
High Accuracy Indicators of Androgen Suppression Therapy Failure for Prostate Cancer—A Modeling Study

 ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
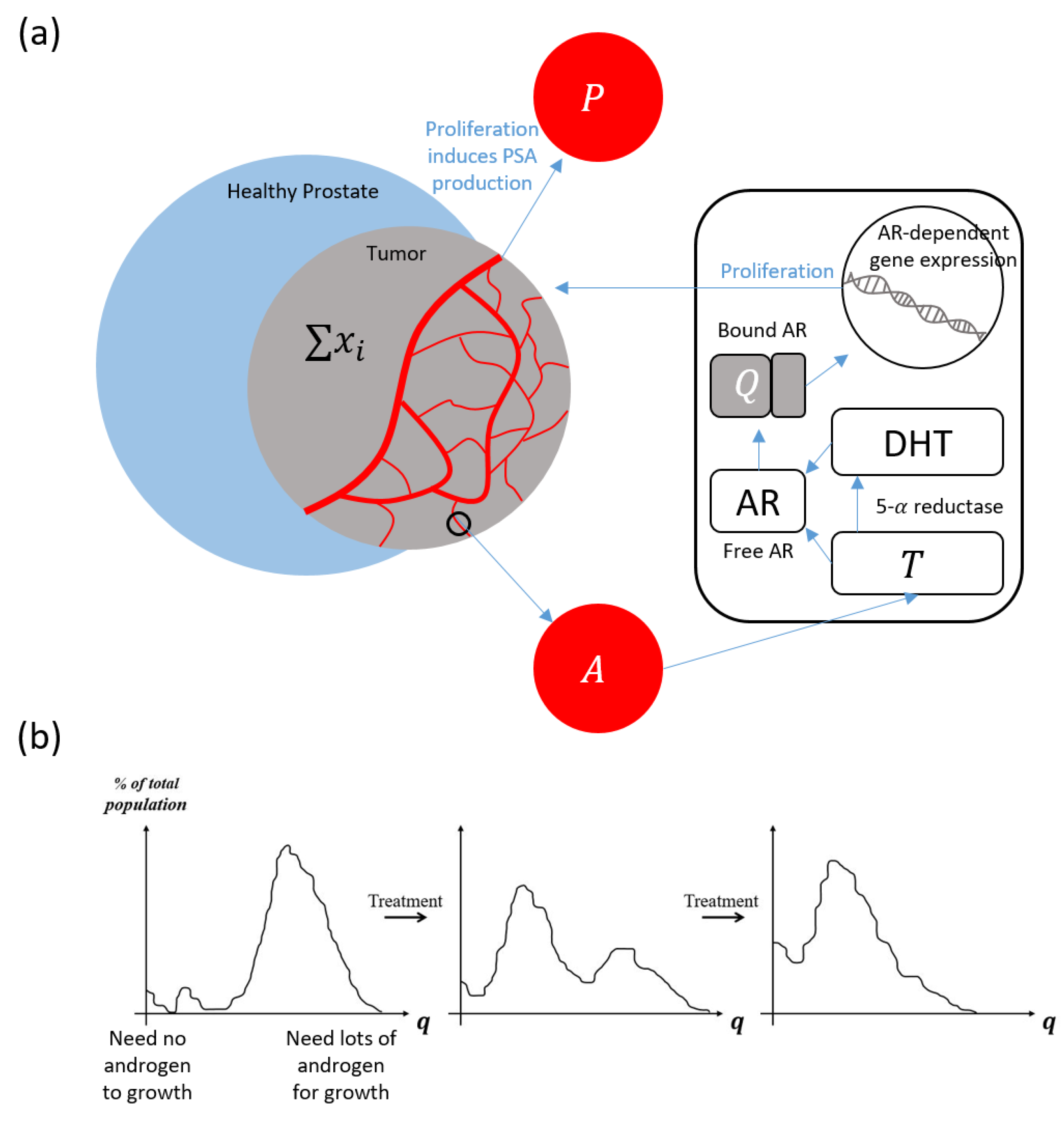
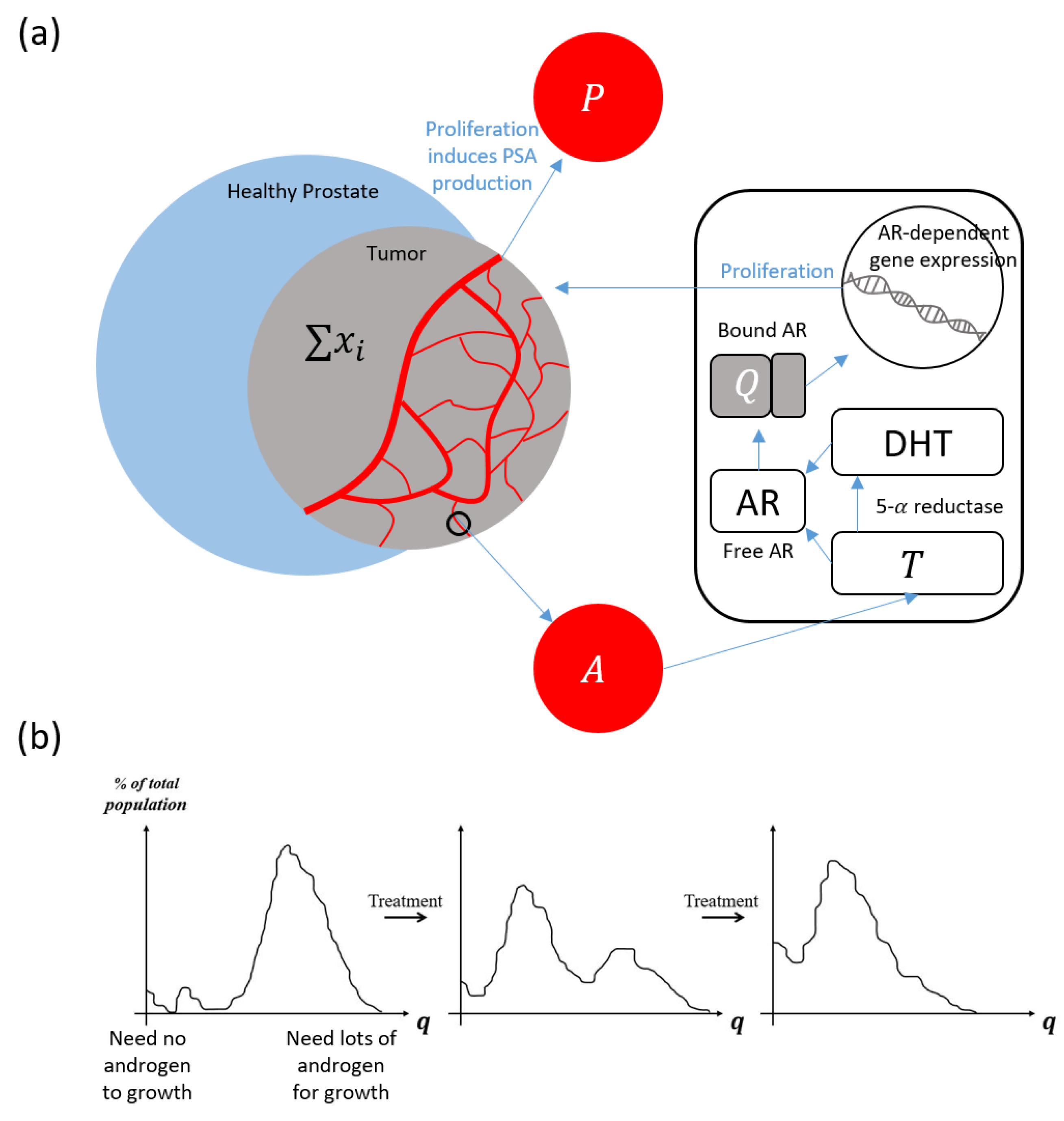
2. Materials and Methods
Model Quick-Guide Box
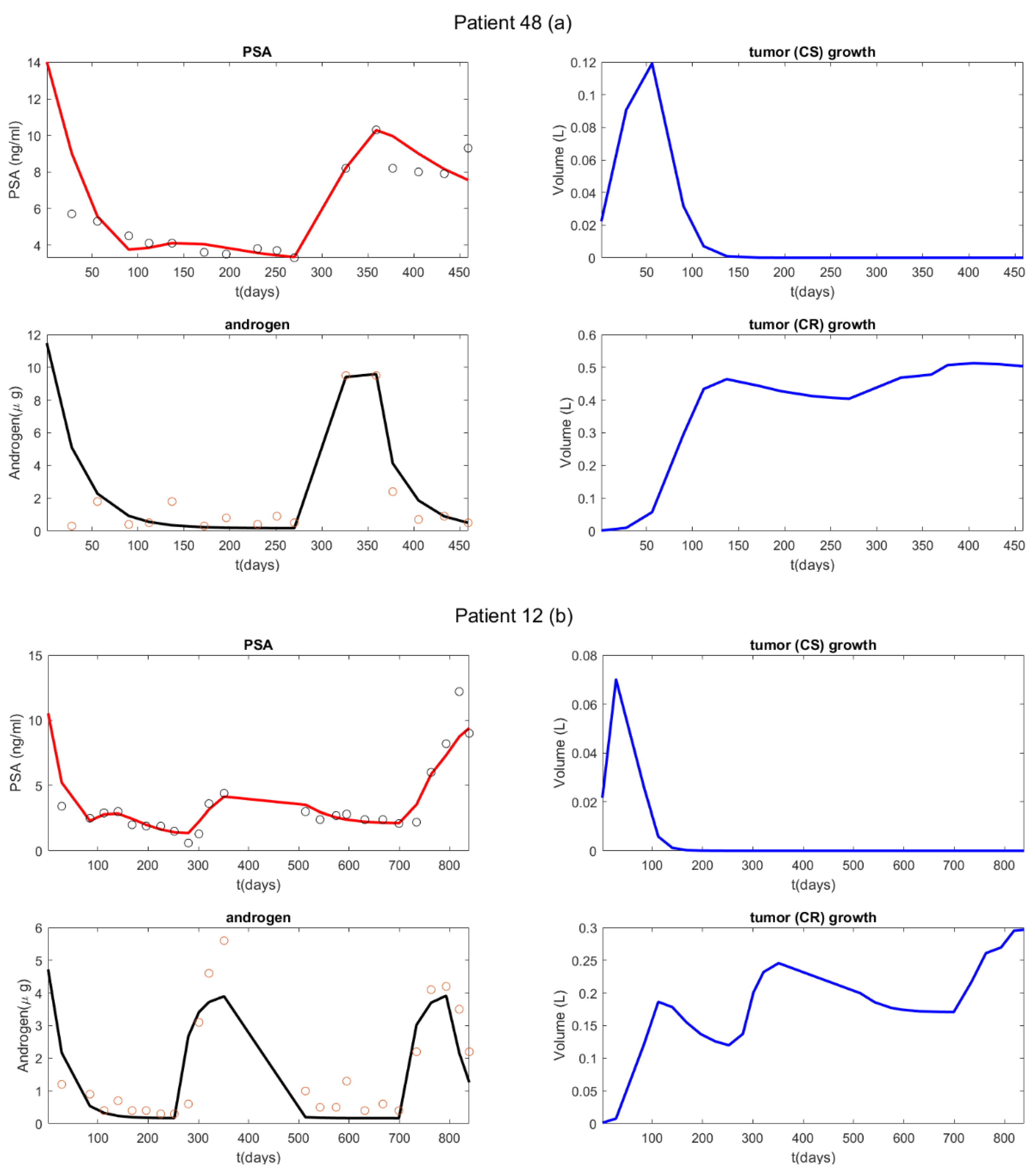
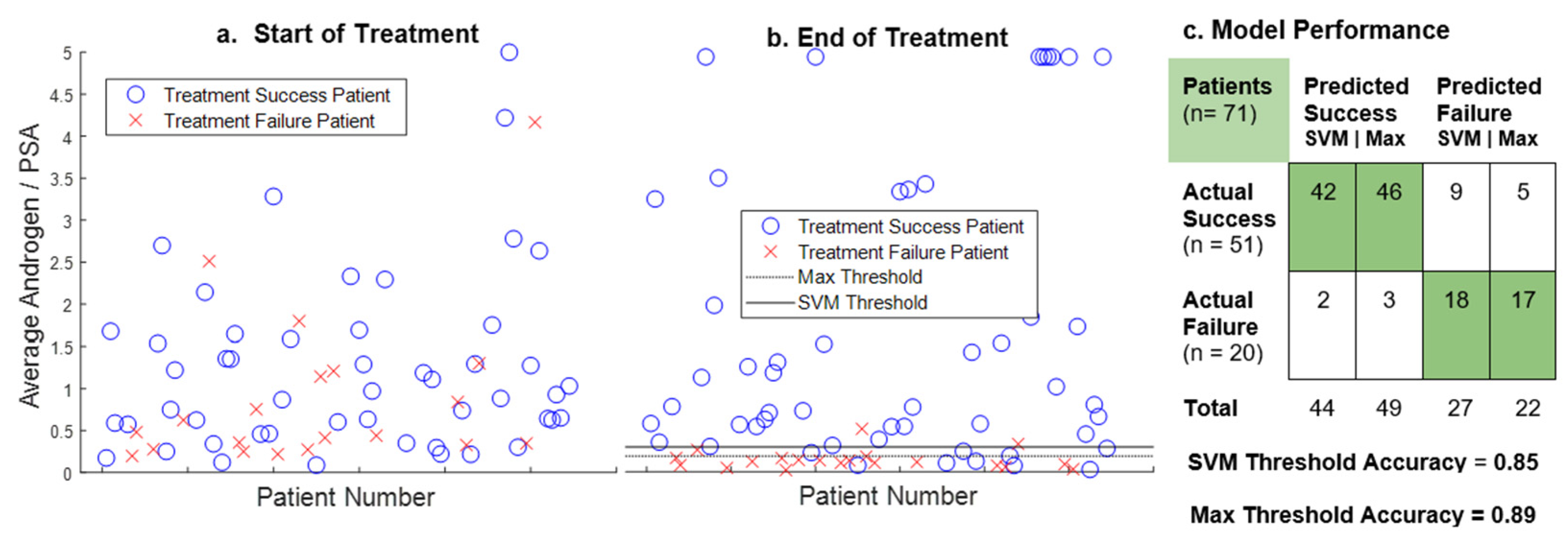
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Sayegh, N.; Swami, U.; Agarwal, N. Recent Advances in the Management of Metastatic Prostate Cancer. J. Oncol. Pr. 2022, 18, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, F.; Dibitetto, F.; Ragonese, M.; Bassi, P. Mechanisms of Resistance to Second-Generation Antiandrogen Therapy for Prostate Cancer: Actual Knowledge and Perspectives. Med. Sci. 2022, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Crook, S.M.; Bryce, A.H.; Maley, C.C.; Kostelich, E.J.; Kuang, Y. Review: Mathematical modeling of prostate cancer and clinical application. Appl. Sci. 2020, 10, 2721. