaccine Efficacy, Safety and Production. Vaccines 2013, 1, 225–249. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Skrzypczynska, K.M.; Fang, Q.; Zhang, W.; O’Brien, S.A.; He, Y.; Wang, L.; Zhang, Q.; Kim, A.; et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell 2020, 181, 442–459. [Google Scholar] [CrossRef]
- Xiao, W.; Ibrahim, M.L.; Redd, P.S.; Klement, J.D.; Lu, C.; Yang, D.; Savage, N.M.; Liu, K. Loss of FAS Expression and Function Is Coupled with Colon Cancer Resistance to Immune Checkpoint Inhibitor Immunotherapy. Mol. Cancer Res. 2018, 17, 420–430. [Google Scholar] [CrossRef]
- Lu, C.; Stewart, D.J.; Lee, J.J.; Ji, L.; Ramesh, R.; Jayachandran, G.; Nunez, M.I.; Wistuba, I.I.; Erasmus, J.J.; Hicks, M.E.; et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS ONE 2012, 7, e34833. [Google Scholar] [CrossRef]
- Meraz, I.M.; Majidi, M.; Cao, X.; Lin, H.; Li, L.; Wang, J.; Baladandayuthapani, V.; Rice, D.; Sepesi, B.; Ji, L.; et al. TUSC2 Immunogene Therapy Synergizes with Anti-PD-1 through Enhanced Proliferation and Infiltration of Natural Killer Cells in Syngeneic Kras-Mutant Mouse Lung Cancer Models. Cancer Immunol. Res. 2018, 6, 163–177. [Google Scholar] [CrossRef]
- Dai, B.; Yan, S.; Lara-Guerra, H.; Kawashima, H.; Sakai, R.; Jayachandran, G.; Majidi, M.; Mehran, R.; Wang, J.; Bekele, B.N.; et al. Exogenous Restoration of TUSC2 Expression Induces Responsiveness to Erlotinib in Wildtype Epidermal Growth Factor Receptor (EGFR) Lung Cancer Cells through Context Specific Pathways Resulting in Enhanced Therapeutic Efficacy. PLoS ONE 2015, 10, e0123967. [Google Scholar] [CrossRef]
- Templeton, N.S.; Lasic, D.D.; Frederik, P.M.; Strey, H.H.; Roberts, D.D.; Pavlakis, G.N. Improved DNA: Liposome complexes for increased systemic delivery and gene expression. Nat. Biotechnol. 1997, 15, 647–652. [Google Scholar] [CrossRef]
- Griswold, D.P.; Corbett, T.H. A colon tumor model for anticancer agent evaluation. Cancer 1975, 36, 2441–2444. [Google Scholar] [CrossRef]
- Castle, J.C.; Loewer, M.; Boegel, S.; de Graaf, J.; Bender, C.; Tadmor, A.D.; Boisguerin, V.; Bukur, T.; Sorn, P.; Paret, C.; et al. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom. 2014, 15, 190. [Google Scholar] [CrossRef]
- Schackert, H.K.; Fidler, I.J. Development of an animal model to study the biology of recurrent colorectal cancer originating from mesenteric lymph system metastases. Int. J. Cancer 1989, 44, 177–181. [Google Scholar] [CrossRef]
- Hewitt, R.E.; McMarlin, A.; Kleiner, D.; Wersto, R.; Martin, P.; Tsokos, M.; Stamp, G.W.; Stetler-Stevenson, W.G.; Tsoskas, M. Validation of a model of colon cancer progression. J. Pathol. 2000, 192, 446–454. [Google Scholar] [CrossRef]
- Fu, Q.; Fu, T.M.; Cruz, A.C.; Sengupta, P.; Thomas, S.K.; Wang, S.; Siegel, R.M.; Wu, H.; Chou, J.J. Structural Basis and Functional Role of Intramembrane Trimerization of the FAS/CD95 Death Receptor. Mol. Cell 2016, 61, 602–613. [Google Scholar] [CrossRef]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-FAS antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Jost, P.J.; Grabow, S.; Gray, D.; McKenzie, M.D.; Nachbur, U.; Huang, D.C.; Bouillet, P.; Thomas, H.E.; Borner, C.; Silke, J.; et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009, 460, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Abrams, S.I.; Coleman, C.N.; Camphausen, K.; Schlom, J.; Hodge, J.W. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 2004, 64, 4328–4337. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, M.; Abrams, S.I.; Camphausen, K.; Liu, K.; Scott, T.; Coleman, C.N.; Hodge, J.W. Irradiation of tumor cells up-regulates FAS and enhances CTL lytic activity and CTL adoptive immunotherapy. J. Immunol. 2003, 170, 6338–6347. [Google Scholar] [CrossRef]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen FAS can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the FAS pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- Singh, N.; Lee, Y.G.; Shestova, O.; Ravikumar, P.; Hayer, K.E.; Hong, S.J.; Lu, X.M.; Pajarillo, R.; Agarwal, S.; Kuramitsu, S.; et al. Impaired Death Receptor Signaling in Leukemia Causes Antigen-Independent Resistance by Inducing CAR T-cell Dysfunction. Cancer Discov. 2020, 10, 552–567. [Google Scholar] [CrossRef]
- Dufva, O.; Koski, J.; Maliniemi, P.; Ianevski, A.; Klievink, J.; Leitner, J.; Polonen, P.; Hohtari, H.; Saeed, K.; Hannunen, T.; et al. Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 2020, 135, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Houston, A.M.; Michael-Robinson, J.M.; Walsh, M.D.; Cummings, M.C.; Ryan, A.E.; Lincoln, D.; Pandeya, N.; Jass, J.R.; Radford-Smith, G.L.; O’Connell, J. The “FAS counterattack” is not an active mode of tumor immune evasion in colorectal cancer with high-level microsatellite instability. Hum. Pathol. 2008, 39, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015, 5, 915–919. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merting, A.D.; Poschel, D.B.; Lu, C.; Klement, J.D.; Yang, D.; Li, H.; Shi, H.; Chapdelaine, E.; Montgomery, M.; Redman, M.T.; et al. Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo. Cancers 2022, 14, 361. https://doi.org/10.3390/cancers14020361
Merting AD, Poschel DB, Lu C, Klement JD, Yang D, Li H, Shi H, Chapdelaine E, Montgomery M, Redman MT, et al. Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo. Cancers. 2022; 14(2):361. https://doi.org/10.3390/cancers14020361
Chicago/Turabian StyleMerting, Alyssa D., Dakota B. Poschel, Chunwan Lu, John D. Klement, Dafeng Yang, Honglin Li, Huidong Shi, Eric Chapdelaine, Mitzi Montgomery, Michael T. Redman, and et al. 2022. "Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo" Cancers 14, no. 2: 361. https://doi.org/10.3390/cancers14020361
APA StyleMerting, A. D., Poschel, D. B., Lu, C., Klement, J. D., Yang, D., Li, H., Shi, H., Chapdelaine, E., Montgomery, M., Redman, M. T., Savage, N. M., Nayak-Kapoor, A., & Liu, K. (2022). Restoring FAS Expression via Lipid-Encapsulated FAS DNA Nanoparticle Delivery Is Sufficient to Suppress Colon Tumor Growth In Vivo. Cancers, 14(2), 361. https://doi.org/10.3390/cancers14020361