
IRF4 as an Oncogenic Master Transcription Factor


{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. IRF4 as a Master Regulator of Immunity
3. Molecular Functions of IRF4
3.1. IRF4 and DNA-Binding Motifs
3.2. Binding Partners and Downstream Targets of IRF4
3.2.1. ETS Family Transcription Factors
3.2.2. AP1 Family Transcription Factors
3.2.3. Other IRF4-Interacting Proteins
3.3. Regulation of IRF4 Activity
3.3.1. Upstream Signaling Pathways of IRF4
3.3.2. Post-Transcriptional Regulation of IRF4
3.4. IRF4 as a Transcriptional Repressor
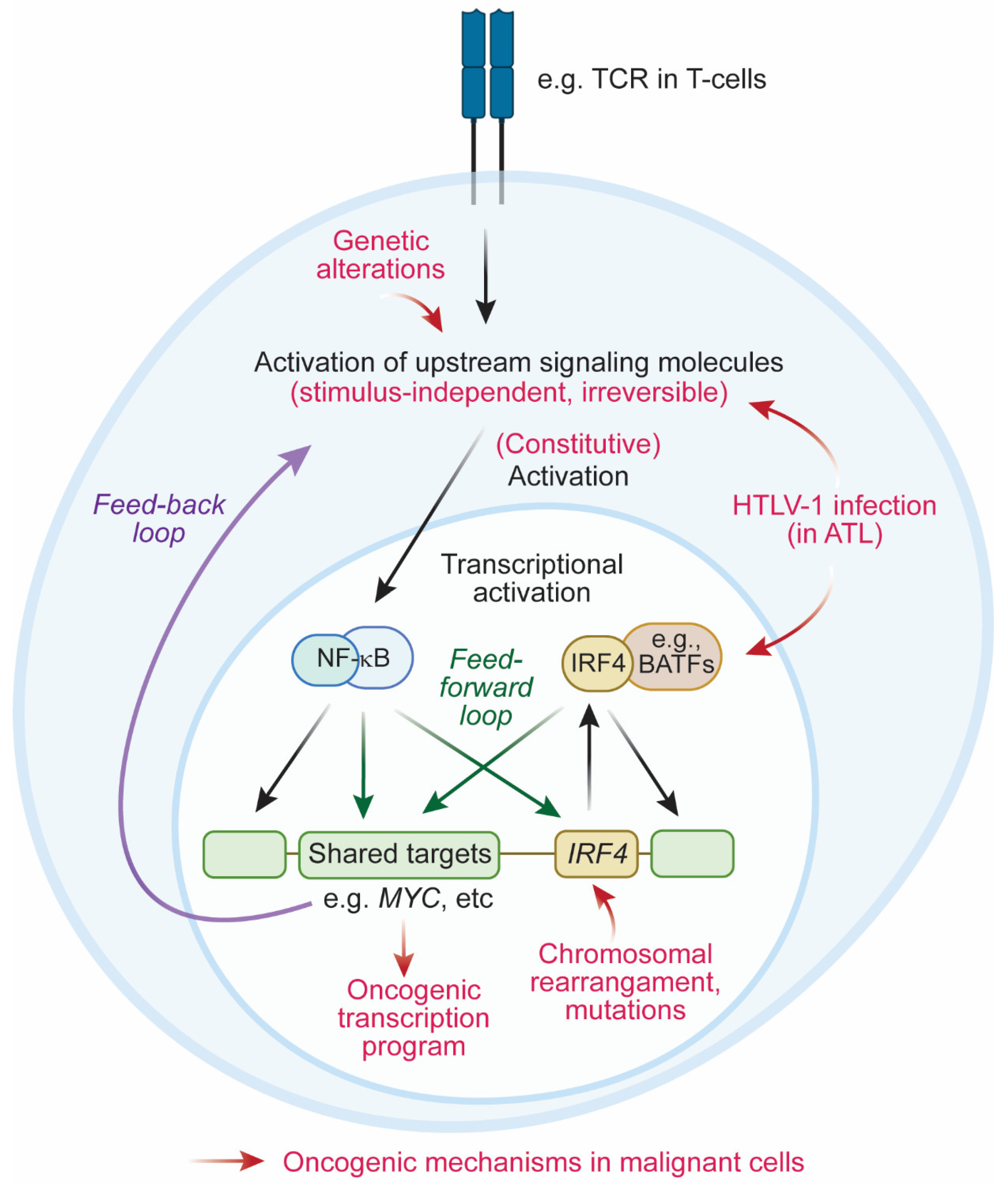
4. Oncogenic Mechanisms Mediated by IRF4
4.1. Activation of IRF4 in Mature Lymphoid Neoplasms
4.2. Oncogenic Transcription Programs Driven by IRF4
4.2.1. MYC Is a Common Downstream Target of IRF4
4.2.2. IRF4 Is an Oncogenic Master Transcription Factor That Characterizes Tumor Type
4.2.3. Feedback Loop between IRF4 and NF-κB
4.3. Coherent Feedforward Loop Mediated by IRF4 and NF-κB
5. Discussion and Future Directions
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Yanai, H.; Negishi, H.; Taniguchi, T. The IRF family of transcription factors: Inception, impact and implications in oncogenesis. OncoImmunology 2012, 1, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Au, W.C.; Moore, P.A.; Lowther, W.; Juang, Y.T.; Pitha, P.M. Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc. Natl. Acad. Sci. USA 1995, 92, 11657–11661. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Grossman, A.; Mittrucker, H.W.; Siderovski, D.P.; Kiefer, F.; Kawakami, T.; Richardson, C.D.; Taniguchi, T.; Yoshinaga, S.K.; Mak, T.W. Molecular cloning of LSIRF, a lymphoid-specific member of the interferon regulatory factor family that binds the interferon-stimulated response element (ISRE). Nucleic Acids Res. 1995, 23, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jiang, M.; Anthony, A.; Pernis, A.B. Lineage-specific modulation of interleukin 4 signaling by interferon regulatory factor 4. J. Exp. Med. 1999, 190, 1837–1848. [Google Scholar] [CrossRef]
- Marecki, S.; Atchison, M.L.; Fenton, M.J. Differential expression and distinct functions of IFN regulatory factor 4 and IFN consensus sequence binding protein in macrophages. J. Immunol. 1999, 163, 2713–2722. [Google Scholar]
- Lehtonen, A.; Veckman, V.; Nikula, T.; Lahesmaa, R.; Kinnunen, L.; Matikainen, S.; Julkunen, I. Differential expression of IFN regulatory factor 4 gene in human monocyte-derived dendritic cells and macrophages. J. Immunol. 2005, 175, 6570–6579. [Google Scholar] [CrossRef]
- Mittrucker, H.W.; Matsuyama, T.; Grossman, A.; Kundig, T.M.; Potter, J.; Shahinian, A.; Wakeham, A.; Patterson, B.; Ohashi, P.S.; Mak, T.W. Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 1997, 275, 540–543. [Google Scholar] [CrossRef]
- Iida, S.; Rao, P.H.; Butler, M.; Corradini, P.; Boccadoro, M.; Klein, B.; Chaganti, R.S.; Dalla-Favera, R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 1997, 17, 226–230. [Google Scholar] [CrossRef]
- Yamagata, T.; Nishida, J.; Tanaka, S.; Sakai, R.; Mitani, K.; Yoshida, M.; Taniguchi, T.; Yazaki, Y.; Hirai, H. A novel interferon regulatory factor family transcription factor, ICSAT/Pip/LSIRF, that negatively regulates the activity of interferon-regulated genes. Mol. Cell Biol. 1996, 16, 1283–1294. [Google Scholar] [CrossRef]
