

MDMX Regulates Transcriptional Activity of p53 and FOXO Proteins to Stimulate Proliferation of Melanoma Cells

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viability Assays
2.2. Manipulation of Cell Lines
2.2.1. Establishment of Inducible MDMX Knockdown Cell Lines
2.2.2. Generation of p53 Knockout MEL202-Derived Cells
2.3. RNA Sequencing
2.4. RNA Isolation, cDNA Synthesis and Real-Time Quantitative PCR
2.5. Western Blot Analysis
2.6. Statistical Analysis
3. Results
3.1. Identification of Genes Transcriptionally Regulated by MDMX
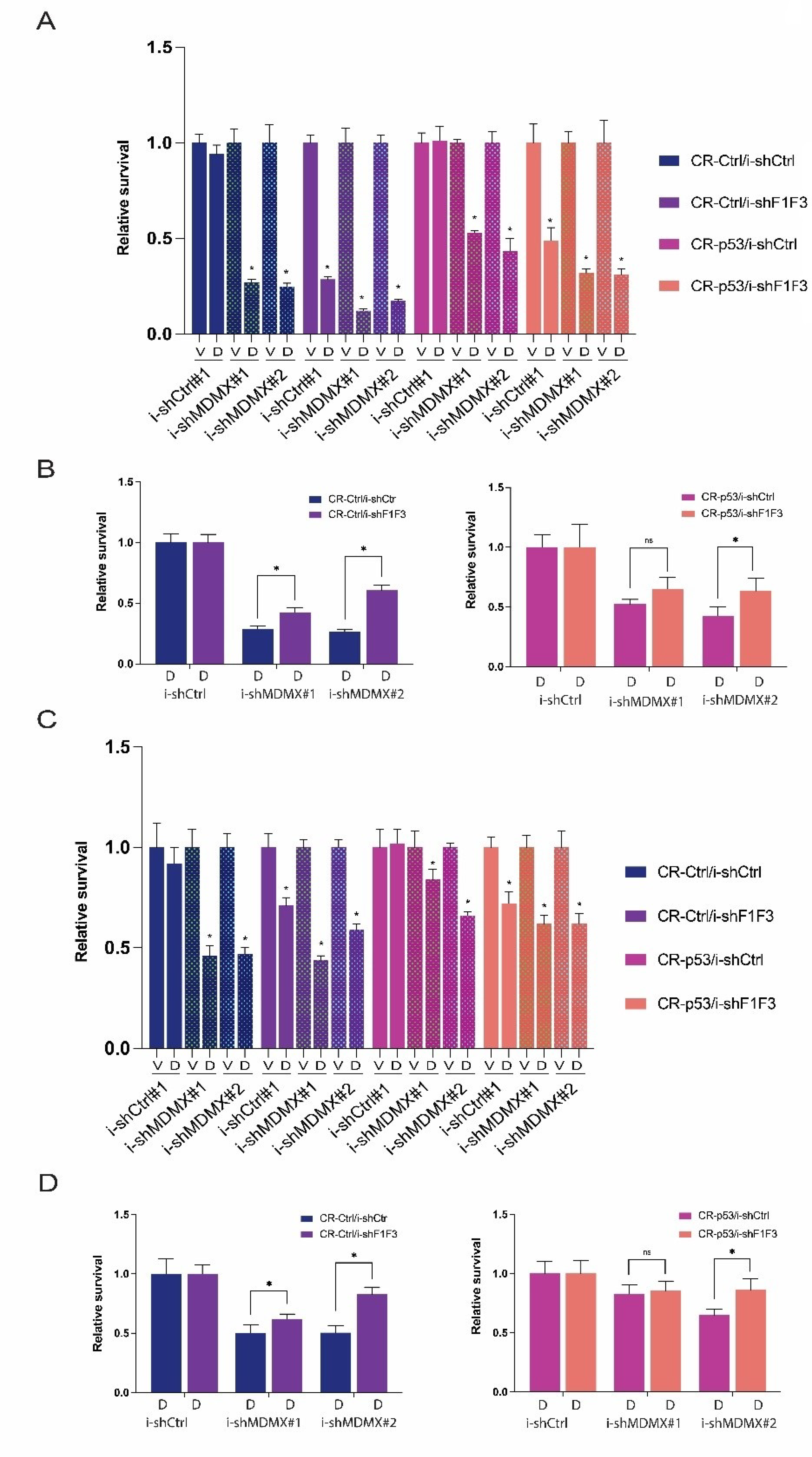
3.2. P53-Dependent and -Independent Effects of MDMX Depletion
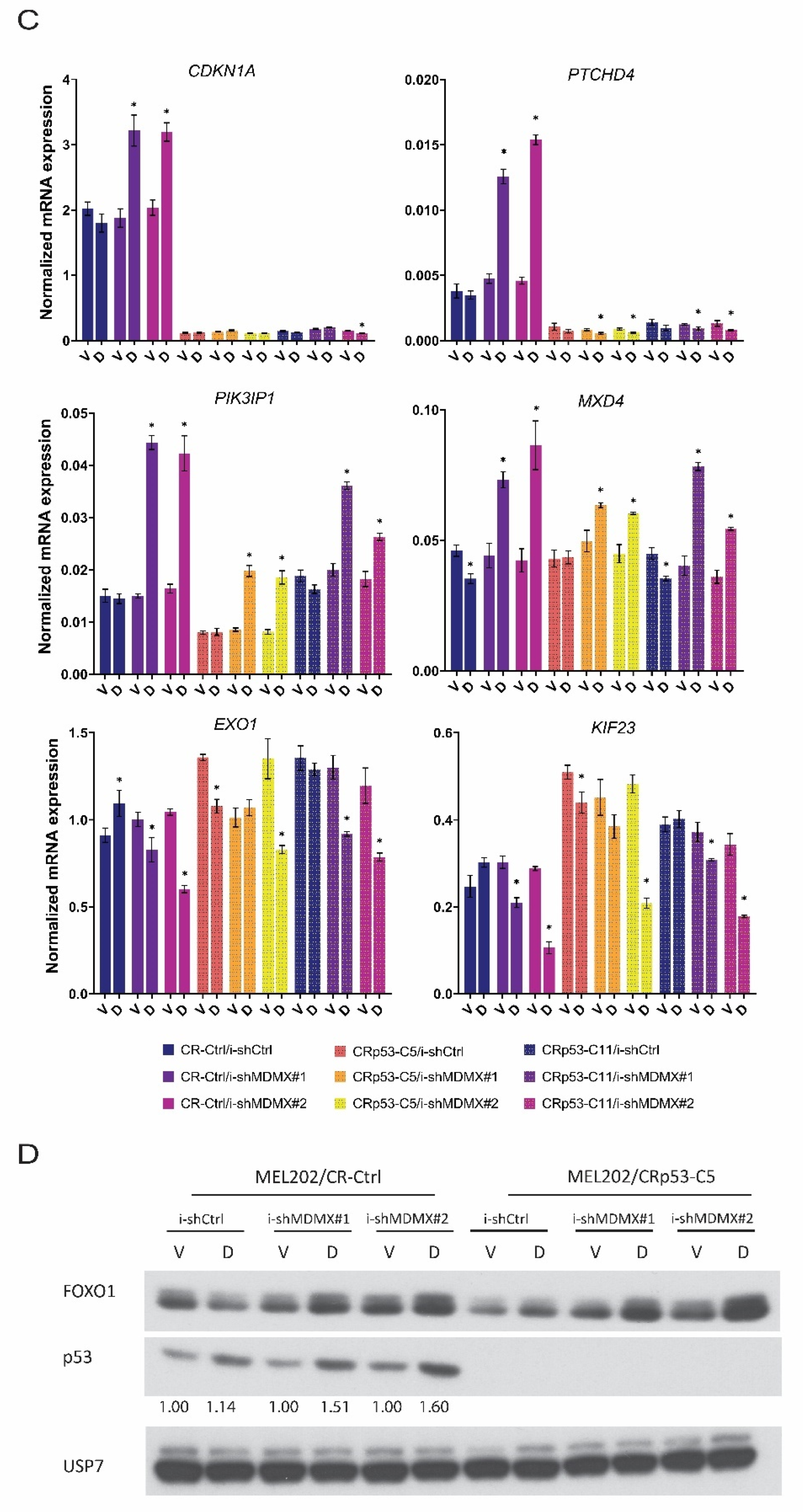
3.2.1. Nutlin-3 Mediated Regulation of MDMX Target Genes
3.2.2. Effects of MDMX-Knockdown in p53-Depleted Cells
3.3. FOXO-Dependent and -Independent Effects upon MDMX Depletion
3.3.1. Effects of FOXO Depletion upon MDMX Knockdown: Transcriptional Consequences
3.3.2. Effects of FOXO Depletion upon MDMX Knockdown: Growth Consequences
3.4. Regulation of RFX7 and RFX7-Target Genes upon MDMX Depletion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Jochemsen, A.G. Mdmx and Mdm2: Brothers in arms? Cell Cycle 2004, 3, 900–904. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef]
- Migliorini, D.; Lazzerini Denchi, E.; Danovi, D.; Jochemsen, A.; Capillo, M.; Gobbi, A.; Helin, K.; Pelicci, P.G.; Marine, J.C. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 2002, 22, 5527–5538. [Google Scholar] [CrossRef]
- Finch, R.A.; Donoviel, D.B.; Potter, D.; Shi, M.; Fan, A.; Freed, D.D.; Wang, C.Y.; Zambrowicz, B.P.; Ramirez-Solis, R.; Sands, A.T.; et al. mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002, 62, 3221–3225. [Google Scholar]
- Francoz, S.; Froment, P.; Bogaerts, S.; De Clercq, S.; Maetens, M.; Doumont, G.; Bellefroid, E.; Marine, J.C. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 3232–3237. [Google Scholar] [CrossRef]
- Marine, J.C.; Francoz, S.; Maetens, M.; Wahl, G.; Toledo, F.; Lozano, G. Keeping p53 in check: Essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ. 2006, 13, 927–934. [Google Scholar] [CrossRef]
- Valentin-Vega, Y.A.; Okano, H.; Lozano, G. The intestinal epithelium compensates for p53-mediated cell death and guarantees organismal survival. Cell Death Differ. 2008, 15, 1772–1781. [Google Scholar] [CrossRef] [PubMed]
