

Risk Stratification for Breast Cancer Patient by Simultaneous Learning of Molecular Subtype and Survival Outcome Using Genetic Algorithm-Based Gene Set Selection †


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breast Cancer Patient Data Collection
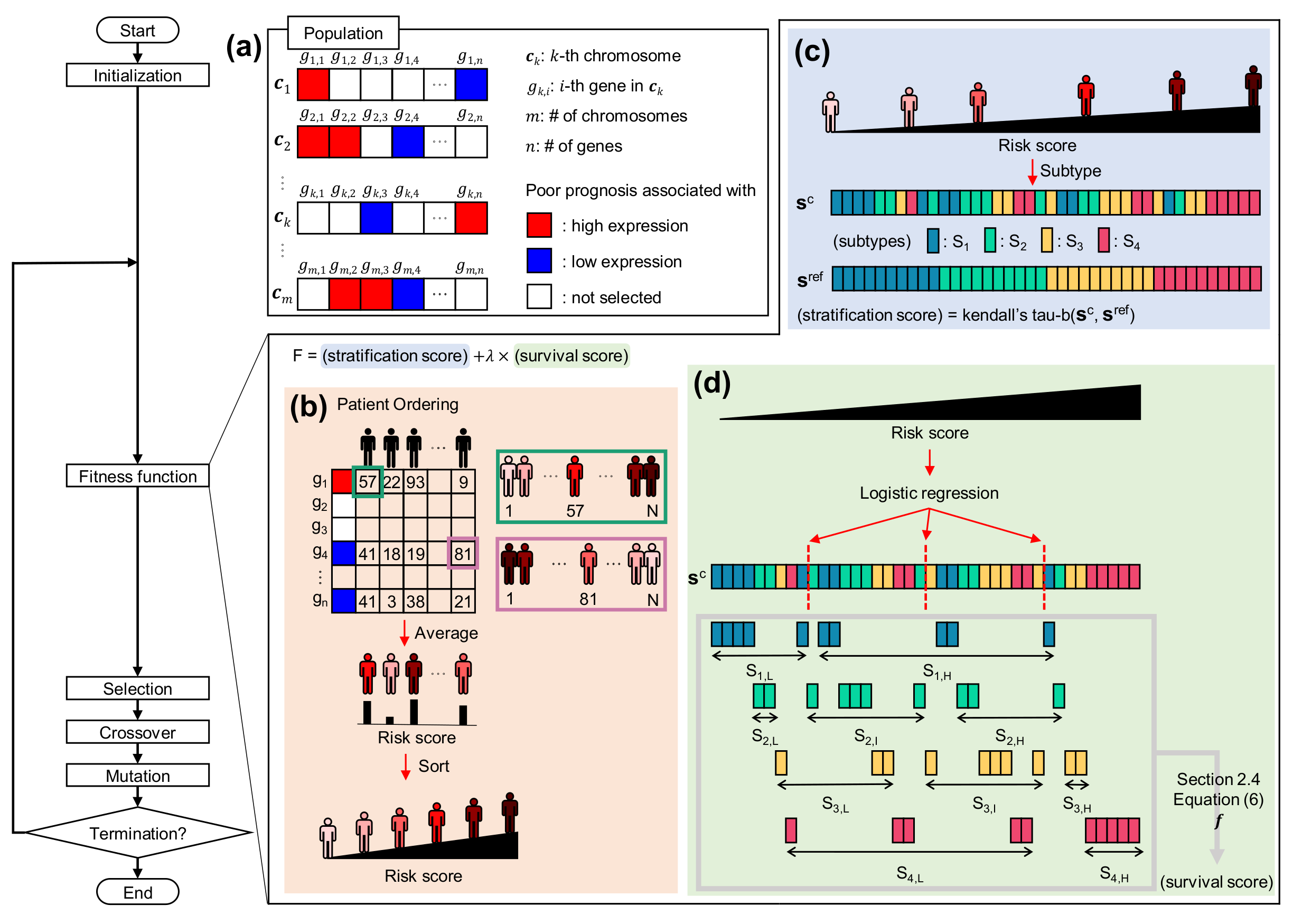
2.2. Chromosome Representation for Gene Combination
2.3. Deriving Patient Risk Score from Chromosome
2.4. Fitness Function for Evaluating Order of Patients
2.5. Biological Operators in GA
2.6. Comparison with Existing Approaches
3. Results
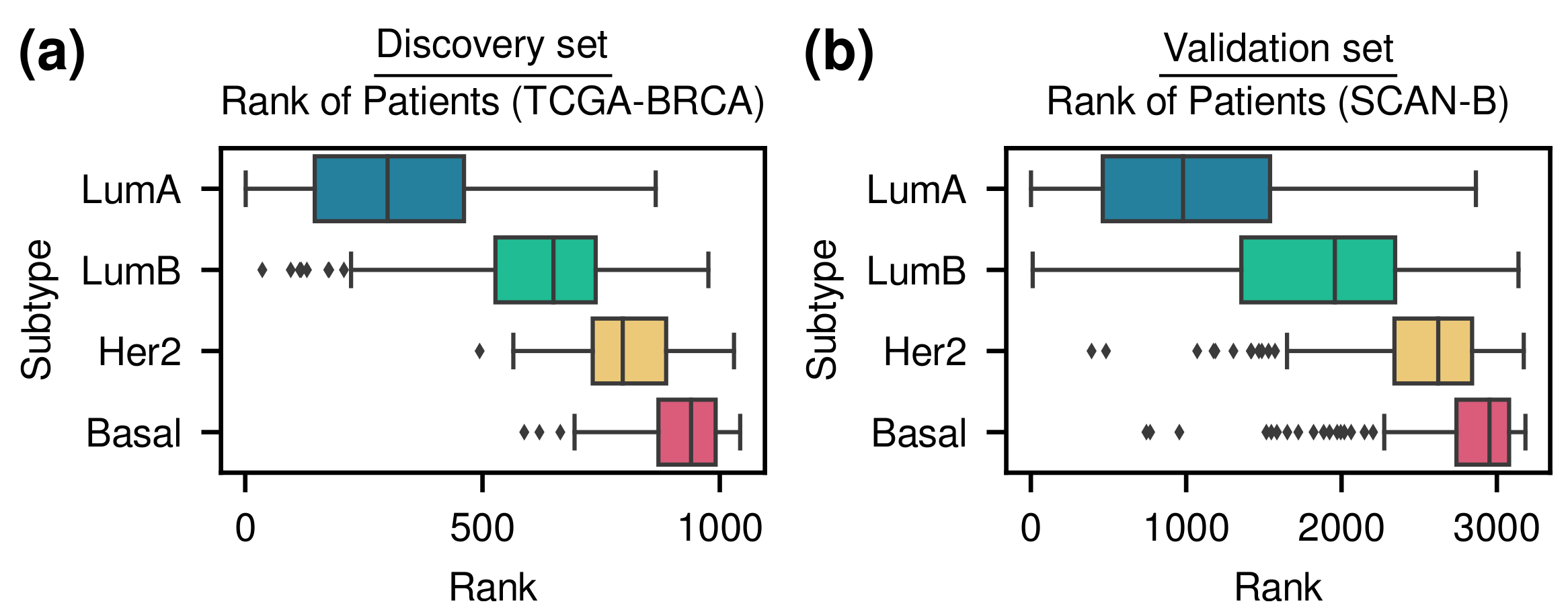
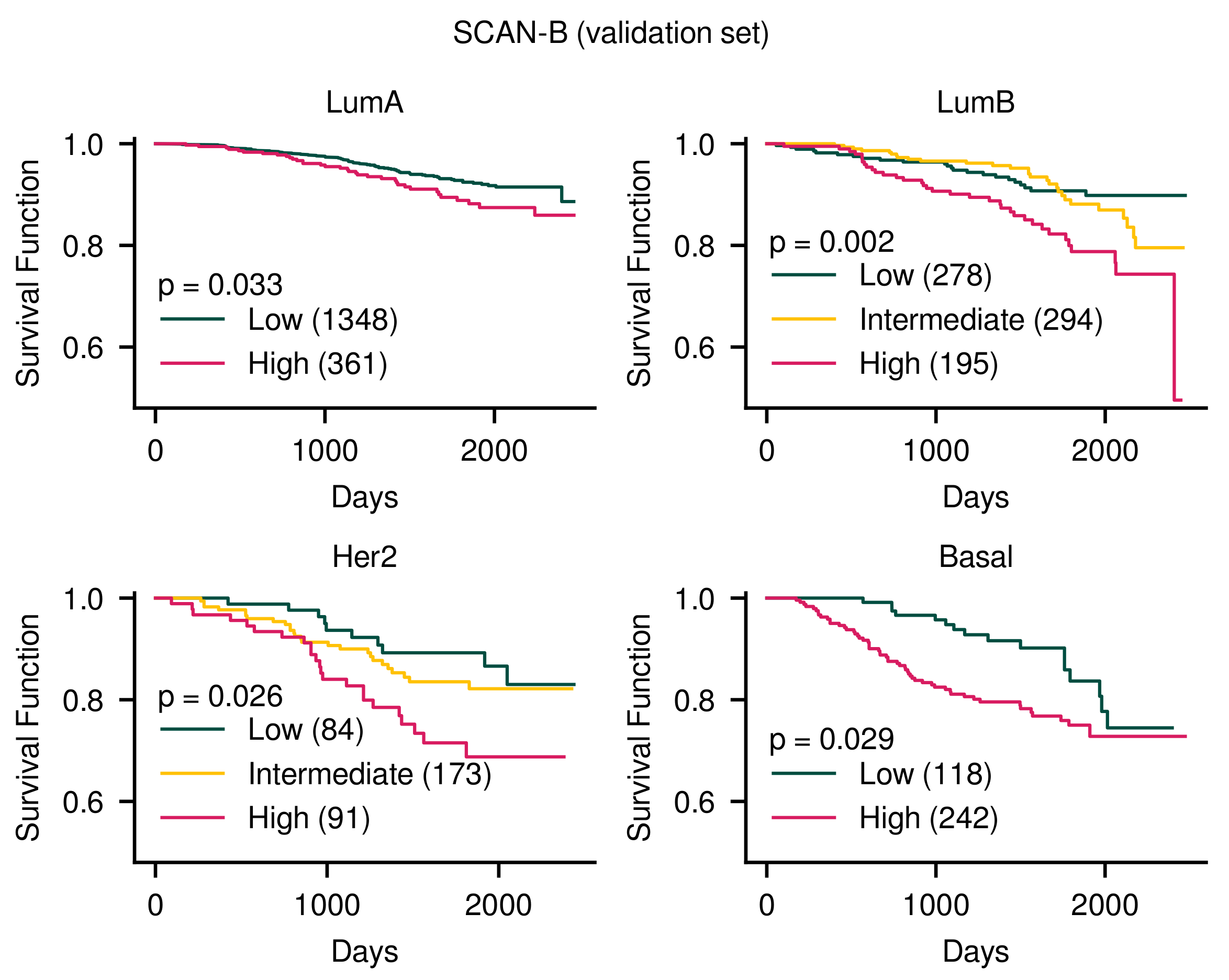
3.1. Patient Stratification Considering Molecular Subtype and Survival Outcome Simultaneously
3.1.1. Our Method Stratified Patients Considering Simultaneously Molecular Subtypes and Survival Outcomes
3.1.2. Previous Methods Do Not Consider Molecular Subtype and Survival Outcome Simultaneously
3.2. Robustness of the Methodology for Constant Patient Ordering
3.3. Usefulness of Fitness Function
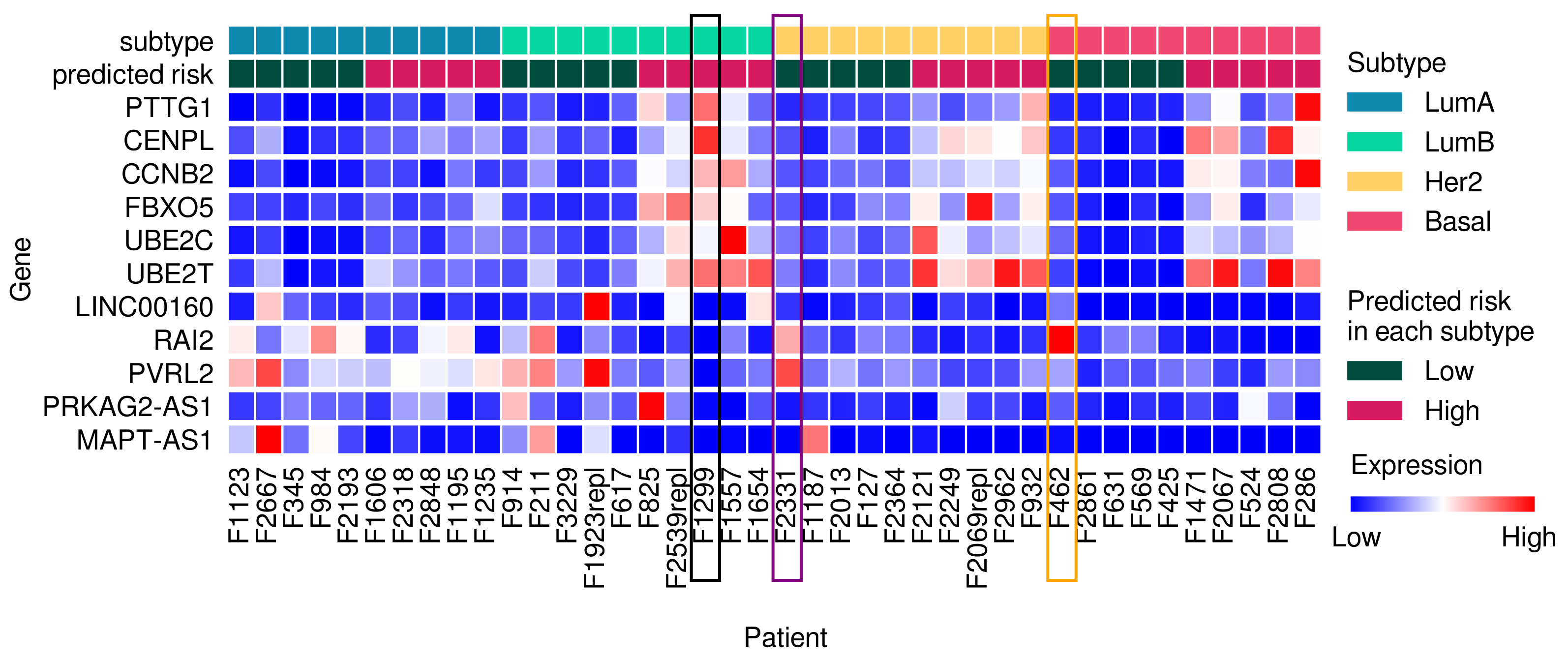
3.4. Frequently Selected Genes
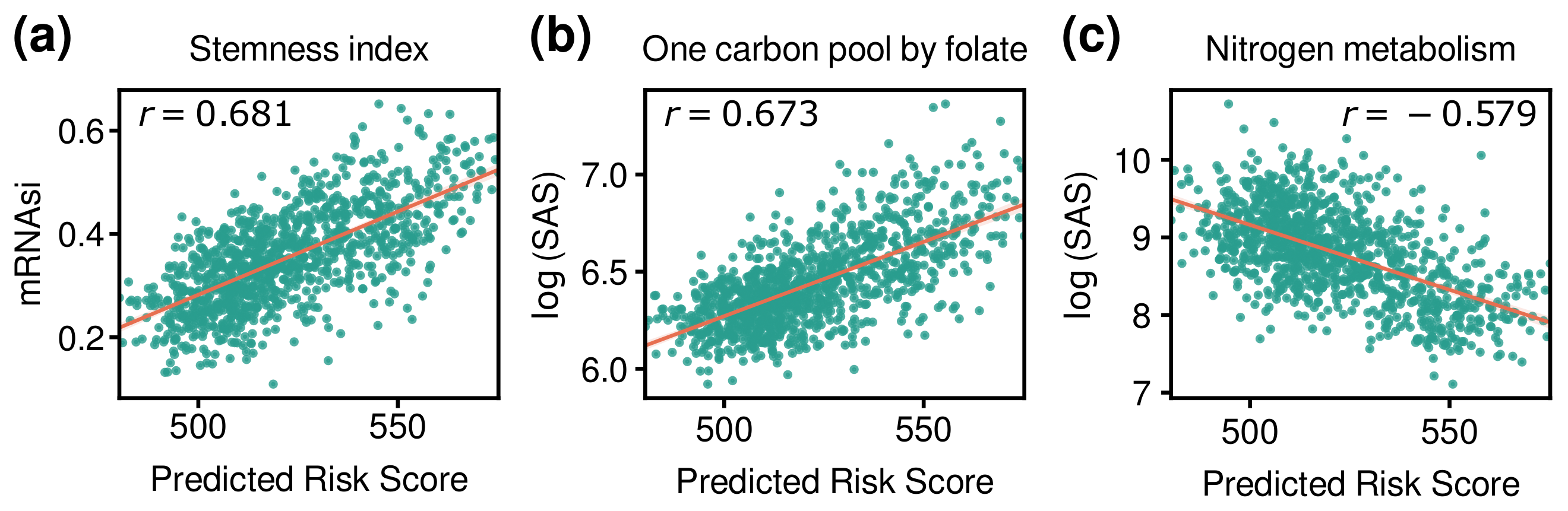
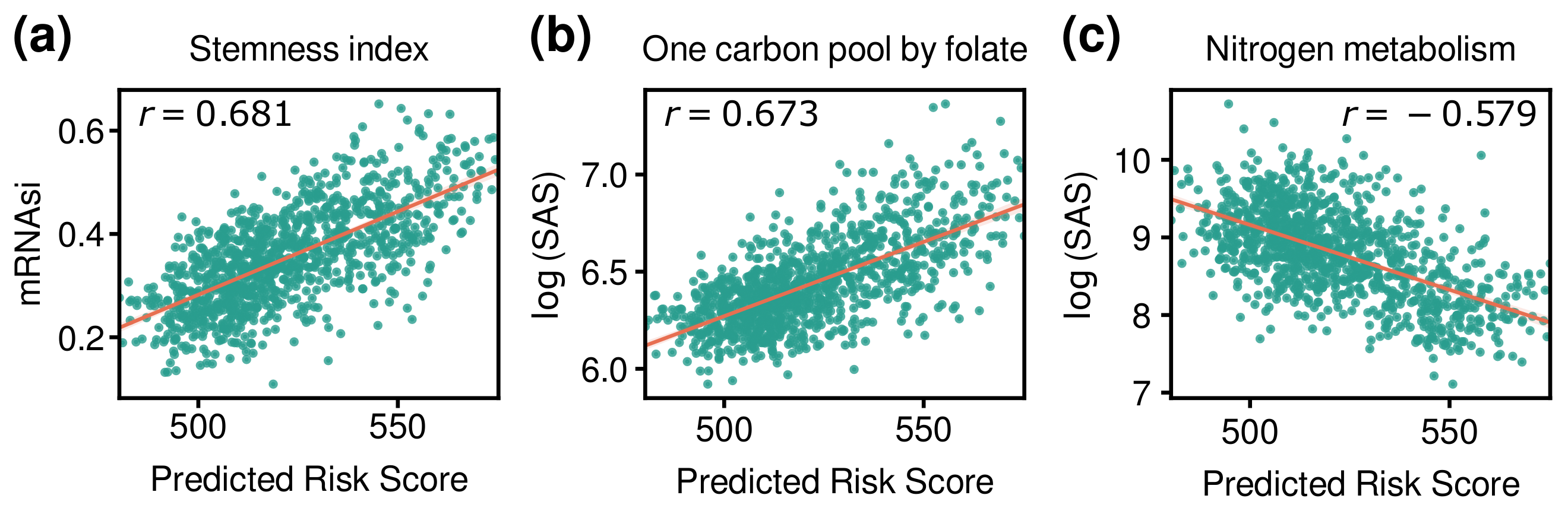
3.5. Comparison of Risk Score with Other Indices
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Basal | Basal-like |
| BRCA | Breast Invasive Carcinoma |
| CCNB2 | Cyclin B2 |
| CENPL | Centromere Protein L |
| CLSPN | Claspin |
| CMS | Consensus Molecular Subtype |
| Cox-filter | Cox-model based filter |
| ER | Estrogen receptor |
| EXO1 | Exonuclease 1 |
| FBXO5 | F-Box Protein 5 |
| FOXM1 | Forkhead Box M1 |
| FPKM | Fragments Per Kilobase of transcript per Million mapped reads |
| GA | Genetic Algorithm |
| GEO | Gene Expression Omnibus |
| GLUD1 | Glutamate Dehydrogenase 1 |
| GO | Gene Ontology |
| High | |
| Her2 | Human epidermal growth factor receptor 2 |
| HR | Hazard Ratio |
| Intermediate | |
| IHC | Immunohistochemistry |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KIF2C | Kinesin Family Member 2C |
| Low | |
| LINC00160 | Long Intergenic Non-Protein Coding RNA 160 |
| LumA | Luminal A |
| LumB | Luminal B |
| lncRNA | long non-coding RNA |
| MAPT | Microtubule Associated Protein Tau |
| MAPT-AS1 | MAPT Antisense RNA 1 |
| mRNA | messenger RNA |
| mRNAsi | mRNA stemness index |
| NAT1 | N-Acetyltransferase 1 |
| NCBI | National Center for Biotechnology Information |
| PAM50 | Prediction Analysis of Microarray 50 |
| PR | Progesterone receptor |