- Eisenbeis, C.F.; Singh, H.; Storb, U. PU.1 is a component of a multiprotein complex which binds an essential site in the murine immunoglobulin lambda 2-4 enhancer. Mol. Cell Biol. 1993, 13, 6452–6461. [Google Scholar] [PubMed]
- Pongubala, J.M.; Nagulapalli, S.; Klemsz, M.J.; McKercher, S.R.; Maki, R.A.; Atchison, M.L. PU.1 recruits a second nuclear factor to a site important for immunoglobulin kappa 3′ enhancer activity. Mol. Cell Biol. 1992, 12, 368–378. [Google Scholar]
- Amanda, S.; Tan, T.K.; Iida, S.; Sanda, T. Lineage- and stage-specific oncogenicity of IRF4. Exp. Hematol. 2022; online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Miasari, M.; Shi, W.; Xin, A.; Henstridge, D.C.; Preston, S.; Pellegrini, M.; Belz, G.T.; Smyth, G.K.; Febbraio, M.A.; et al. The transcription factor IRF4 is essential for TCR affinity-mediated metabolic programming and clonal expansion of T cells. Nat. Immunol. 2013, 14, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Durai, V.; Tussiwand, R.; Briseno, C.G.; Wu, X.; Grajales-Reyes, G.E.; Egawa, T.; Murphy, T.L.; Murphy, K.M. Quality of TCR signaling determined by differential affinities of enhancers for the composite BATF-IRF4 transcription factor complex. Nat. Immunol. 2017, 18, 563–572. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Kannanganat, S.; Maienschein-Cline, M.; Cook, S.L.; Chen, J.; Bahroos, N.; Sievert, E.; Corse, E.; Chong, A.; Sciammas, R. The IRF4 Gene Regulatory Module Functions as a Read-Write Integrator to Dynamically Coordinate T Helper Cell Fate. Immunity 2017, 47, 481–497.e7. [Google Scholar] [CrossRef]
- Conley, J.M.; Gallagher, M.P.; Rao, A.; Berg, L.J. Activation of the Tec Kinase ITK Controls Graded IRF4 Expression in Response to Variations in TCR Signal Strength. J. Immunol. 2020, 205, 335–345. [Google Scholar] [CrossRef]
- Lohoff, M.; Mittrucker, H.W.; Prechtl, S.; Bischof, S.; Sommer, F.; Kock, S.; Ferrick, D.A.; Duncan, G.S.; Gessner, A.; Mak, T.W. Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc. Natl. Acad. Sci. USA 2002, 99, 11808–11812. [Google Scholar] [CrossRef]
- Tominaga, N.; Ohkusu-Tsukada, K.; Udono, H.; Abe, R.; Matsuyama, T.; Yui, K. Development of Th1 and not Th2 immune responses in mice lacking IFN-regulatory factor-4. Int. Immunol. 2003, 15, 1–10. [Google Scholar] [CrossRef]
- Huber, M.; Brustle, A.; Reinhard, K.; Guralnik, A.; Walter, G.; Mahiny, A.; von Low, E.; Lohoff, M. IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc. Natl. Acad. Sci. USA 2008, 105, 20846–20851. [Google Scholar] [CrossRef]
- Lu, R.; Medina, K.L.; Lancki, D.W.; Singh, H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003, 17, 1703–1708. [Google Scholar] [CrossRef]
- Sciammas, R.; Shaffer, A.L.; Schatz, J.H.; Zhao, H.; Staudt, L.M.; Singh, H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity 2006, 25, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef]
- Ochiai, K.; Maienschein-Cline, M.; Simonetti, G.; Chen, J.; Rosenthal, R.; Brink, R.; Chong, A.S.; Klein, U.; Dinner, A.R.; Singh, H.; et al. Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 2013, 38, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Klein, U.; Casola, S.; Cattoretti, G.; Shen, Q.; Lia, M.; Mo, T.; Ludwig, T.; Rajewsky, K.; Dalla-Favera, R. Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat. Immunol. 2006, 7, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, G.; Carette, A.; Silva, K.; Wang, H.; De Silva, N.S.; Heise, N.; Siebel, C.W.; Shlomchik, M.J.; Klein, U. IRF4 controls the positioning of mature B cells in the lymphoid microenvironments by regulating NOTCH2 expression and activity. J. Exp. Med. 2013, 210, 2887–2902. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K.; et al. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 2003, 18, 675–685. [Google Scholar] [CrossRef]