- Valentin-Vega, Y.A.; Box, N.; Terzian, T.; Lozano, G. Mdm4 loss in the intestinal epithelium leads to compartmentalized cell death but no tissue abnormalities. Differentiation 2009, 77, 442–449. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grier, J.D.; Xiong, S.; Elizondo-Fraire, A.C.; Parant, J.M.; Lozano, G. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol. Cell. Biol. 2006, 26, 192–198. [Google Scholar] [CrossRef]
- Ringshausen, I.; O’Shea, C.C.; Finch, A.J.; Swigart, L.B.; Evan, G.I. Mdm2 is critically and continuously required to suppress lethal p53 activity in vivo. Cancer Cell 2006, 10, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Moyer, S.M.; Larsson, C.A.; Lozano, G. Mdm proteins: Critical regulators of embry ogenesis and homeostasis. J. Mol. Cell Biol. 2017, 9, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Oren, M. Enhanced binding of a 95 kDa protein to p53 in cells undergoing p53-mediated growth arrest. EMBO J. 1992, 11, 2115–2121. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Bottger, V.; Bottger, A.; Garcia-Echeverria, C.; Ramos, Y.F.; van der Eb, A.J.; Jochemsen, A.G.; Lane, D.P. Comparative study of the p53-mdm2 and p53-MDMX interfaces. Oncogene 1999, 18, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Sharp, D.A.; Kratowicz, S.A.; Sank, M.J.; George, D.L. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem. 1999, 274, 38189–38196. [Google Scholar] [CrossRef] [PubMed]
- Linares, L.K.; Hengstermann, A.; Ciechanover, A.; Muller, S.; Scheffner, M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc. Natl. Acad. Sci. USA 2003, 100, 12009–12014. [Google Scholar] [CrossRef]
- Little, N.A.; Jochemsen, A.G. Hdmx and Mdm2 can repress transcription activation by p53 but not by p63. Oncogene 2001, 20, 4576–4580. [Google Scholar] [CrossRef]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004, 23, 1547–1556. [Google Scholar] [CrossRef]
- Pereg, Y.; Shkedy, D.; de Graaf, P.; Meulmeester, E.; Edelson-Averbukh, M.; Salek, M.; Biton, S.; Teunisse, A.F.; Lehmann, W.D.; Jochemsen, A.G.; et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc. Natl. Acad. Sci. USA 2005, 102, 5056–5061. [Google Scholar] [CrossRef]
- Okamoto, K.; Kashima, K.; Pereg, Y.; Ishida, M.; Yamazaki, S.; Nota, A.; Teunisse, A.; Migliorini, D.; Kitabayashi, I.; Marine, J.C.; et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol. Cell. Biol. 2005, 25, 9608–9620. [Google Scholar] [CrossRef] [PubMed]