| PRKAG2 | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 |
| PRKAG2-AS1 | PRKAG2 Antisense RNA 1 |
| RAI2 | Retinoic Acid Induced 2 |
| RNA | Ribonucleic acid |
| PTTG1 | Pituitary Tumor Transforming Gene 1 |
| PVRL2 | Poliovirus Receptor-related 2 |
| SAS | Subsystem Activation Score |
| SCAN-B | Sweden Cancerome Analysis Network - Breast |
| SCC | Spearman’s Correlation Coefficient |
| sparse LR | sparse Logistic Regression |
| TCGA | The Cancer Genome Atlas |
| TP53 | Tumor Protein P53 |
| UBE2C | Ubiquitin Conjugating Enzyme E2 C |
| UBE2T | Ubiquitin Conjugating Enzyme E2 T |
References
- Beaulieu-Jones, B.K.; Yuan, W.; Brat, G.A.; Beam, A.L.; Weber, G.; Ruffin, M.; Kohane, I.S. Machine learning for patient risk stratification: Standing on, or looking over, the shoulders of clinicians? NPJ Digit. Med. 2021, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Sim, W.; Lee, J.; Choi, C. Robust method for identification of prognostic gene signatures from gene expression profiles. Sci. Rep. 2017, 7, 16926. [Google Scholar] [CrossRef]
- Nagy, Á.; Munkácsy, G.; Györffy, B. Pancancer survival analysis of cancer hallmark genes. Sci. Rep. 2021, 11, 6047. [Google Scholar] [CrossRef]
- Shevade, S.K.; Keerthi, S.S. A simple and efficient algorithm for gene selection using sparse logistic regression. Bioinformatics 2003, 19, 2246–2253. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, C.; Luan, X.Z.; Leung, K.S.; Chan, T.M.; Xu, Z.B.; Zhang, H. Sparse logistic regression with a L1/2 penalty for gene selection in cancer classification. BMC Bioinform. 2013, 14, 198. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, H.; Shen, H.; Yang, Z. Gene selection in cancer classification using sparse logistic regression with L1/2 regularization. Appl. Sci. 2018, 8, 1569. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Y.; Liang, Y.; Zhang, H.; Chai, H.; Zhang, B.; Peng, C. Robust Sparse Logistic Regression with the Lq(0 < q < 1) Regularization for Feature Selection Using Gene Expression Data. IEEE Access 2018, 6, 68586–68595. [Google Scholar]
- Li, L.; Liu, Z.P. Biomarker discovery for predicting spontaneous preterm birth from gene expression data by regularized logistic regression. Comput. Struct. Biotechnol. J. 2020, 18, 3434–3446. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Lee, V.H.; Ng, M.K.; Yan, H.; Bijlsma, M.F. Molecular subtyping of cancer: Current status and moving toward clinical applications. Briefings Bioinform. 2019, 20, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Venkatraman, S.E.; Meyerson, M.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study: Director’s Challenge Consortium for the molecular classification of lung adenocarcinoma. Nat. Med. 2008, 14, 822–827. [Google Scholar]