- Tanigaki, K.; Han, H.; Yamamoto, N.; Tashiro, K.; Ikegawa, M.; Kuroda, K.; Suzuki, A.; Nakano, T.; Honjo, T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 2002, 3, 443–450. [Google Scholar] [CrossRef]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Negishi, H.; Ohba, Y.; Yanai, H.; Takaoka, A.; Honma, K.; Yui, K.; Matsuyama, T.; Taniguchi, T.; Honda, K. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 2005, 102, 15989–15994. [Google Scholar] [CrossRef]
- Suzuki, S.; Honma, K.; Matsuyama, T.; Suzuki, K.; Toriyama, K.; Akitoyo, I.; Yamamoto, K.; Suematsu, T.; Nakamura, M.; Yui, K.; et al. Critical roles of interferon regulatory factor 4 in CD11bhighCD8alpha- dendritic cell development. Proc. Natl. Acad. Sci. USA 2004, 101, 8981–8986. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Tjota, M.Y.; Clay, B.S.; Vander Lugt, B.; Bandukwala, H.S.; Hrusch, C.L.; Decker, D.C.; Blaine, K.M.; Fixsen, B.R.; Singh, H.; et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat. Commun. 2013, 4, 2990. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Kehrli, E.; Eisenbeis, C.F.; Storb, U.; Singh, H. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev. 1996, 10, 2335–2347. [Google Scholar] [CrossRef] [PubMed]
- Glasmacher, E.; Agrawal, S.; Chang, A.B.; Murphy, T.L.; Zeng, W.; Vander Lugt, B.; Khan, A.A.; Ciofani, M.; Spooner, C.J.; Rutz, S.; et al. A genomic regulatory element that directs assembly and function of immune-specific AP-1-IRF complexes. Science 2012, 338, 975–980. [Google Scholar] [CrossRef]
- Pongubala, J.M.; Van Beveren, C.; Nagulapalli, S.; Klemsz, M.J.; McKercher, S.R.; Maki, R.A.; Atchison, M.L. Effect of PU.1 phosphorylation on interaction with NF-EM5 and transcriptional activation. Science 1993, 259, 1622–1625. [Google Scholar] [CrossRef]
- Escalante, C.R.; Brass, A.L.; Pongubala, J.M.; Shatova, E.; Shen, L.; Singh, H.; Aggarwal, A.K. Crystal structure of PU.1/IRF-4/DNA ternary complex. Mol. Cell 2002, 10, 1097–1105. [Google Scholar] [CrossRef]
- Himmelmann, A.; Riva, A.; Wilson, G.L.; Lucas, B.P.; Thevenin, C.; Kehrl, J.H. PU.1/Pip and basic helix loop helix zipper transcription factors interact with binding sites in the CD20 promoter to help confer lineage- and stage-specific expression of CD20 in B lymphocytes. Blood 1997, 90, 3984–3995. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kato, T.; Hotta, C.; Nishiyama, A.; Kurotaki, D.; Yoshinari, M.; Takami, M.; Ichino, M.; Nakazawa, M.; Matsuyama, T.; et al. Shared and distinct functions of the transcription factors IRF4 and IRF8 in myeloid cell development. PLoS ONE 2011, 6, e25812. [Google Scholar] [CrossRef]
- Rao, S.; Matsumura, A.; Yoon, J.; Simon, M.C. SPI-B activates transcription via a unique proline, serine, and threonine domain and exhibits DNA binding affinity differences from PU.1. J. Biol. Chem. 1999, 274, 11115–11124. [Google Scholar] [CrossRef]
- Li, P.; Spolski, R.; Liao, W.; Wang, L.; Murphy, T.L.; Murphy, K.M.; Leonard, W.J. BATF-JUN is critical for IRF4-mediated transcription in T cells. Nature 2012, 490, 543–546. [Google Scholar] [CrossRef]