- Meulmeester, E.; Maurice, M.M.; Boutell, C.; Teunisse, A.F.; Ovaa, H.; Abraham, T.E.; Dirks, R.W.; Jochemsen, A.G. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol. Cell 2005, 18, 565–576. [Google Scholar] [CrossRef]
- Cheng, Q.; Chen, L.; Li, Z.; Lane, W.S.; Chen, J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009, 28, 3857–3867. [Google Scholar] [CrossRef]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef]
- Phillips, A.; Teunisse, A.; Lam, S.; Lodder, K.; Darley, M.; Emaduddin, M.; Wolf, A.; Richter, J.; de Lange, J.; Verlaan-de Vries, M.; et al. HDMX-L is expressed from a functional p53-responsive promoter in the first intron of the HDMX gene and participates in an autoregulatory feedback loop to control p53 activity. J. Biol. Chem. 2010, 285, 29111–29127. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef]
- Hollstein, M.; Hergenhahn, M.; Yang, Q.; Bartsch, H.; Wang, Z.Q.; Hainaut, P. New approaches to understanding p53 gene tumor mutation spectra. Mutat. Res. 1999, 431, 199–209. [Google Scholar] [CrossRef]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Sanz, G.; Singh, M.; Peuget, S.; Selivanova, G. Inhibition of p53 inhibitors: Progress, challenges and perspectives. J. Mol. Cell Biol. 2019, 11, 586–599. [Google Scholar] [CrossRef]
- Biswas, S.; Killick, E.; Jochemsen, A.G.; Lunec, J. The clinical development of p53-reactivating drugs in sarcomas—Charting future therapeutic approaches and understanding the clinical molecular toxicology of Nutlins. Expert Opin. Investig. Drugs 2014, 23, 629–645. [Google Scholar] [CrossRef]
- Haupt, S.; Mejia-Hernandez, J.O.; Vijayakumaran, R.; Keam, S.P.; Haupt, Y. The long and the short of it: The MDM4 tail so far. J. Mol. Cell Biol. 2019, 11, 231–244. [Google Scholar] [CrossRef]
- Dewaele, M.; Tabaglio, T.; Willekens, K.; Bezzi, M.; Teo, S.X.; Low, D.H.; Koh, C.M.; Rambow, F.; Fiers, M.; Rogiers, A.; et al. Antisense oligonucleotide-mediated MDM4 exon 6 skipping impairs tumor growth. J. Clin. Investig. 2016, 126, 68–84. [Google Scholar] [CrossRef] [PubMed]