- Tian, S.; Wang, C.; An, M.W. Test on existence of histology subtype-specific prognostic signatures among early stage lung adenocarcinoma and squamous cell carcinoma patients using a Cox-model based filter. Biol. Direct 2015, 10, 15. [Google Scholar] [CrossRef]
- Ronen, J.; Hayat, S.; Akalin, A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Hao, J.; Kim, Y.; Mallavarapu, T.; Oh, J.H.; Kang, M. Interpretable deep neural network for cancer survival analysis by integrating genomic and clinical data. BMC Med. Genom. 2019, 12, 189. [Google Scholar] [CrossRef]
- Vale-Silva, L.A.; Rohr, K. Long-term cancer survival prediction using multimodal deep learning. Sci. Rep. 2021, 11, 13505. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.; Chen, W.; Zhang, C.; Wei, Z. A convolutional neural network model for survival prediction based on prognosis-related cascaded Wx feature selection. Lab. Investig. 2022, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hennigs, A.; Riedel, F.; Gondos, A.; Sinn, P.; Schirmacher, P.; Marmé, F.; Jäger, D.; Kauczor, H.U.; Stieber, A.; Lindel, K.; et al. Prognosis of breast cancer molecular subtypes in routine clinical care: A large prospective cohort study. BMC Cancer 2016, 16, 734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallahpour, S.; Navaneelan, T.; De, P.; Borgo, A. Breast cancer survival by molecular subtype: A population-based analysis of cancer registry data. Can. Med. Assoc. Open Access J. 2017, 5, E734–E739. [Google Scholar] [CrossRef]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Katoch, S.; Chauhan, S.S.; Kumar, V. A review on genetic algorithm: Past, present, and future. Multimed. Tools Appl. 2021, 80, 8091–8126. [Google Scholar] [CrossRef]
- Koboldt, D.; Fulton, R.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.; Fulton, L.; Dooling, D.; Ding, L.; Mardis, E.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Brueffer, C.; Vallon-Christersson, J.; Grabau, D.; Ehinger, A.; Häkkinen, J.; Hegardt, C.; Malina, J.; Chen, Y.; Bendahl, P.O.; Manjer, J.; et al. Clinical value of RNA sequencing–based classifiers for prediction of the five conventional breast cancer biomarkers: A report from the population-based multicenter Sweden Cancerome Analysis Network—Breast Initiative. JCO Precis. Oncol. 2018, 2, 1–18. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell 2018, 33, 690–705. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 2018, 173, 400–416. [Google Scholar] [PubMed]