- Tussiwand, R.; Lee, W.L.; Murphy, T.L.; Mashayekhi, M.; Kc, W.; Albring, J.C.; Satpathy, A.T.; Rotondo, J.A.; Edelson, B.T.; Kretzer, N.M.; et al. Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 2012, 490, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Rengarajan, J.; Mowen, K.A.; McBride, K.D.; Smith, E.D.; Singh, H.; Glimcher, L.H. Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J. Exp. Med. 2002, 195, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Chaudhry, A.; Kas, A.; deRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Thierry-Mieg, D.; Thierry-Mieg, J.; Kim, H.P.; Oh, J.; Tunyaplin, C.; Carotta, S.; Donovan, C.E.; Goldman, M.L.; Tailor, P.; et al. Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 2009, 31, 941–952. [Google Scholar] [CrossRef]
- Ochiai, K.; Kondo, H.; Okamura, Y.; Shima, H.; Kurokochi, Y.; Kimura, K.; Funayama, R.; Nagashima, T.; Nakayama, K.; Yui, K.; et al. Zinc finger-IRF composite elements bound by Ikaros/IRF4 complexes function as gene repression in plasma cell. Blood Adv. 2018, 2, 883–894. [Google Scholar] [CrossRef]
- Grumont, R.J.; Gerondakis, S. Rel induces interferon regulatory factor 4 (IRF-4) expression in lymphocytes: Modulation of interferon-regulated gene expression by rel/nuclear factor kappaB. J. Exp. Med. 2000, 191, 1281–1292. [Google Scholar] [CrossRef]
- Pridans, C.; Holmes, M.L.; Polli, M.; Wettenhall, J.M.; Dakic, A.; Corcoran, L.M.; Smyth, G.K.; Nutt, S.L. Identification of Pax5 target genes in early B cell differentiation. J. Immunol. 2008, 180, 1719–1728. [Google Scholar] [CrossRef]
- Chen, Q.; Yang, W.; Gupta, S.; Biswas, P.; Smith, P.; Bhagat, G.; Pernis, A.B. IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 2008, 29, 899–911. [Google Scholar] [CrossRef]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Investig. 2010, 120, 3280–3295. [Google Scholar] [CrossRef]
- Biswas, P.S.; Gupta, S.; Stirzaker, R.A.; Kumar, V.; Jessberger, R.; Lu, T.T.; Bhagat, G.; Pernis, A.B. Dual regulation of IRF4 function in T and B cells is required for the coordination of T-B cell interactions and the prevention of autoimmunity. J. Exp. Med. 2012, 209, 581–596. [Google Scholar] [CrossRef]
- Mamane, Y.; Sharma, S.; Petropoulos, L.; Lin, R.; Hiscott, J. Posttranslational regulation of IRF-4 activity by the immunophilin FKBP52. Immunity 2000, 12, 129–140. [Google Scholar] [CrossRef]
- Rosenbauer, F.; Waring, J.F.; Foerster, J.; Wietstruk, M.; Philipp, D.; Horak, I. Interferon consensus sequence binding protein and interferon regulatory factor-4/Pip form a complex that represses the expression of the interferon-stimulated gene-15 in macrophages. Blood 1999, 94, 4274–4281. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Gao, J.; Basso, K.; Kitagawa, Y.; Smith, P.M.; Bhagat, G.; Pernis, A.; Pasqualucci, L.; Dalla-Favera, R. A signaling pathway mediating downregulation of BCL6 in germinal center B cells is blocked by BCL6 gene alterations in B cell lymphoma. Cancer Cell 2007, 12, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Nakazawa, N.; Iida, S.; Hayami, Y.; Sato, S.; Wakita, A.; Shimizu, S.; Taniwaki, M.; Ueda, R. Detection of MUM1/IRF4-IgH fusion in multiple myeloma. Leukemia 1999, 13, 1812–1816. [Google Scholar] [CrossRef]
