
The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collecting and Disposing Data
2.2. Identification of mRLs Associated with OV Prognosis
2.3. RNA Isolation and qRT-PCR
2.4. Establish and Prove an mRL Prognostic Signature for OV
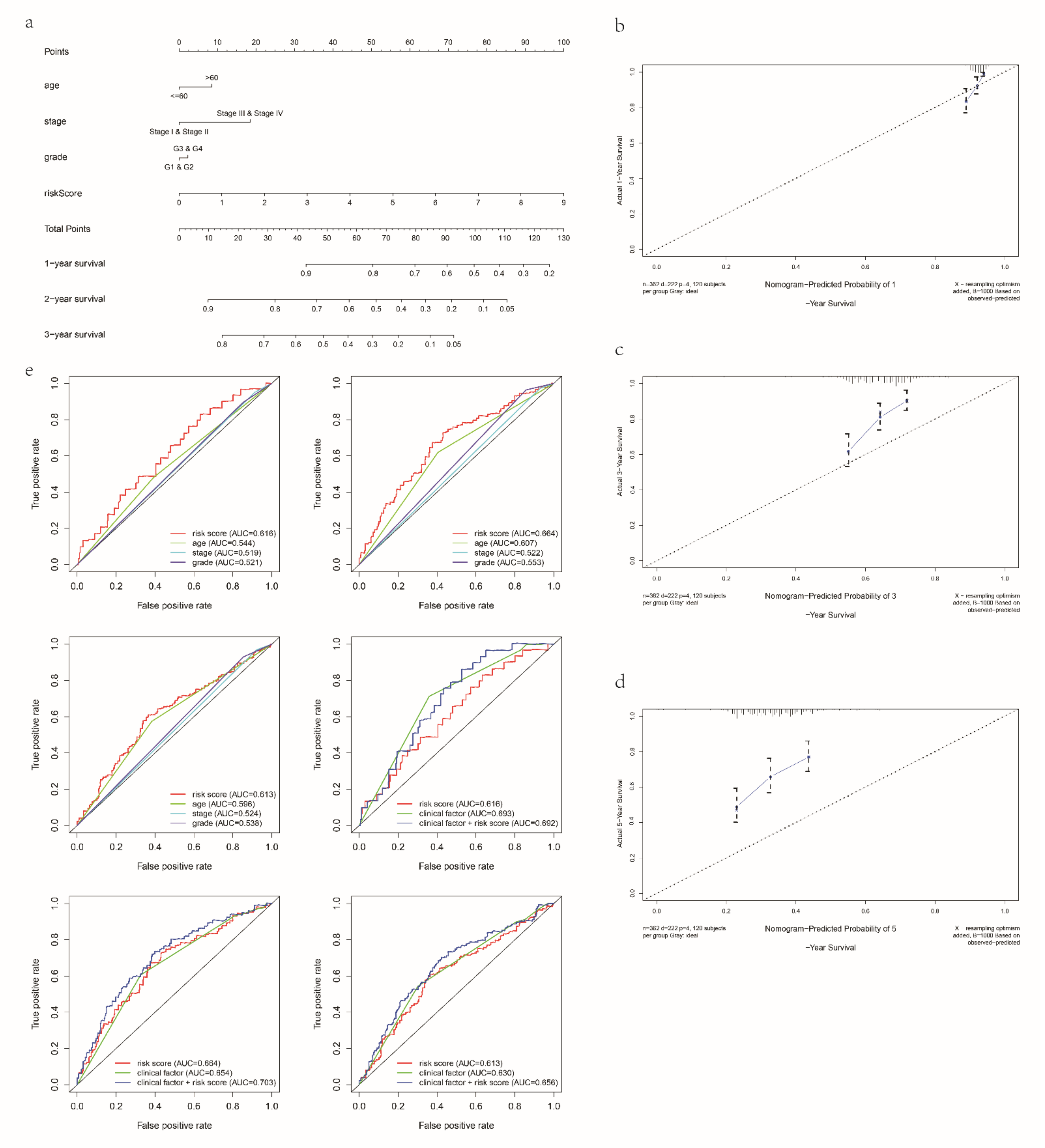
2.5. Establishment and Validation of a Nomogram
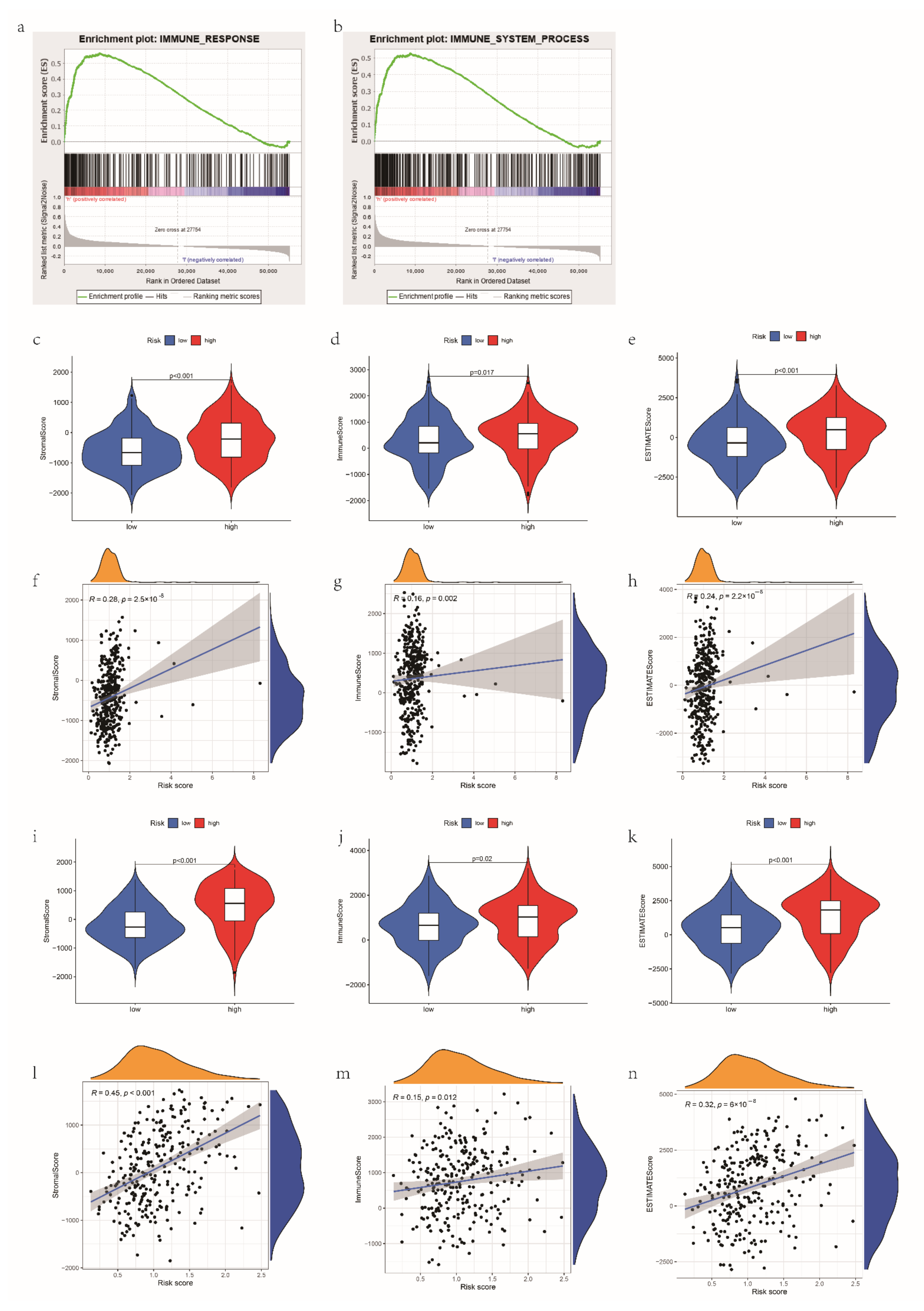
2.6. Gene Set Enrichment Analysis
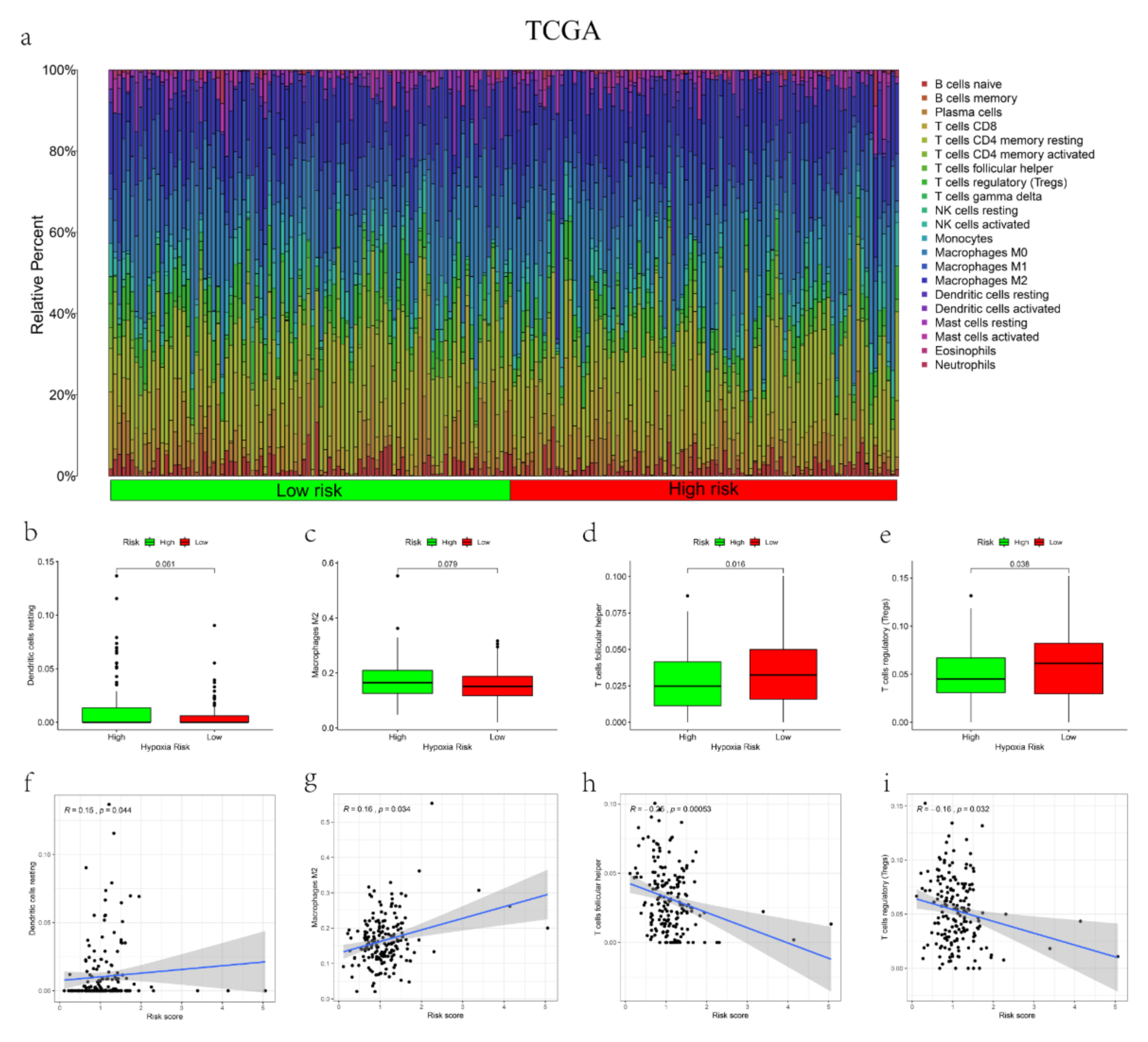
2.7. The Correlation between the Signature and Tumor Infiltration Immune Cells (TIICs)
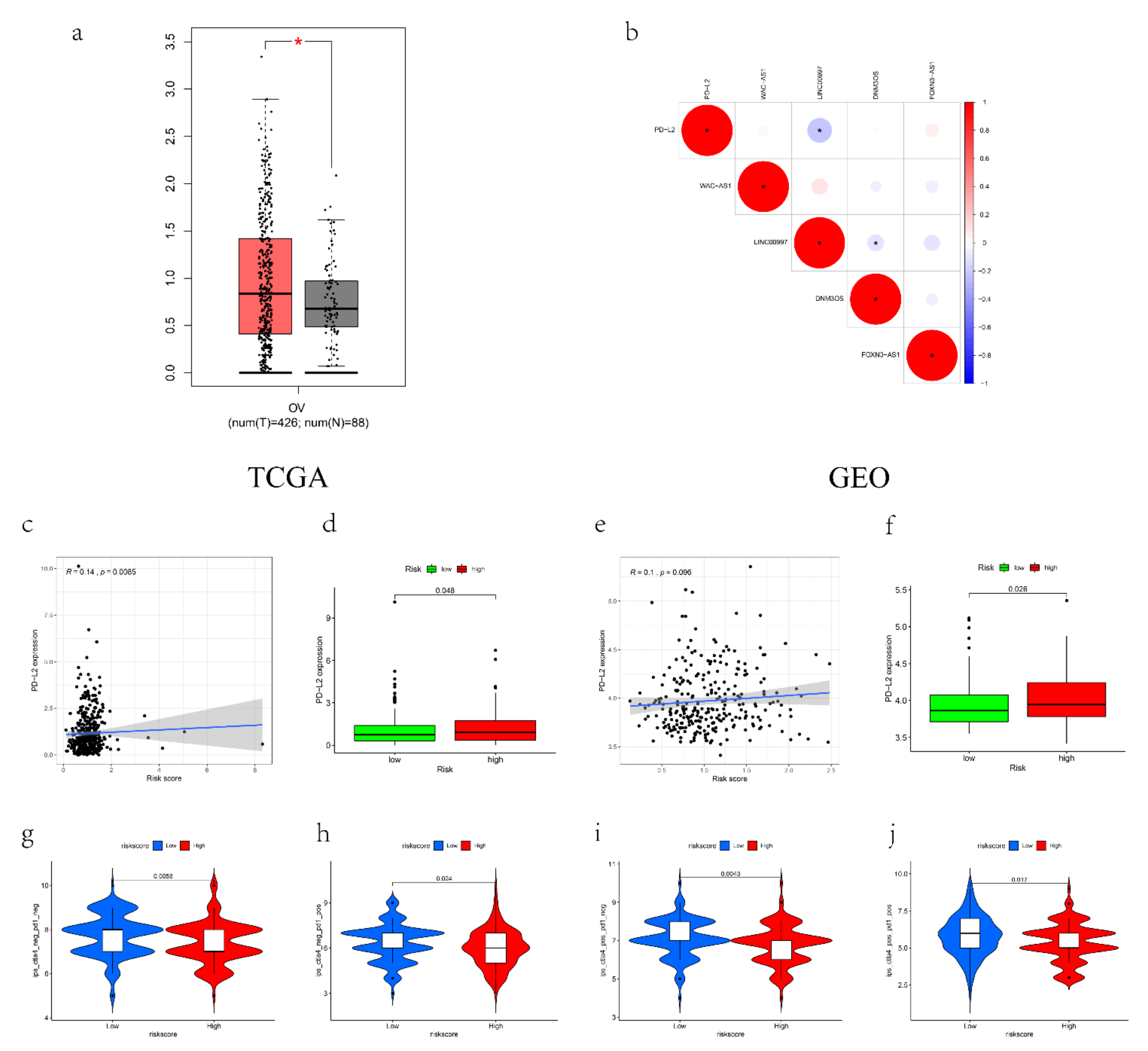
2.8. The Correlation to Other Immune Features
2.9. Immunotherapy Response Prediction
2.10. Assess Drug Sensitivity
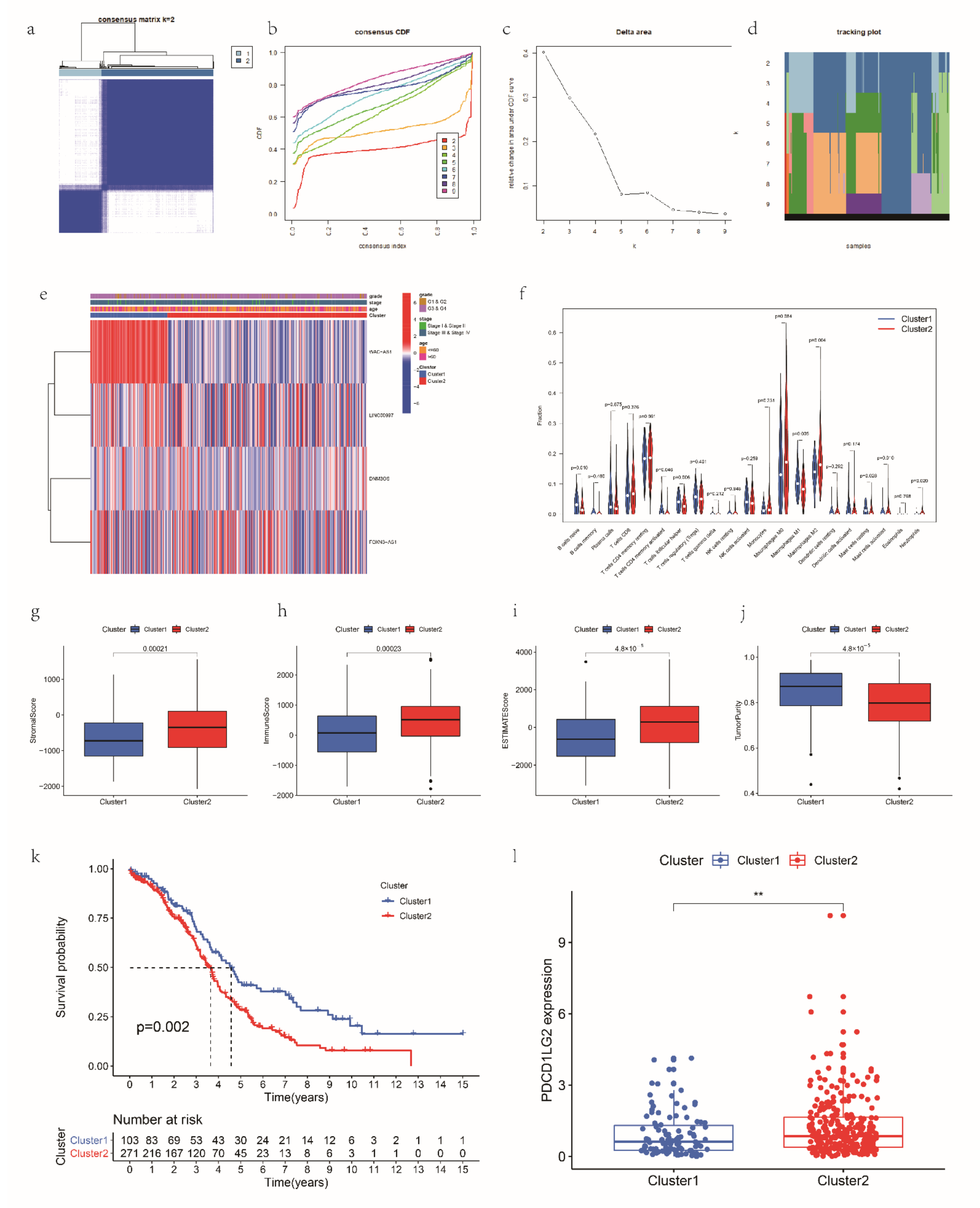
2.11. Consensus Clustering
2.12. Statistical Analysis
3. Results
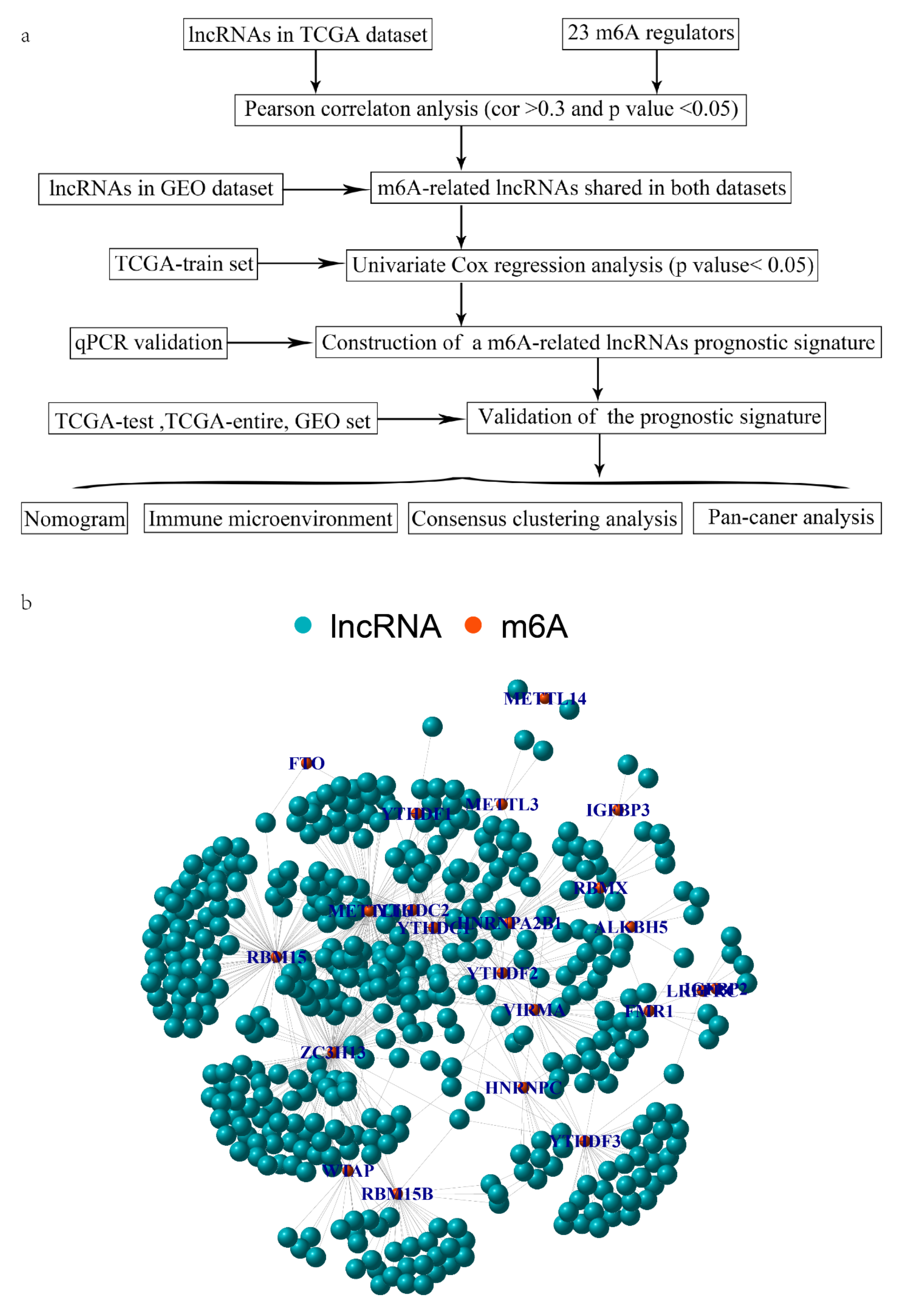
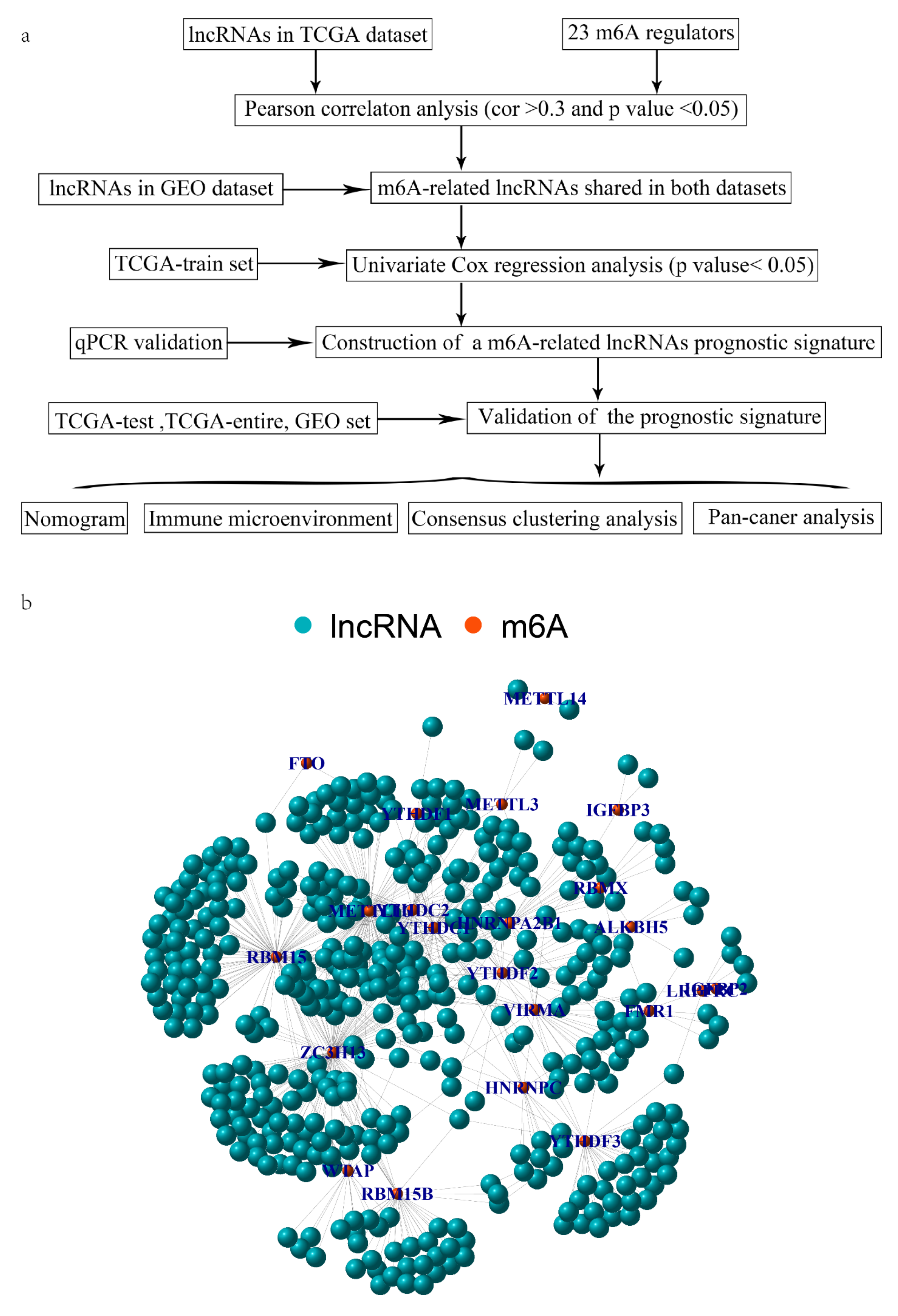
3.1. Identification of mRLs in OV
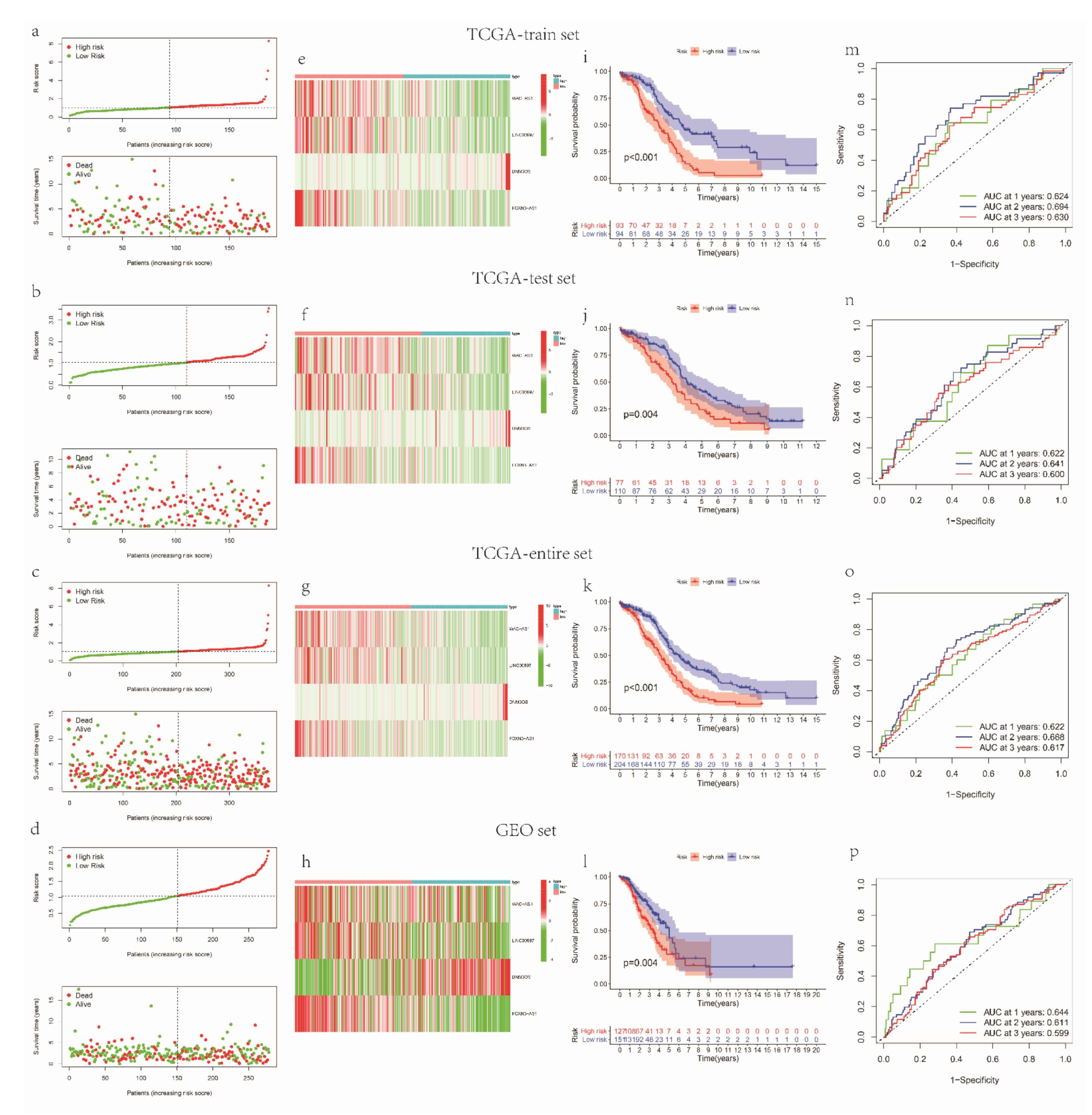
3.2. Establish and Verify a Prognostic Signature Based on mRLs in OV Patients
3.3. The Relationships between the Model and Clinical Factors
3.4. Clinical Application of the Signature
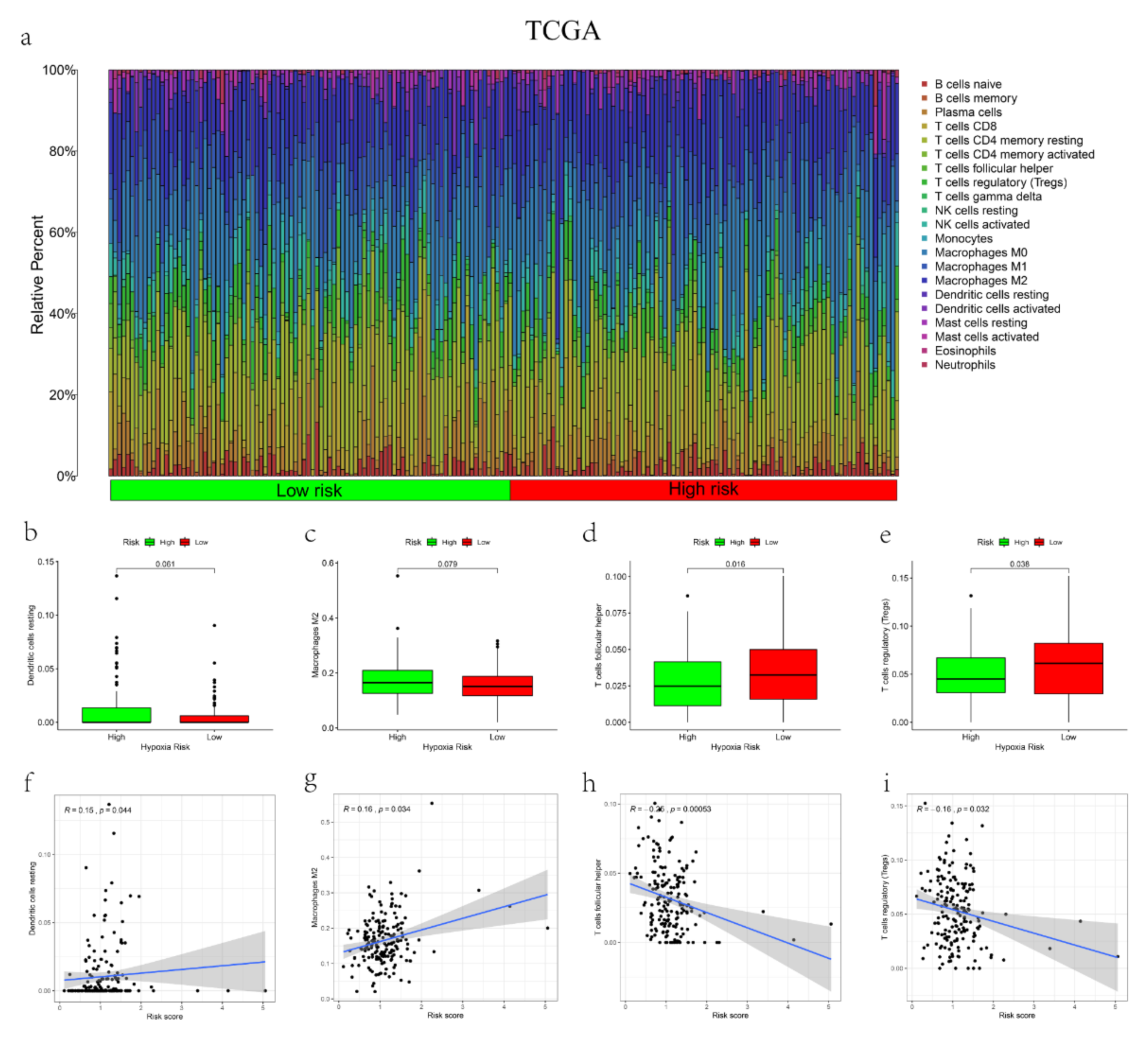
3.5. The Correlations between the mRL Prognostic Signature and TME
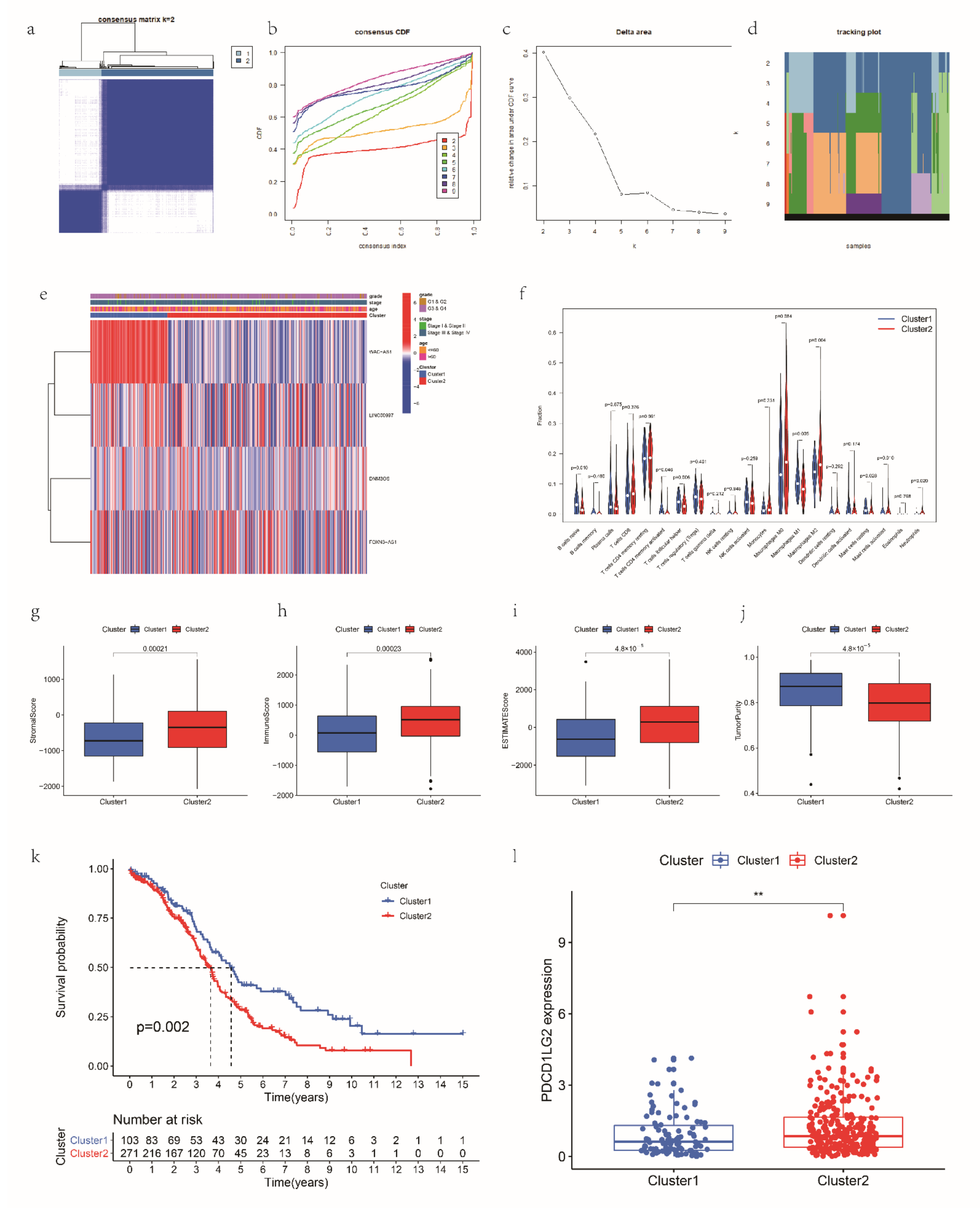
3.6. Consensus Clustering for mRLs Related to OV Prognosis and TME
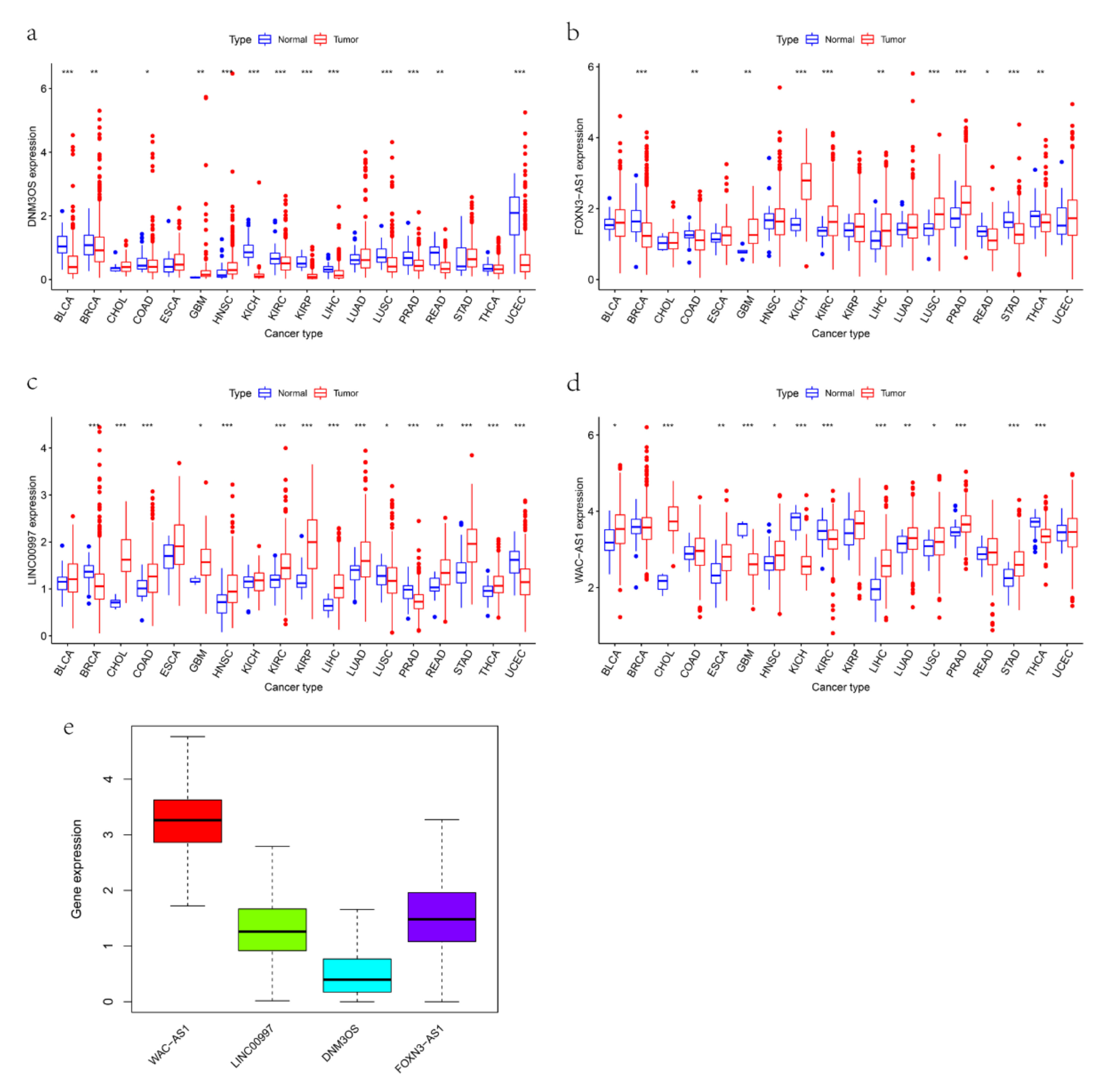
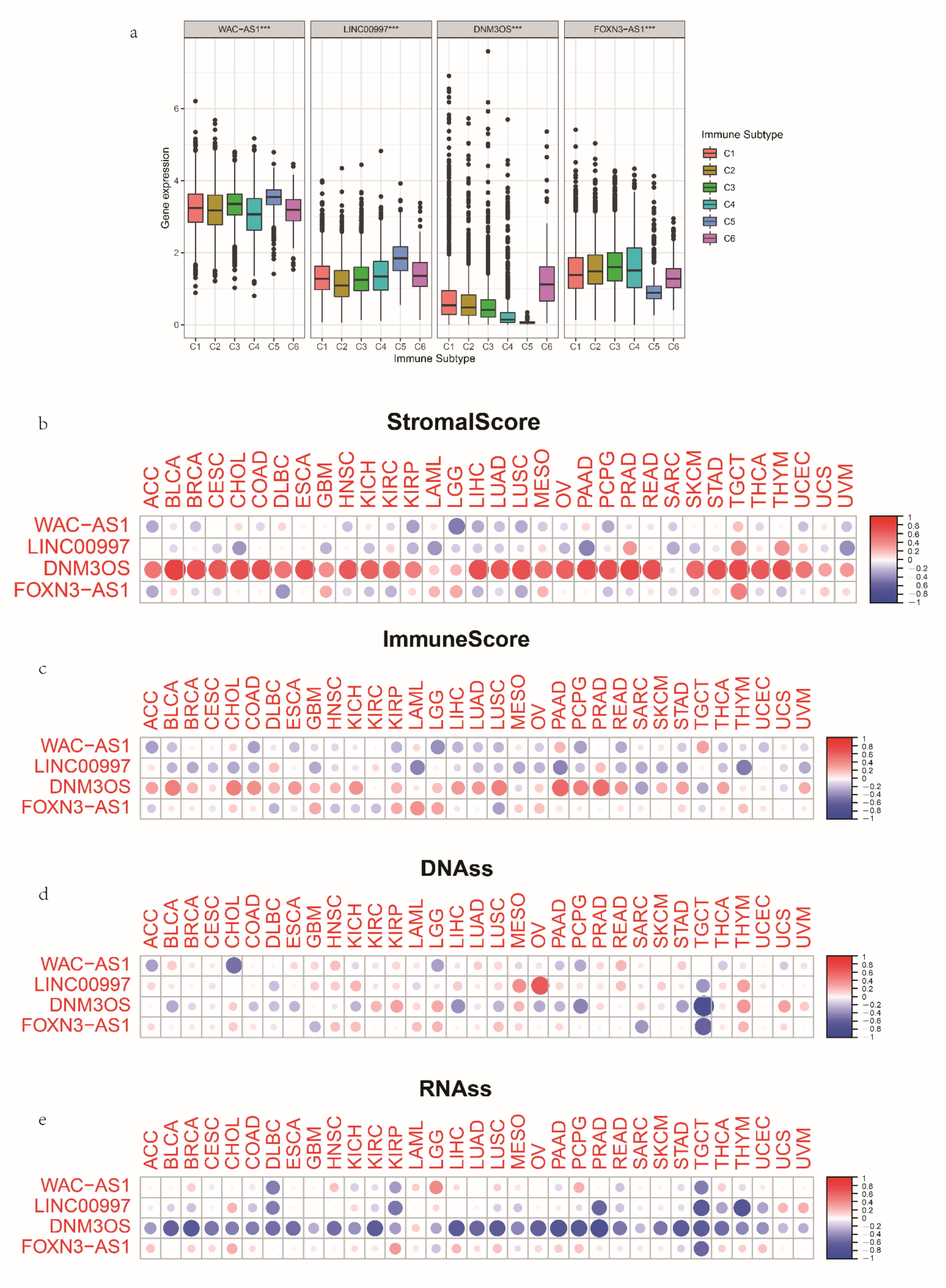
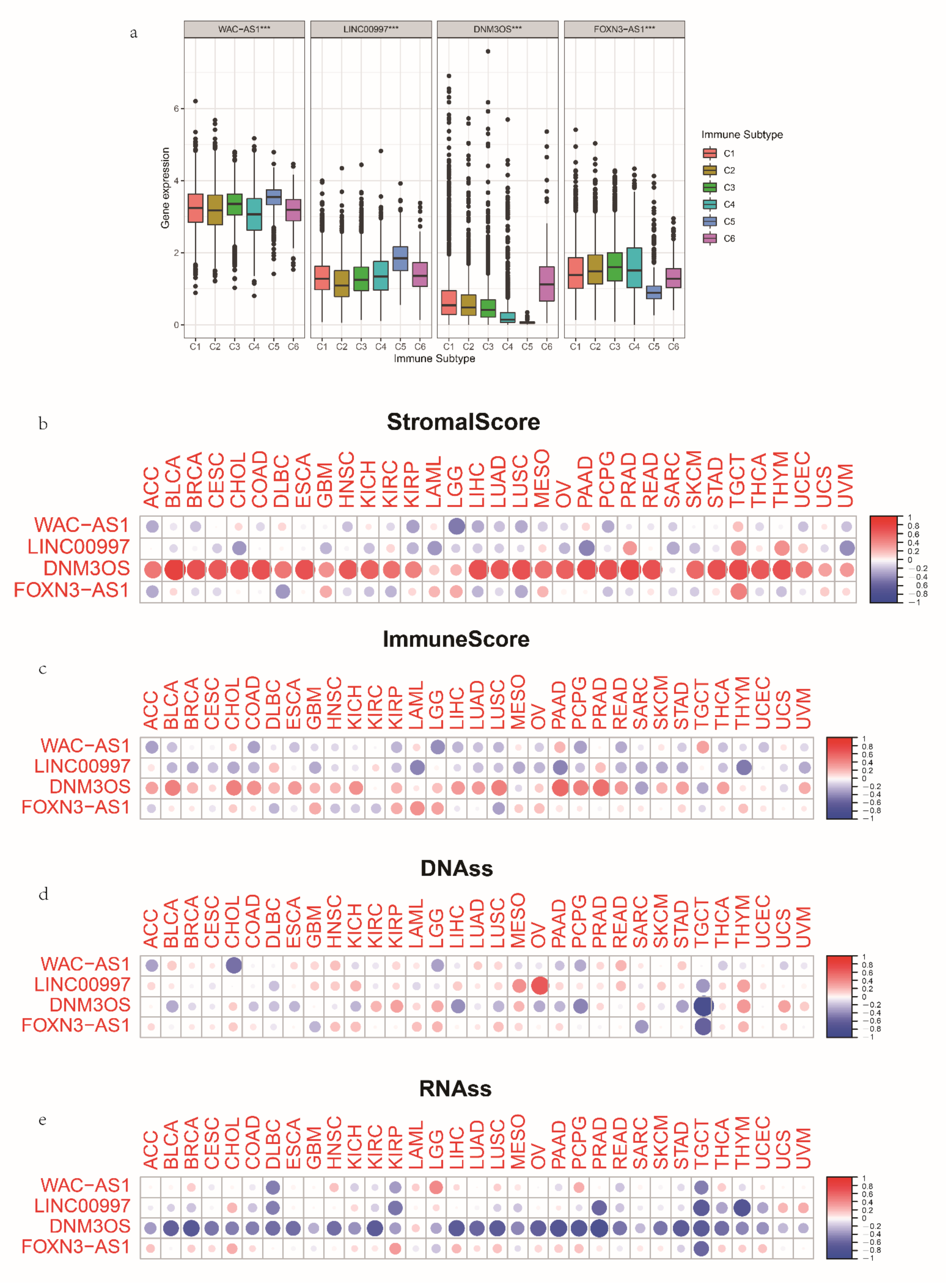
3.7. The Expression and Immune Status of lncRNAs in Pan-Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | analysis of variance |
| AUC | area under the curve |
| CI | confidence interval |
| DNAss | stemness scores based on DNA methylation |
| GEO | Gene Expression Omnibus |
| GSEA | gene set enrichment analysis |
| HR | hazard ratio |
| ICIs | immune checkpoint inhibitors |
| IPSs | immunophenoscores |
| LASSO | least absolute shrinkage and selection operator |
| lncRNAs | long noncoding RNAs |
| m6A | N6-methyladenosine |
| mRLs | N6-methyladenosine-related long noncoding RNAs |