- Ein-Dor, L.; Kela, I.; Getz, G.; Givol, D.; Domany, E. Outcome signature genes in breast cancer: Is there a unique set? Bioinformatics 2005, 21, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Geng, R.; Min, N.; Zheng, Y.; Hong, C.; Wu, R.; Wu, H.; Wei, Y.; Zhang, Y.; Li, X. External verification and improvement of the Neo-Bioscore staging system in a Chinese cohort. Ann. Transl. Med. 2022, 10, 626. [Google Scholar] [CrossRef] [PubMed]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer. Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Langerød, A.; Zhao, H.; Borgan, Ø.; Nesland, J.M.; Bukholm, I.R.; Ikdahl, T.; Kåresen, R.; Børresen-Dale, A.L.; Jeffrey, S.S. TP53 mutation status and gene expression profiles are powerful prognostic markers of breast cancer. Breast Cancer Res. 2007, 9, R30. [Google Scholar] [CrossRef]
- Kendall, M.G. The treatment of ties in ranking problems. Biometrika 1945, 33, 239–251. [Google Scholar] [CrossRef]
- Breslow, N.E. Analysis of survival data under the proportional hazards model. Int. Stat. Rev. Int. Stat. 1975, 43, 45–57. [Google Scholar] [CrossRef]
- Li, L.; Darden, T.A.; Weingberg, C.; Levine, A.; Pedersen, L.G. Gene assessment and sample classification for gene expression data using a genetic algorithm/k-nearest neighbor method. Comb. Chem. High Throughput Screen. 2001, 4, 727–739. [Google Scholar] [CrossRef]
- Filipits, M.; Rudas, M.; Jakesz, R.; Dubsky, P.; Fitzal, F.; Singer, C.F.; Dietze, O.; Greil, R.; Jelen, A.; Sevelda, P.; et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin. Cancer Res. 2011, 17, 6012–6020. [Google Scholar] [CrossRef]
- Van’t Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Klijn, J.G.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [PubMed]
- Haibe-Kains, B.; Desmedt, C.; Rothé, F.; Piccart, M.; Sotiriou, C.; Bontempi, G. A fuzzy gene expression-based computational approach improves breast cancer prognostication. Genome Biol. 2010, 11, R18. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Meng, C.; Zou, Y.; Hong, W.; Bao, C.; Jia, X. Estrogen-regulated PTTG1 promotes breast cancer progression by regulating cyclin kinase expression. Mol. Med. 2020, 26, 33. [Google Scholar] [CrossRef]