- Laurie, N.A.; Donovan, S.L.; Shih, C.S.; Zhang, J.; Mills, N.; Fuller, C.; Teunisse, A.; Lam, S.; Ramos, Y.; Mohan, A.; et al. Inactivation of the p53 pathway in retinoblastoma. Nature 2006, 444, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gembarska, A.; Luciani, F.; Fedele, C.; Russell, E.A.; Dewaele, M.; Villar, S.; Zwolinska, A.; Haupt, S.; de Lange, J.; Yip, D.; et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 2012, 18, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- de Lange, J.; Teunisse, A.F.; Vries, M.V.; Lodder, K.; Lam, S.; Luyten, G.P.; Bernal, F.; Jager, M.J.; Jochemsen, A.G. High levels of Hdmx promote cell growth in a subset of uveal melanomas. Am. J. Cancer Res. 2012, 2, 492–507. [Google Scholar]
- Heijkants, R.C.; Nieveen, M.; Hart, K.C.; Teunisse, A.; Jochemsen, A.G. Targeting MDMX and PKCdelta to improve current uveal melanoma therapeutic strategies. Oncogenesis 2018, 7, 33. [Google Scholar] [CrossRef]
- Haupt, S.; Buckley, D.; Pang, J.M.; Panimaya, J.; Paul, P.J.; Gamell, C.; Takano, E.A.; Lee, Y.Y.; Hiddingh, S.; Rogers, T.M.; et al. Targeting Mdmx to treat breast cancers with wild-type p53. Cell Death Dis. 2015, 6, e1821. [Google Scholar] [CrossRef]
- Jeffreena Miranda, P.; Buckley, D.; Raghu, D.; Pang, J.B.; Takano, E.A.; Vijayakumaran, R.; Teunisse, A.F.; Posner, A.; Procter, T.; Herold, M.J.; et al. MDM4 is a rational target for treating breast cancers with mutant p53. J. Pathol. 2017, 241, 661–670. [Google Scholar] [CrossRef]
- Carrillo, A.M.; Bouska, A.; Arrate, M.P.; Eischen, C.M. Mdmx promotes genomic instability independent of p53 and Mdm2. Oncogene 2015, 34, 846–856. [Google Scholar] [CrossRef]
- Klein, A.M.; de Queiroz, R.M.; Venkatesh, D.; Prives, C. The roles and regulation of MDM2 and MDMX: It is not just about p53. Genes Dev. 2021, 35, 575–601. [Google Scholar] [CrossRef]
- Chen, P.W.; Murray, T.G.; Uno, T.; Salgaller, M.L.; Reddy, R.; Ksander, B.R. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin. Exp. Metastasis 1997, 15, 509–518. [Google Scholar] [CrossRef]
- De Waard-Siebinga, I.; Blom, D.J.; Griffioen, M.; Schrier, P.I.; Hoogendoorn, E.; Beverstock, G.; Danen, E.H.; Jager, M.J. Establishment and characterization of an uveal-melanoma cell line. Int. J. Cancer 1995, 62, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Herold, M.J.; van den Brandt, J.; Seibler, J.; Reichardt, H.M. Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc. Natl. Acad. Sci. USA 2008, 105, 18507–18512. [Google Scholar] [CrossRef] [PubMed]