| OS | overall survival |
| OV | ovarian serous cystadenocarcinoma |
| PCA | principal component analysis |
| qRT-PCR | quantitative real-time PCR |
| RNAss | stemness scores based on mRNA |
| ROC | receiver-operating characteristic curves |
| ssGSEA | Single Sample Gene Set Enrichment Analysis |
| TCGA | The Cancer Genome Atlas |
| TCIA | The Cancer Immunome Atlas |
| TIICs | tumor-infiltration immune cells |
| TME | tumor microenvironment |
| Treg | regulatory T cells |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics. CA Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Sikora, M.J.; Boisen, M.M.; Ma, T.; Christie, A.; Tseng, G.; Park, Y.; Luthra, S.; Chandran, U.; Haluska, P.; et al. Active Estrogen Receptor-alpha Signaling in Ovarian Cancer Models and Clinical Specimens. Clin. Cancer Res. 2017, 23, 3802–3812. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Zhou, J.-Q.; Liao, Y.-S. Autophagy-related long non-coding RNA signature for ovarian cancer. J. Int. Med. Res. 2020, 48, 300060520970761. [Google Scholar] [CrossRef]
- Henderson, J.T.; Webber, E.M.; Sawaya, G.F. Screening for Ovarian Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 595–606. [Google Scholar] [CrossRef]
- Huang, H.; Weng, H.; Chen, J. m6A Modification in Coding and Non-coding RNAs: Roles and Therapeutic Implications in Cancer. Cancer Cell 2020, 37, 270–288. [Google Scholar] [CrossRef]
- Shulman, Z.; Stern-Ginossar, N. The RNA modification N6-methyladenosine as a novel regulator of the immune system. Nat. Immunol. 2020, 21, 501–512. [Google Scholar] [CrossRef]
- Roundtree, I.A.; Evans, M.E.; Pan, T.; He, C. Dynamic RNA Modifications in Gene Expression Regulation. Cell 2017, 169, 1187–1200. [Google Scholar] [CrossRef]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef]
- Botti, G.; Scognamiglio, G.; Aquino, G.; Liguori, G.; Cantile, M. LncRNA HOTAIR in Tumor Microenvironment: What Role? Int. J. Mol. Sci. 2019, 20, 2279. [Google Scholar] [CrossRef]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, Y.; Shu, Y.; He, J.; Gao, W. Interaction between N6-methyladenosine (m6A) modification and noncoding RNAs in cancer. Mol. Cancer 2020, 19, 94. [Google Scholar] [CrossRef]