- Xiea, Y.; Wangb, R. Pttg1 promotes growth of breast cancer through P27 nuclear exclusion. Cell. Physiol. Biochem. 2016, 38, 393–400. [Google Scholar] [CrossRef]
- Wu, C.C.; Ekanem, T.I.; Phan, N.N.; Loan, D.T.T.; Hou, S.Y.; Lee, K.H.; Wang, C.Y. Gene signatures and prognostic analyses of the Tob/BTG pituitary tumor-transforming gene (PTTG) family in clinical breast cancer patients. Int. J. Med. Sci. 2020, 17, 3112–3124. [Google Scholar] [CrossRef]
- Yin, J.; Lin, C.; Jiang, M.; Tang, X.; Xie, D.; Chen, J.; Ke, R. CENPL, ISG20L2, LSM4, MRPL3 are four novel hub genes and may serve as diagnostic and prognostic markers in breast cancer. Sci. Rep. 2021, 11, 15610. [Google Scholar] [CrossRef]
- Shubbar, E.; Kovács, A.; Hajizadeh, S.; Parris, T.Z.; Nemes, S.; Gunnarsdóttir, K.; Einbeigi, Z.; Karlsson, P.; Helou, K. Elevated cyclin B2 expression in invasive breast carcinoma is associated with unfavorable clinical outcome. BMC Cancer 2013, 13, 1. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, T.; Zhang, S.; Shan, J. Prognostic values of F-box members in breast cancer: An online database analysis and literature review. Biosci. Rep. 2019, 39, BSR20180949. [Google Scholar] [CrossRef]
- Kim, Y.J.; Lee, G.; Han, J.; Song, K.; Choi, J.S.; Choi, Y.L.; Shin, Y.K. UBE2C overexpression aggravates patient outcome by promoting estrogen-dependent/independent cell proliferation in early hormone receptor-positive and HER2-negative breast cancer. Front. Oncol. 2020, 9, 1574. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Park, J.H.; Nishidate, T.; Kijima, K.; Hirata, K.; Nakamura, Y.; Katagiri, T. Ubiquitination and downregulation of BRCA1 by ubiquitin-conjugating enzyme E2T overexpression in human breast cancer cells. Cancer Res. 2009, 69, 8752–8760. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Dong, C.; Ma, B. UBE2T promotes proliferation, invasion and glycolysis of breast cancer cells by regualting the PI3K/AKT signaling pathway. J. Recept. Signal Transduct. 2020, 42, 151–159. [Google Scholar]
- Zhang, Z.; Yu, W.; Tang, D.; Zhou, Y.; Bi, M.; Wang, H.; Zheng, Y.; Chen, M.; Li, L.; Xu, X.; et al. Epigenomics-based identification of oestrogen-regulated long noncoding RNAs in ER+ breast cancer. RNA Biol. 2020, 17, 1590–1602. [Google Scholar] [CrossRef]