- Chapman, M.A.; Lawrence, M.S.; Keats, J.J.; Cibulskis, K.; Sougnez, C.; Schinzel, A.C.; Harview, C.L.; Brunet, J.P.; Ahmann, G.J.; Adli, M.; et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011, 471, 467–472. [Google Scholar] [CrossRef]
- Lohr, J.G.; Stojanov, P.; Carter, S.L.; Cruz-Gordillo, P.; Lawrence, M.S.; Auclair, D.; Sougnez, C.; Knoechel, B.; Gould, J.; Saksena, G.; et al. Widespread genetic heterogeneity in multiple myeloma: Implications for targeted therapy. Cancer Cell 2014, 25, 91–101. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Kronke, J.; Udeshi, N.D.; Narla, A.; Grauman, P.; Hurst, S.N.; McConkey, M.; Svinkina, T.; Heckl, D.; Comer, E.; Li, X.; et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014, 343, 301–305. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; de Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9, 19. [Google Scholar] [CrossRef]
- Misiewicz-Krzeminska, I.; de Ramon, C.; Corchete, L.A.; Krzeminski, P.; Rojas, E.A.; Isidro, I.; Garcia-Sanz, R.; Martinez-Lopez, J.; Oriol, A.; Blade, J.; et al. Quantitative expression of Ikaros, IRF4, and PSMD10 proteins predicts survival in VRD-treated patients with multiple myeloma. Blood Adv. 2020, 4, 6023–6033. [Google Scholar] [CrossRef]
- Tsuboi, K.; Iida, S.; Inagaki, H.; Kato, M.; Hayami, Y.; Hanamura, I.; Miura, K.; Harada, S.; Kikuchi, M.; Komatsu, H.; et al. MUM1/IRF4 expression as a frequent event in mature lymphoid malignancies. Leukemia 2000, 14, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Natkunam, Y.; Warnke, R.A.; Montgomery, K.; Falini, B.; Van De Rijn, M. Analysis of MUM1/IRF4 protein expression using tissue microarrays and immunohistochemistry. Mod. Pathol. 2001, 14, 686–694. [Google Scholar] [CrossRef]
- Frauenfeld, L.; Castrejon-de-Anta, N.; Ramis-Zaldivar, J.E.; Streich, S.; Salmeron-Villalobos, J.; Otto, F.; Mayer, A.K.; Steinhilber, J.; Pinyol, M.; Mankel, B.; et al. Diffuse large B-cell lymphomas in adults with aberrant coexpression of CD10, BCL6, and MUM1 are enriched in IRF4 rearrangements. Blood Adv. 2022, 6, 2361–2372. [Google Scholar] [CrossRef]
- Salaverria, I.; Philipp, C.; Oschlies, I.; Kohler, C.W.; Kreuz, M.; Szczepanowski, M.; Burkhardt, B.; Trautmann, H.; Gesk, S.; Andrusiewicz, M.; et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011, 118, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of A20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef]
- Yang, Y.; Shaffer, A.L.; Emre, N.C.T.; Ceribelli, M.; Zhang, M.; Wright, G.; Xiao, W.; Powell, J.; Platig, J.; Kohlhammer, H.; et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 21, 723–737. [Google Scholar] [CrossRef]
- Zhang, L.H.; Kosek, J.; Wang, M.; Heise, C.; Schafer, P.H.; Chopra, R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br. J. Haematol. 2013, 160, 487–502. [Google Scholar] [CrossRef]
- Hernandez-Ilizaliturri, F.J.; Deeb, G.; Zinzani, P.L.; Pileri, S.A.; Malik, F.; Macon, W.R.; Goy, A.; Witzig, T.E.; Czuczman, M.S. Higher response to lenalidomide in relapsed/refractory diffuse large B-cell lymphoma in nongerminal center B-cell-like than in germinal center B-cell-like phenotype. Cancer 2011, 117, 5058–5066. [Google Scholar] [CrossRef]
- Mondello, P.; Steiner, N.; Willenbacher, W.; Ferrero, S.; Ghione, P.; Marabese, A.; Pitini, V.; Cuzzocrea, S.; Mian, M. Lenalidomide in Relapsed or Refractory Diffuse Large B-Cell Lymphoma: Is It a Valid Treatment Option? Oncologist 2016, 21, 1107–1112. [Google Scholar] [CrossRef]