- Coker, H.; Wei, G.; Brockdorff, N. m6A modification of non-coding RNA and the control of mammalian gene expression. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 310–318. [Google Scholar] [CrossRef]
- Yi, Y.-C.; Chen, X.-Y.; Zhang, J.; Zhu, J.-S. Novel insights into the interplay between m6A modification and noncoding RNAs in cancer. Mol. Cancer 2020, 19, 121. [Google Scholar] [CrossRef]
- Liu, H.; Xu, Y.; Yao, B.; Sui, T.; Lai, L.; Li, Z. A novel N6-methyladenosine (m6A)-dependent fate decision for the lncRNA THOR. Cell Death Dis. 2020, 11, 613. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Chen, Z.; Tian, L.; Jiang, G.; Chen, F.; Li, J.; An, P.; Lu, L.; Luo, N.; et al. m6A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol. Cancer 2019, 18, 87. [Google Scholar] [CrossRef]
- Dai, F.; Wu, Y.; Lu, Y.; An, C.; Zheng, X.; Dai, L.; Guo, Y.; Zhang, L.; Li, H.; Xu, W.; et al. Crosstalk between RNA m6A Modification and Non-coding RNA Contributes to Cancer Growth and Progression. Mol. Ther.-Nucleic Acids 2020, 22, 62–71. [Google Scholar] [CrossRef]
- Song, P.; Yang, F.; Jin, H.; Wang, X. The regulation of protein translation and its implications for cancer. Signal Transduct. Target. Ther. 2021, 6, 68. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.-H.; Wu, Q.-N.; Jin, Y.; Wang, D.-S.; Chen, Y.-X.; Liu, J.; Luo, X.-J.; Meng, Q.; Pu, H.-Y.; et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol. Cancer 2019, 18, 174. [Google Scholar] [CrossRef]
- Wang, J.; Ding, W.; Xu, Y.; Tao, E.; Mo, M.; Xu, W.; Cai, X.; Chen, X.; Yuan, J.; Wu, X. Long non-coding RNA RHPN1-AS1 promotes tumorigenesis and metastasis of ovarian cancer by acting as a ceRNA against miR-596 and upregulating LETM1. Aging 2020, 12, 4558–4572. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Goldman, M.J.; Zhang, J.; Fonseca, N.A.; Cortés-Ciriano, I.; Xiang, Q.; Craft, B.; Piñeiro-Yáñez, E.; O’Connor, B.D.; Bazant, W.; Barrera, E.; et al. A user guide for the online exploration and visualization of PCAWG data. Nat. Commun. 2020, 11, 3400. [Google Scholar] [CrossRef]