- Jonsson, P.; Coarfa, C.; Mesmar, F.; Raz, T.; Rajapakshe, K.; Thompson, J.F.; Gunaratne, P.H.; Williams, C. Single-molecule sequencing reveals estrogen-regulated clinically relevant lncRNAs in breast cancer. Mol. Endocrinol. 2015, 29, 1634–1645. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Wu, K.; Herman, J.G.; Xu, X.; Yang, Y.; Dai, G.; Guo, M. Retinoic acid-induced 2 (RAI2) is a novel tumor suppressor, and promoter region methylation of RAI2 is a poor prognostic marker in colorectal cancer. Clin. Epigenetics 2018, 10, 69. [Google Scholar]
- Nishikawa, S.; Uemoto, Y.; Kim, T.S.; Hisada, T.; Kondo, N.; Wanifuchi-Endo, Y.; Fujita, T.; Asano, T.; Katagiri, Y.; Terada, M.; et al. Low RAI2 expression is a marker of poor prognosis in breast cancer. Breast Cancer Res. Treat. 2021, 187, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Qu, P.; Chen, Y.; Zhou, X.; Wu, Y.; Liu, F.; Wang, D.; Zhang, J.; An, J. Low expression of CD112 is associated with poor overall survival in patients with hepatocellular carcinoma. Hum. Pathol. 2014, 45, 1944–1950. [Google Scholar]
- Ou, Y.; Deng, Y.; Wang, H.; Zhang, Q.; Luo, H.; Hu, P. Targeting antisense lncRNA PRKAG2-AS1, as a therapeutic target, suppresses malignant behaviors of hepatocellular carcinoma cells. Front. Med. 2021, 8, 374. [Google Scholar] [CrossRef]
- Wang, D.; Li, J.; Cai, F.; Xu, Z.; Li, L.; Zhu, H.; Liu, W.; Xu, Q.; Cao, J.; Sun, J.; et al. Overexpression of MAPT-AS1 is associated with better patient survival in breast cancer. Biochem. Cell Biol. 2019, 97, 158–164. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Bianco, J.N.; Bergoglio, V.; Lin, Y.L.; Pillaire, M.J.; Schmitz, A.L.; Gilhodes, J.; Lusque, A.; Mazières, J.; Lacroix-Triki, M.; Roumeliotis, T.I.; et al. Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat. Commun. 2019, 10, 910. [Google Scholar] [PubMed]
- Saba, R.; Alsayed, A.; Zacny, J.P.; Dudek, A.Z. The role of forkhead box protein M1 in breast cancer progression and resistance to therapy. Int. J. Breast Cancer 2016, 2016, 9768183. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.; Sim, J.; Abdul, R.; Chung, M.S.; Paik, S.S.; Oh, Y.H.; Park, C.K.; Jang, K. Increased expression of forkhead box M1 is associated with aggressive phenotype and poor prognosis in estrogen receptor-positive breast cancer. J. Korean Med. Sci. 2015, 30, 390–397. [Google Scholar] [CrossRef]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef]
- Lim, S.; Park, Y.; Hur, B.; Kim, M.; Han, W.; Kim, S. Protein interaction network (pin)-based breast cancer subsystem identification and activation measurement for prognostic modeling. Methods 2016, 110, 81–89. [Google Scholar]