- Carlotti, F.; Bazuine, M.; Kekarainen, T.; Seppen, J.; Pognonec, P.; Maassen, J.A.; Hoeben, R.C. Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol. Ther. 2004, 9, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Rinsma, M.; Janssen, J.M.; Liu, J.; Maggio, I.; Goncalves, M.A. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016, 44, 6482–6492. [Google Scholar] [CrossRef]
- Benedict, B.; van Schie, J.J.M.; Oostra, A.B.; Balk, J.A.; Wolthuis, R.M.F.; Riele, H.T.; de Lange, J. WAPL-Dependent Repair of Damaged DNA Replication Forks Underlies Oncogene-Induced Loss of Sister Chromatid Cohesion. Dev. Cell 2020, 52, 683–698.e7. [Google Scholar] [CrossRef]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Goncalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef]
- Holkers, M.; Maggio, I.; Henriques, S.F.; Janssen, J.M.; Cathomen, T.; Goncalves, M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef]
- Szuhai, K.; Bezrookove, V.; Wiegant, J.; Vrolijk, J.; Dirks, R.W.; Rosenberg, C.; Raap, A.K.; Tanke, H.J. Simultaneous molecular karyotyping and mapping of viral DNA integration sites by 25-color COBRA-FISH. Genes Chromosomes Cancer 2000, 28, 92–97. [Google Scholar] [CrossRef]
- Szuhai, K.; Tanke, H.J. COBRA: Combined binary ratio labeling of nucleic-acid probes for multi-color fluorescence in situ hybridization karyotyping. Nat. Protoc. 2006, 1, 264–275. [Google Scholar] [CrossRef]
- Janky, R.; Verfaillie, A.; Imrichova, H.; Van de Sande, B.; Standaert, L.; Christiaens, V.; Hulselmans, G.; Herten, K.; Naval Sanchez, M.; Potier, D.; et al. iRegulon: From a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014, 10, e1003731. [Google Scholar] [CrossRef]
- Fischer, M. Census and evaluation of p53 target genes. Oncogene 2017, 36, 3943–3956. [Google Scholar] [CrossRef] [PubMed]
- Sammons, M.A.; Nguyen, T.T.; McDade, S.S.; Fischer, M. Tumor suppressor p53: From engaging DNA to target gene regulation. Nucleic Acids Res. 2020, 48, 8848–8869. [Google Scholar] [CrossRef] [PubMed]
- Jochemsen, A.G. Reactivation of p53 as therapeutic intervention for malignant melanoma. Curr. Opin. Oncol. 2014, 26, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Grossmann, P.; Padi, M.; DeCaprio, J.A. Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 2016, 44, 6070–6086. [Google Scholar] [CrossRef]