- Gualco, G.; Queiroga, E.M.; Weiss, L.M.; Klumb, C.E.; Harrington, W.J., Jr.; Bacchi, C.E. Frequent expression of multiple myeloma 1/interferon regulatory factor 4 in Burkitt lymphoma. Hum. Pathol. 2009, 40, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Uccini, S.; Al-Jadiry, M.F.; Cippitelli, C.; Talerico, C.; Scarpino, S.; Al-Darraji, A.F.; Al-Badri, S.A.F.; Alsaadawi, A.R.; Al-Hadad, S.A.; Ruco, L. Burkitt lymphoma in Iraqi children: A distinctive form of sporadic disease with high incidence of EBV(+) cases and more frequent expression of MUM1/IRF4 protein in cases with head and neck presentation. Pediatr. Blood Cancer 2018, 65, e27399. [Google Scholar] [CrossRef]
- Kridel, R.; Mottok, A.; Farinha, P.; Ben-Neriah, S.; Ennishi, D.; Zheng, Y.; Chavez, E.A.; Shulha, H.P.; Tan, K.; Chan, F.C.; et al. Cell of origin of transformed follicular lymphoma. Blood 2015, 126, 2118–2127. [Google Scholar] [CrossRef]
- Naresh, K.N. MUM1 expression dichotomises follicular lymphoma into predominantly, MUM1-negative low-grade and MUM1-positive high-grade subtypes. Haematologica 2007, 92, 267–268. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pham-Ledard, A.; Prochazkova-Carlotti, M.; Laharanne, E.; Vergier, B.; Jouary, T.; Beylot-Barry, M.; Merlio, J.P. IRF4 gene rearrangements define a subgroup of CD30-positive cutaneous T-cell lymphoma: A study of 54 cases. J. Investig. Dermatol. 2010, 130, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Feldman, A.L.; Law, M.; Remstein, E.D.; Macon, W.R.; Erickson, L.A.; Grogg, K.L.; Kurtin, P.J.; Dogan, A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia 2009, 23, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Kiran, T.; Demirkesen, C.; Eker, C.; Kumusoglu, H.; Tuzuner, N. The significance of MUM1/IRF4 protein expression and IRF4 translocation of CD30(+) cutaneous T-cell lymphoproliferative disorders: A study of 53 cases. Leuk. Res. 2013, 37, 396–400. [Google Scholar] [CrossRef]
- Wada, D.A.; Law, M.E.; Hsi, E.D.; Dicaudo, D.J.; Ma, L.; Lim, M.S.; De Souza, A.; Comfere, N.I.; Weenig, R.H.; MacOn, W.R.; et al. Specificity of IRF4 translocations for primary cutaneous anaplastic large cell lymphoma: A multicenter study of 204 skin biopsies. Mod. Pathol. 2011, 24, 596–605. [Google Scholar] [CrossRef]
- Lim, M.S.; Bailey, N.G.; King, R.L.; Piris, M. Molecular Genetics in the Diagnosis and Biology of Lymphoid Neoplasms. Am. J. Clin. Pathol. 2019, 152, 277–301. [Google Scholar] [CrossRef]
- Sciallis, A.P.; Law, M.E.; Inwards, D.J.; McClure, R.F.; Macon, W.R.; Kurtin, P.J.; Dogan, A.; Feldman, A.L. Mucosal CD30-positive T-cell lymphoproliferations of the head and neck show a clinicopathologic spectrum similar to cutaneous CD30-positive T-cell lymphoproliferative disorders. Mod. Pathol. 2012, 25, 983–992. [Google Scholar] [CrossRef]
- Xing, X.; Flotte, T.J.; Law, M.E.; Blahnik, A.J.; Chng, W.J.; Huang, G.; Knudson, R.A.; Ketterling, R.P.; Porcher, J.C.; Ansell, S.M.; et al. Expression of the chemokine receptor gene, CCR8, is associated With DUSP22 rearrangements in anaplastic large cell lymphoma. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.J.; Tan, T.K.; Amanda, S.; Ngoc, P.C.T.; Leong, W.Z.; Tan, S.H.; Asamitsu, K.; Hibi, Y.; Ueda, R.; Okamoto, T.; et al. Feed-forward regulatory loop driven by IRF4 and NF-kappaB in adult T-cell leukemia/lymphoma. Blood 2020, 135, 934–947. [Google Scholar] [CrossRef]