- Park, A.K.; Lee, J.Y.; Cheong, H.; Ramaswamy, V.; Park, S.H.; Kool, M.; Phi, J.H.; Choi, S.A.; Cavalli, F.; Taylor, M.D.; et al. Subgroup-specific prognostic signaling and metabolic pathways in pediatric medulloblastoma. BMC Cancer 2019, 19, 571. [Google Scholar] [CrossRef]
- Kurmi, K.; Haigis, M.C. Nitrogen metabolism in cancer and immunity. Trends Cell Biol. 2020, 30, 408–424. [Google Scholar] [CrossRef]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar]
- Cooper, A.J. Role of glutamine in cerebral nitrogen metabolism and ammonia neurotoxicity. Ment. Retard. Dev. Disabil. Res. Rev. 2001, 7, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1320. [Google Scholar] [CrossRef] [PubMed]
- Craze, M.L.; El-Ansari, R.; Aleskandarany, M.A.; Cheng, K.W.; Alfarsi, L.; Masisi, B.; Diez-Rodriguez, M.; Nolan, C.C.; Ellis, I.O.; Rakha, E.A.; et al. Glutamate dehydrogenase (GLUD1) expression in breast cancer. Breast Cancer Res. Treat. 2019, 174, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Coloff, J.L.; Murphy, J.P.; Braun, C.R.; Harris, I.S.; Shelton, L.M.; Kami, K.; Gygi, S.P.; Selfors, L.M.; Brugge, J.S. Differential glutamate metabolism in proliferating and quiescent mammary epithelial cells. Cell Metab. 2016, 23, 867–880. [Google Scholar] [CrossRef] [PubMed]
- de Souza, B.F.; de Carvalho, A.C. Gene selection using genetic algorithms. In Proceedings of the International Symposium on Biological and Medical Data Analysis; Springer: Berlin/Heidelberg, Germany, 2004; pp. 479–490. [Google Scholar]
- Alanni, R.; Hou, J.; Azzawi, H.; Xiang, Y. A novel gene selection algorithm for cancer classification using microarray datasets. BMC Med. Genom. 2019, 12, 10. [Google Scholar]
- Masoudi-Sobhanzadeh, Y.; Motieghader, H.; Omidi, Y.; Masoudi-Nejad, A. A machine learning method based on the genetic and world competitive contests algorithms for selecting genes or features in biological applications. Sci. Rep. 2021, 11, 3349. [Google Scholar] [PubMed]
- Kim, H.K.; Park, K.H.; Kim, Y.; Park, S.E.; Lee, H.S.; Lim, S.W.; Cho, J.H.; Kim, J.Y.; Lee, J.E.; Ahn, J.S.; et al. Discordance of the PAM50 intrinsic subtypes compared with immunohistochemistry-based surrogate in breast cancer patients: Potential implication of genomic alterations of discordance. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2019, 51, 737–747. [Google Scholar] [CrossRef]