- Zaccara, S.; Ries, R.J.; Jaffrey, S.R. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 608–624. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Yu, X.; Jin, F. Identification and validation of stemness-related lncRNA prognostic signature for breast cancer. J. Transl. Med. 2020, 18, 331. [Google Scholar] [CrossRef]
- Liu, J.; Xu, W.; Li, S.; Sun, R.; Cheng, W. Multi-omics analysis of tumor mutational burden combined with prognostic assessment in epithelial ovarian cancer based on TCGA database. Int. J. Med. Sci. 2020, 17, 3200–3213. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef]
- Reinhold, W.C.; Sunshine, M.; Varma, S.; Doroshow, J.H.; Pommier, Y. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin. Cancer Res. 2015, 21, 3841–3852. [Google Scholar] [CrossRef] [PubMed]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830.e14. [Google Scholar] [CrossRef] [PubMed]
- Webb, P.M.; Jordan, S.J. Epidemiology of epithelial ovarian cancer. Best Pract. Res. Clin. Obstet. Gynaecol. 2017, 41, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Wang, G.; Zhang, R.; Zhao, Y.; Yu, H.; Wei, Y.; Chen, F. Development and validation of an immune gene-set based Prognostic signature in ovarian cancer. eBioMedicine 2019, 40, 318–326. [Google Scholar] [CrossRef]
- Millstein, J.; Budden, T.; Goode, E.; Anglesio, M.; Talhouk, A.; Intermaggio, M.; Leong, H.; Chen, S.; Elatre, W.; Gilks, B.; et al. Prognostic gene expression signature for high-grade serous ovarian cancer. Ann. Oncol. 2020, 31, 1240–1250. [Google Scholar] [CrossRef]
- Liu, J.; Meng, H.; Nie, S.; Sun, Y.; Jiang, P.; Li, S.; Yang, J.; Sun, R.; Cheng, W. Identification of a prognostic signature of epithelial ovarian cancer based on tumor immune microenvironment exploration. Genomics 2020, 112, 4827–4841. [Google Scholar] [CrossRef]