- Good, L.; Sun, S.C. Persistent activation of NF-kappa B/Rel by human T-cell leukemia virus type 1 tax involves degradation of I kappa B beta. J. Virol. 1996, 70, 2730–2735. [Google Scholar] [CrossRef]
- Sun, S.C.; Yamaoka, S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene 2005, 24, 5952–5964. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Suzuki, T.; Fujisawa, J.; Inoue, J.; Yoshida, M. Tax protein of human T-cell leukemia virus type I binds to the ankyrin motifs of inhibitory factor kappa B and induces nuclear translocation of transcription factor NF-kappa B proteins for transcriptional activation. Proc. Natl. Acad. Sci. USA 1994, 91, 3584–3588. [Google Scholar] [CrossRef]
- Hironaka, N.; Mochida, K.; Mori, N.; Maeda, M.; Yamamoto, N.; Yamaoka, S. Tax-independent constitutive IkappaB kinase activation in adult T-cell leukemia cells. Neoplasia 2004, 6, 266–278. [Google Scholar] [CrossRef]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Sanda, T.; Asamitsu, K.; Ogura, H.; Iida, S.; Utsunomiya, A.; Ueda, R.; Okamoto, T. Induction of cell death in adult T-cell leukemia cells by a novel IkappaB kinase inhibitor. Leukemia 2006, 20, 590–598. [Google Scholar] [CrossRef]
- Xiao, G.; Cvijic, M.E.; Fong, A.; Harhaj, E.W.; Uhlik, M.T.; Waterfield, M.; Sun, S.C. Retroviral oncoprotein Tax induces processing of NF-kappaB2/p100 in T cells: Evidence for the involvement of IKKalpha. EMBO J. 2001, 20, 6805–6815. [Google Scholar] [CrossRef] [PubMed]
- Bandini, C.; Pupuleku, A.; Spaccarotella, E.; Pellegrino, E.; Wang, R.; Vitale, N.; Duval, C.; Cantarella, D.; Rinaldi, A.; Provero, P.; et al. IRF4 Mediates the Oncogenic Effects of STAT3 in Anaplastic Large Cell Lymphomas. Cancers 2018, 10, 21. [Google Scholar] [CrossRef]
- Weilemann, A.; Grau, M.; Erdmann, T.; Merkel, O.; Sobhiafshar, U.; Anagnostopoulos, I.; Hummel, M.; Siegert, A.; Hayford, C.; Madle, H.; et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood 2015, 125, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Shaffer, A.L.; Ceribelli, M.; Zhang, M.; Wright, G.; Huang, D.W.; Xiao, W.; Powell, J.; Petrus, M.N.; Yang, Y.; et al. Targeting the HTLV-I-Regulated BATF3/IRF4 Transcriptional Network in Adult T Cell Leukemia/Lymphoma. Cancer Cell 2018, 34, 286–297.e10. [Google Scholar] [CrossRef]
- Boddicker, R.L.; Kip, N.S.; Xing, X.; Zeng, Y.; Yang, Z.Z.; Lee, J.H.; Almada, L.L.; Elsawa, S.F.; Knudson, R.A.; Law, M.E.; et al. The oncogenic transcription factor IRF4 is regulated by a novel CD30/NF-kB positive feedback loop in peripheral T-cell lymphoma. Blood 2015, 125, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Amanda, S.; Tan, T.K.; Ong, J.Z.L.; Theardy, M.S.; Wong, R.W.J.; Huang, X.Z.; Ali, M.Z.; Li, Y.; Gong, Z.; Inagaki, H.; et al. IRF4 drives clonal evolution and lineage choice in a zebrafish model of T-cell lymphoma. Nat. Commun. 2022, 13, 2420. [Google Scholar] [CrossRef]
- Ballard, D.W.; Bohnlein, E.; Lowenthal, J.W.; Wano, Y.; Franza, B.R.; Greene, W.C. HTLV-I tax induces cellular proteins that activate the kappa B element in the IL-2 receptor alpha gene. Science 1988, 241, 1652–1655. [Google Scholar] [CrossRef] [PubMed]
- Iha, H.; Kibler, K.V.; Yedavalli, V.R.; Peloponese, J.M.; Haller, K.; Miyazato, A.; Kasai, T.; Jeang, K.T. Segregation of NF-kappaB activation through NEMO/IKKgamma by Tax and TNFalpha: Implications for stimulus-specific interruption of oncogenic signaling. Oncogene 2003, 22, 8912–8923. [Google Scholar] [CrossRef]