- Islam, M.M.; Mohammed, N.; Wang, Y.; Hu, P. Differential Private Deep Learning Models for Analyzing Breast Cancer Omics Data. Front. Oncol. 2022, 12, 879607. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subtype | ER and/or PR | HER2 | Ki-67 |
|---|---|---|---|
| Luminal A (LumA) | ER+ or PR+ | HER2− | Ki-67− |
| Luminal B (LumB) | ER+ or PR+ | any | Ki-67+ |
| Her2-enriched (Her2) | ER− and PR− | HER2+ | any |
| Basal-like (Basal) | ER− and PR− | HER2− | any |
| Subtype | TCGA-BRCA | SCAN-B |
|---|---|---|
| LumA | 563 (53.98%) | 1709 (53.67%) |
| LumB | 206 (19.75%) | 767 (24.09%) |
| Her2 | 82 (7.86%) | 348 (10.93%) |
| Basal | 192 (18.41%) | 360 (11.31%) |
| Total | 1043 | 3184 |
| Log-Rank Test (p-Value) | ||||
|---|---|---|---|---|
| LumA | LumB | Her2 | Basal | |
| GA (Ours) | 0.033 | 0.002 | 0.026 | 0.029 |
| PAM50 [14] | 0.248 | 0.267 | 0.780 | 0.894 |
| sparse LR [8] | 0.166 | 0.802 | 0.803 | 0.571 |
| Cox-filter [18] | 0.330 | 0.823 | 0.012 | 0.196 |
| EndoPredict [40] | 0.120 | 0.159 | 0.171 | 0.033 |
| GENE70 [41] | 0.140 | 0.094 | 0.845 | 0.570 |
| GENE76 [42] | 0.082 | 0.061 | 0.995 | 0.414 |
| GENIUS M1 [43] | 0.452 | 0.019 | 0.014 | 0.285 |
| GENIUS M2 [43] | 0.515 | 0.371 | 0.253 | 0.063 |
| GENIUS M3 [43] | 0.050 | 0.544 | 0.529 | 0.788 |
| GGI [44] | 0.282 | 0.637 | 0.810 | 0.584 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koo, B.; Lee, D.; Lee, S.; Sung, I.; Kim, S.; Lee, S. Risk Stratification for Breast Cancer Patient by Simultaneous Learning of Molecular Subtype and Survival Outcome Using Genetic Algorithm-Based Gene Set Selection. Cancers 2022, 14, 4120. https://doi.org/10.3390/cancers14174120
Koo B, Lee D, Lee S, Sung I, Kim S, Lee S. Risk Stratification for Breast Cancer Patient by Simultaneous Learning of Molecular Subtype and Survival Outcome Using Genetic Algorithm-Based Gene Set Selection. Cancers. 2022; 14(17):4120. https://doi.org/10.3390/cancers14174120
Chicago/Turabian StyleKoo, Bonil, Dohoon Lee, Sangseon Lee, Inyoung Sung, Sun Kim, and Sunho Lee. 2022. "Risk Stratification for Breast Cancer Patient by Simultaneous Learning of Molecular Subtype and Survival Outcome Using Genetic Algorithm-Based Gene Set Selection" Cancers 14, no. 17: 4120. https://doi.org/10.3390/cancers14174120
APA StyleKoo, B., Lee, D., Lee, S., Sung, I., Kim, S., & Lee, S. (2022). Risk Stratification for Breast Cancer Patient by Simultaneous Learning of Molecular Subtype and Survival Outcome Using Genetic Algorithm-Based Gene Set Selection. Cancers, 14(17), 4120. https://doi.org/10.3390/cancers14174120