- Zheng, M.; Hu, Y.; Gou, R.; Nie, X.; Li, X.; Liu, J.; Lin, B. Identification three LncRNA prognostic signature of ovarian cancer based on genome-wide copy number variation. Biomed. Pharmacother. 2020, 124, 109810. [Google Scholar] [CrossRef]
- Fan, L.; Lin, Y.; Lei, H.; Shu, G.; He, L.; Yan, Z.; Rihan, H.; Yin, G. A newly defined risk signature, consisting of three m(6)A RNA methylation regulators, predicts the prognosis of ovarian cancer. Aging 2020, 12, 18453–18475. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, C.; Ding, Q.; Zhao, Y.; Wang, Z.; Chen, J.; Jiang, Z.; Zhang, Y.; Xu, G.; Zhang, J.; et al. METTL3-mediated m6A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut 2020, 69, 1193–1205. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Hüttelmaier, S.; Liu, T. The Critical Role of RNA m6A Methylation in Cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef]
- He, L.; He, G. DNM3OS Facilitates Ovarian Cancer Progression by Regulating miR-193a-3p/MAP3K3 Axis. Yonsei Med. J. 2021, 62, 535–544. [Google Scholar] [CrossRef]
- Xia, X.; Zhang, H.; Xia, P.; Zhu, Y.; Liu, J.; Xu, K.; Yuan, Y. Identification of Glycolysis-Related lncRNAs and the Novel lncRNA WAC-AS1 Promotes Glycolysis and Tumor Progression in Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 733595. [Google Scholar] [CrossRef]
- Zheng, J.; Zhou, Z.; Qiu, Y.; Wang, M.; Yu, H.; Wu, Z.; Wang, X.; Jiang, X. A Prognostic Ferroptosis-Related lncRNAs Signature Associated With Immune Landscape and Radiotherapy Response in Glioma. Front. Cell Dev. Biol. 2021, 9, 675555. [Google Scholar] [CrossRef]
- Shi, Z.; Shen, C.; Yu, C.; Yang, X.; Shao, J.; Guo, J.; Zhu, X.; Zhou, G. Long non-coding RNA LINC00997 silencing inhibits the progression and metastasis of colorectal cancer by sponging miR-512-3p. Bioengineered 2021, 12, 627–639. [Google Scholar] [CrossRef]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. S8), viii1–viii7. [Google Scholar] [CrossRef]