- Cherian, M.A.; Olson, S.; Sundaramoorthi, H.; Cates, K.; Cheng, X.; Harding, J.; Martens, A.; Challen, G.A.; Tyagi, M.; Ratner, L.; et al. An activating mutation of interferon regulatory factor 4 (IRF4) in adult T-cell leukemia. J. Biol. Chem. 2018, 293, 6844–6858. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Lu, L.; Kang, J.; Hagner, P.R.; Havens, C.G.; Amatangelo, M.; Wang, M.; Ren, Y.; Couto, S.; Breider, M.; et al. Rate of CRL4CRBN substrate Ikaros and Aiolos degradation underlies differential activity of lenalidomide and pomalidomide in multiple myeloma cells by regulation of c-Myc and IRF4. Blood Cancer J. 2015, 5, e354. [Google Scholar] [CrossRef]
- Aldinucci, D.; Celegato, M.; Borghese, C.; Colombatti, A.; Carbone, A. IRF4 silencing inhibits Hodgkin lymphoma cell proliferation, survival and CCL5 secretion. Br. J. Haematol. 2011, 152, 182–190. [Google Scholar] [CrossRef]
- Teng, Y.; Takahashi, Y.; Yamada, M.; Kurosu, T.; Koyama, T.; Miura, O.; Miki, T. IRF4 negatively regulates proliferation of germinal center B cell-derived Burkitt’s lymphoma cell lines and induces differentiation toward plasma cells. Eur. J. Cell Biol. 2007, 86, 581–589. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-Andre, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.W.J.; Ngoc, P.C.T.; Leong, W.Z.; Yam, A.W.Y.; Zhang, T.; Asamitsu, K.; Iida, S.; Okamoto, T.; Ueda, R.; Gray, N.S. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood J. Am. Soc. Hematol. 2017, 130, 2326–2338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, L.; Stupack, D.G.; Bai, N.; Xun, J.; Ren, G.; Han, J.; Li, L.; Luo, Y.; Xiang, R.; et al. JMJD3 promotes survival of diffuse large B-cell lymphoma subtypes via distinct mechanisms. Oncotarget 2016, 7, 29387–29399. [Google Scholar] [CrossRef]
- Mangan, S.; Zaslaver, A.; Alon, U. The Coherent Feedforward Loop Serves as a Sign-sensitive Delay Element in Transcription Networks. J. Mol. Biol. 2003, 334, 197–204. [Google Scholar] [CrossRef]
- Perkins, N.D. The diverse and complex roles of NF-kappaB subunits in cancer. Nat. Rev. Cancer 2012, 12, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Mondala, P.K.; Vora, A.A.; Zhou, T.; Lazzari, E.; Ladel, L.; Luo, X.; Kim, Y.; Costello, C.; MacLeod, A.R.; Jamieson, C.H.M.; et al. Selective antisense oligonucleotide inhibition of human IRF4 prevents malignant myeloma regeneration via cell cycle disruption. Cell Stem Cell 2021, 28, 623–636.e9. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, R.W.J.; Ong, J.Z.L.; Theardy, M.S.; Sanda, T. IRF4 as an Oncogenic Master Transcription Factor. Cancers 2022, 14, 4314. https://doi.org/10.3390/cancers14174314
Wong RWJ, Ong JZL, Theardy MS, Sanda T. IRF4 as an Oncogenic Master Transcription Factor. Cancers. 2022; 14(17):4314. https://doi.org/10.3390/cancers14174314
Chicago/Turabian StyleWong, Regina Wan Ju, Jolynn Zu Lin Ong, Madelaine Skolastika Theardy, and Takaomi Sanda. 2022. "IRF4 as an Oncogenic Master Transcription Factor" Cancers 14, no. 17: 4314. https://doi.org/10.3390/cancers14174314
APA StyleWong, R. W. J., Ong, J. Z. L., Theardy, M. S., & Sanda, T. (2022). IRF4 as an Oncogenic Master Transcription Factor. Cancers, 14(17), 4314. https://doi.org/10.3390/cancers14174314