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive Therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortunato, A.; Boddy, A.; Mallo, D.; Aktipis, A.; Maley, C.C.; Pepper, J.W. Natural Selection in Cancer Biology: From Molecular Snowflakes to Trait Hallmarks. Cold Spring Harb. Perspect. Med. 2017, 7, a029652. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Lloyd, M.; Cunningham, J.; Bui, M.; Gillies, R.; Brown, J.; Gatenby, R. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [Green Version]
- Morken, J.D.; Packer, A.; Everett, R.A.; Nagy, J.D.; Kuang, Y. Mechanisms of resistance to intermittent androgen deprivation in patients with prostate cancer identified by a novel computational method. Cancer Res. 2014, 74, 3673–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Cunningham, J.J.; Brown, J.S.; Gatenby, R.A. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat. Commun. 2017, 8, 1816. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer. JAMA 2017, 317, 2532. [Google Scholar] [CrossRef] [PubMed]
- Magnan, S.; Zarychanski, R.; Pilote, L.; Bernier, L.; Shemilt, M.; Vigneault, E.; Fradet, V.; Turgeon, A.F. Intermittent vs. Continuous Androgen Deprivation Therapy for Prostate Cancer. JAMA Oncol. 2015, 1, 1261. [Google Scholar] [CrossRef] [Green Version]
- Reckell, T.; Nguyen, K.; Phan, T.; Crook, S.; Kostelich, E.J.; Kuang, Y. Modeling the synergistic properties of drugs in hormonal treatment for prostate cancer. J. Theor. Biol. 2021, 514, 110570. [Google Scholar] [CrossRef]
- Bruchovsky, N.; Klotz, L.; Crook, J.; Malone, S.; Ludgate, C.; Morris, W.J.; Gleave, M.E.; Goldenberf, S.L. Final results of the Canadian prospective phase II trial of intermittent androgen suppression for men in biochemical recurrence after radiotherapy for locally advanced prostate cancer. Cancer 2006, 107, 389–395. [Google Scholar] [CrossRef]
- Spry, N.A.; Kristjanson, L.; Hooton, B.; Hayden, L.; Neerhut, G.; Gurney, H.; Corica, T.; Korbel, E.; Weinstein, S.; McCaul, K. Adverse effects to quality of life arising