- Hartnett, E.G.; Knight, J.; Radolec, M.; Buckanovich, R.J.; Edwards, R.P.; Vlad, A.M. Immunotherapy Advances for Epithelial Ovarian Cancer. Cancers 2020, 12, 3733. [Google Scholar] [CrossRef]
- Osorio Ovarian Tumor Tissue Analysis (OTTA) Consortium; Goode, E.L.; Block, M.S.; Kalli, K.R.; Vierkant, R.A.; Chen, W.; Fogarty, Z.C.; Gentry-Maharaj, A.; Toloczko, A.; Hein, A.; et al. Dose-Response Association of CD8+ Tumor-Infiltrating Lymphocytes and Survival Time in High-Grade Serous Ovarian Cancer. JAMA Oncol. 2017, 3, e173290. [Google Scholar] [CrossRef]
- Tamborero, D.; Rubio-Perez, C.; Muiños, F.; Sabarinathan, R.; Piulats, J.M.; Muntasell, A.; Dienstmann, R.; Lopez-Bigas, N.; Gonzalez-Perez, A. A Pan-cancer Landscape of Interactions between Solid Tumors and Infiltrating Immune Cell Populations. Clin. Cancer Res. 2018, 24, 3717–3728. [Google Scholar] [CrossRef]
- Kreuzinger, C.; Geroldinger, A.; Smeets, D.; Braicu, E.I.; Sehouli, J.; Koller, J.; Wolf, A.; Darb-Esfahani, S.; Joehrens, K.; Vergote, I.; et al. A Complex Network of Tumor Microenvironment in Human High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2017, 23, 7621–7632. [Google Scholar] [CrossRef]
- Zheng, J.; Guo, J.; Cao, B.; Zhou, Y.; Tong, J. Identification and validation of lncRNAs involved in m6A regulation for patients with ovarian cancer. Cancer Cell Int. 2021, 21, 363. [Google Scholar] [CrossRef]
- Wei, Q.; Yang, D.; Liu, X.; Zhao, H.; Yang, Y.; Xu, J.; Liu, T.; Yi, P. Exploration of the Role of m6 A RNA Methylation Regulators in Malignant Progression and Clinical Prognosis of Ovarian Cancer. Front. Genet. 2021, 12, 650554. [Google Scholar] [CrossRef]
- Olbrecht, S.; Busschaert, P.; Qian, J.; Vanderstichele, A.; Loverix, L.; Van Gorp, T.; Van Nieuwenhuysen, E.; Han, S.; Van den Broeck, A.; Coosemans, A.; et al. High-grade serous tubo-ovarian cancer refined with single-cell RNA sequencing: Specific cell subtypes influence survival and determine molecular subtype classification. Genome Med. 2021, 13, 111. [Google Scholar] [CrossRef]