- Schmidt-Strassburger, U.; Schips, T.G.; Maier, H.J.; Kloiber, K.; Mannella, F.; Braunstein, K.E.; Holzmann, K.; Ushmorov, A.; Liebau, S.; Boeckers, T.M.; et al. Expression of constitutively active FoxO3 in murine forebrain leads to a loss of neural progenitors. FASEB J. 2012, 26, 4990–5001. [Google Scholar] [CrossRef]
- de Lange, J.; Ly, L.V.; Lodder, K.; Verlaan-de Vries, M.; Teunisse, A.F.; Jager, M.J.; Jochemsen, A.G. Synergistic growth inhibition based on small-molecule p53 activation as treatment for intraocular melanoma. Oncogene 2012, 31, 1105–1116. [Google Scholar] [CrossRef]
- Hornsveld, M.; Smits, L.M.M.; Meerlo, M.; van Amersfoort, M.; Groot Koerkamp, M.J.A.; van Leenen, D.; Kloet, D.E.A.; Holstege, F.C.P.; Derksen, P.W.B.; Burgering, B.M.T.; et al. FOXO Transcription Factors Both Suppress and Support Breast Cancer Progression. Cancer Res. 2018, 78, 2356–2369. [Google Scholar] [CrossRef]
- Varmeh-Ziaie, S.; Okan, I.; Wang, Y.; Magnusson, K.P.; Warthoe, P.; Strauss, M.; Wiman, K.G. Wig-1, a new p53-induced gene encoding a zinc finger protein. Oncogene 1997, 15, 2699–2704. [Google Scholar] [CrossRef]
- Coronel, L.; Riege, K.; Schwab, K.; Forste, S.; Hackes, D.; Semerau, L.; Bernhart, S.H.; Siebert, R.; Hoffmann, S.; Fischer, M. Transcription factor RFX7 governs a tumor suppressor network in response to p53 and stress. Nucleic Acids Res. 2021, 49, 7437–7456. [Google Scholar] [CrossRef]
- Fischer, M.; Schwarz, R.; Riege, K.; DeCaprio, J.A.; Hoffmann, S. TargetGeneReg 2.0: A comprehensive web-atlas for p53, p63, and cell cycle-dependent gene regulation. Nucleic Acids Res. Cancer 2022, 4, zcac009. [Google Scholar] [CrossRef]
- McDonel, P.; Demmers, J.; Tan, D.W.; Watt, F.; Hendrich, B.D. Sin3a is essential for the genome integrity and viability of pluripotent cells. Dev. Biol. 2012, 363, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Dannenberg, J.H.; David, G.; Zhong, S.; van der Torre, J.; Wong, W.H.; Depinho, R.A. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005, 19, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Collado, M.; Villasante, A.; Matheu, A.; Lynch, C.J.; Canamero, M.; Rizzoti, K.; Carneiro, C.; Martinez, G.; Vidal, A.; et al. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell 2012, 11, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Quaas, M.; Steiner, L.; Engeland, K. The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res. 2016, 44, 164–174. [Google Scholar] [CrossRef]