- Tan, Q.; Liu, H.; Xu, J.; Mo, Y.; Dai, F. Integrated analysis of tumor-associated macrophage infiltration and prognosis in ovarian cancer. Aging 2021, 13, 23210–23232. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, Y.; Zhao, S.; Qian, L.; Song, T.; Zheng, J.; Zhang, J.; Chen, B. Growth differentiation factor-15 promotes immune escape of ovarian cancer via targeting CD44 in dendritic cells. Exp. Cell Res. 2021, 402, 112522. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F.; et al. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Klingemann, H.; Boissel, L.; Toneguzzo, F. Natural Killer Cells for Immunotherapy—Advantages of the NK-92 Cell Line over Blood NK Cells. Front. Immunol. 2016, 7, 91. [Google Scholar] [CrossRef]
- Motz, G.T.; Coukos, G. Deciphering and Reversing Tumor Immune Suppression. Immunity 2013, 39, 61–73. [Google Scholar] [CrossRef]
- Banchereau, R.; Leng, N.; Zill, O.; Sokol, E.; Liu, G.; Pavlick, D.; Maund, S.; Liu, L.F.; Kadel, E., 3rd; Baldwin, N.; et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat. Commun. 2021, 12, 3969. [Google Scholar] [CrossRef] [PubMed]
- Maiorano, B.A.; Maiorano, M.F.P.; Lorusso, D.; Maiello, E. Ovarian Cancer in the Era of Immune Checkpoint Inhibitors: State of the Art and Future Perspectives. Cancers 2021, 13, 4438. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Xia, B.-R.; Zhang, Z.-C.; Zhang, Y.-J.; Lou, G.; Jin, W.-L. Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front. Immunol. 2020, 11, 577869. [Google Scholar] [CrossRef]
- Miao, Y.R.; Thakkar, K.N.; Qian, J.; Kariolis, M.S.; Huang, W.; Nandagopal, S.; Yang, T.T.C.; Diep, A.N.; Cherf, G.M.; Xu, Y.; et al. Neutralization of PD-L2 is Essential for Overcoming Immune Checkpoint Blockade Resistance in Ovarian Cancer. Clin. Cancer Res. 2021, 27, 4435–4448. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Sun, Z.-J. Turning cold tumors into hot tumors by improving T-cell infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Hegde, P.S.; Karanikas, V.; Evers, S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin. Cancer Res. 2016, 22, 1865–1874. [Google Scholar] [CrossRef]
- Gu, J.; Bi, F. Significance of N6-Methyladenosine RNA Methylation Regulators in Immune Infiltrates of Ovarian Cancer. Front. Genet. 2021, 12, 671179. [Google Scholar] [CrossRef]
- Jiang, Y.; Wan, Y.; Gong, M.; Zhou, S.; Qiu, J.; Cheng, W. RNA demethylase ALKBH5 promotes ovarian carcinogenesis in a simulated tumour microenvironment through stimulating NF-kappaB pathway. J. Cell Mol. Med. 2020, 24, 6137–6148. [Google Scholar] [CrossRef]
- Das, S.; Reddy, M.A.; Senapati, P.; Stapleton, K.; Lanting, L.; Wang, M.; Amaram, V.; Ganguly, R.; Zhang, L.; Devaraj, S.; et al. Diabetes Mellitus–Induced Long Noncoding RNA Dnm3os Regulates Macrophage Functions and Inflammation via Nuclear Mechanisms. Arter. Thromb. Vasc. Biol. 2018, 38, 1806–1820. [Google Scholar] [CrossRef] [PubMed]
- Lakhia, R.; Yheskel, M.; Flaten, A.; Ramalingam, H.; Aboudehen, K.; Ferrè, S.; Biggers, L.; Mishra, A.; Chaney, C.; Wallace, D.P.; et al. Interstitial microRNA miR-214 attenuates inflammation and polycystic kidney disease progression. JCI Insight 2020, 5, e133785. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geng, R.; Chen, T.; Zhong, Z.; Ni, S.; Bai, J.; Liu, J. The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer. Cancers 2022, 14, 4056. https://doi.org/10.3390/cancers14164056
Geng R, Chen T, Zhong Z, Ni S, Bai J, Liu J. The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer. Cancers. 2022; 14(16):4056. https://doi.org/10.3390/cancers14164056
Chicago/Turabian StyleGeng, Rui, Tian Chen, Zihang Zhong, Senmiao Ni, Jianling Bai, and Jinhui Liu. 2022. "The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer" Cancers 14, no. 16: 4056. https://doi.org/10.3390/cancers14164056
APA StyleGeng, R., Chen, T., Zhong, Z., Ni, S., Bai, J., & Liu, J. (2022). The m6A-Related Long Noncoding RNA Signature Predicts Prognosis and Indicates Tumor Immune Infiltration in Ovarian Cancer. Cancers, 14(16), 4056. https://doi.org/10.3390/cancers14164056