- Fischer, M.; Steiner, L.; Engeland, K. The transcription factor p53: Not a repressor, solely an activator. Cell Cycle 2014, 13, 3037–3058. [Google Scholar] [CrossRef]
- He, X.; Zhu, Z.; Johnson, C.; Stoops, J.; Eaker, A.E.; Bowen, W.; DeFrances, M.C. PIK3IP1, a negative regulator of PI3K, suppresses the development of hepatocellular carcinoma. Cancer Res. 2008, 68, 5591–5598. [Google Scholar] [CrossRef]
- Gaviraghi, M.; Vivori, C.; Pareja Sanchez, Y.; Invernizzi, F.; Cattaneo, A.; Santoliquido, B.M.; Frenquelli, M.; Segalla, S.; Bachi, A.; Doglioni, C.; et al. Tumor suppressor PNRC1 blocks rRNA maturation by recruiting the decapping complex to the nucleolus. EMBO J. 2018, 37, e99179. [Google Scholar] [CrossRef]
- Hurlin, P.J.; Queva, C.; Koskinen, P.J.; Steingrimsson, E.; Ayer, D.E.; Copeland, N.G.; Jenkins, N.A.; Eisenman, R.N. Mad3 and Mad4: Novel Max-interacting transcriptional repressors that suppress c-myc dependent transformation and are expressed during neural and epidermal differentiation. EMBO J. 1995, 14, 5646–5659. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heijkants, R.C.; Teunisse, A.F.A.S.; de Jong, D.; Glinkina, K.; Mei, H.; Kielbasa, S.M.; Szuhai, K.; Jochemsen, A.G. MDMX Regulates Transcriptional Activity of p53 and FOXO Proteins to Stimulate Proliferation of Melanoma Cells. Cancers 2022, 14, 4482. https://doi.org/10.3390/cancers14184482
Heijkants RC, Teunisse AFAS, de Jong D, Glinkina K, Mei H, Kielbasa SM, Szuhai K, Jochemsen AG. MDMX Regulates Transcriptional Activity of p53 and FOXO Proteins to Stimulate Proliferation of Melanoma Cells. Cancers. 2022; 14(18):4482. https://doi.org/10.3390/cancers14184482
Chicago/Turabian StyleHeijkants, Renier C., Amina F. A. S. Teunisse, Danielle de Jong, Kseniya Glinkina, Hailiang Mei, Szymon M. Kielbasa, Karoly Szuhai, and Aart G. Jochemsen. 2022. "MDMX Regulates Transcriptional Activity of p53 and FOXO Proteins to Stimulate Proliferation of Melanoma Cells" Cancers 14, no. 18: 4482. https://doi.org/10.3390/cancers14184482
APA StyleHeijkants, R. C., Teunisse, A. F. A. S., de Jong, D., Glinkina, K., Mei, H., Kielbasa, S. M., Szuhai, K., & Jochemsen, A. G. (2022). MDMX Regulates Transcriptional Activity of p53 and FOXO Proteins to Stimulate Proliferation of Melanoma Cells. Cancers, 14(18), 4482. https://doi.org/10.3390/cancers14184482