from treatment can recover with intermittent androgen suppression in men with prostate cancer. Eur. J. Cancer 2006, 42, 1083–1092. [Google Scholar] [CrossRef]
- Shore, N.D.; Crawford, E.D. Intermittent androgen deprivation therapy: Redefining the standard of care? Rev. Urol. 2010, 12, 1–11. [Google Scholar]
- Barnaby, J.P.; Sorribes, I.C.; Jain, H.V. Relating prostate-specific antigen leakage with vascular tumor growth in a mathematical model of prostate cancer response to androgen deprivation. Comput. Syst. Oncol. 2021, 1, e1014. [Google Scholar] [CrossRef]
- Saini, S. PSA and beyond: Alternative prostate cancer biomarkers. Cell Oncol. 2016, 39, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, T.L. A mathematical model of prostate tumor growth and androgen-independent relapse. Discret. Cont. Dyn. Syst. Ser. B 2003, 4, 187–201. [Google Scholar] [CrossRef]
- Baez, J.; Kuang, Y. Mathematical models of androgen resistance in prostate cancer patients under intermittent androgen suppression therapy. Appl. Sci. 2016, 6, 352. [Google Scholar] [CrossRef] [Green Version]
- Portz, T.; Kuang, Y.; Nagy, J.D. A clinical data validated mathematical model of prostate cancer growth under intermittent androgen suppression therapy. AIP Adv. 2012, 2, 011002. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.; He, C.; Martinez, A.; Kuang, Y. Dynamics and implications of models for intermittent androgen suppression therapy. Math. Biosci. Eng. 2019, 16, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Nguyen, K.; Sharma, P.; Kuang, Y. The impact of intermittent androgen suppression therapy in prostate cancer modeling. Appl. Sci. 2018, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Phan, T.; Baez, J.; Kuang, Y.; Kostelich, E.J. Predictability and identifiability assessment of models for prostate cancer under androgen suppression therapy. Math. Biosci. Eng. 2019, 16, 3512–3536. [Google Scholar] [CrossRef]
- Brady-Nicholls, R.; Nagy, J.D.; Gerke, T.A.; Zhang, T.; Wang, A.Z.; Zhang, J.; Gatenby, A.; Enderling, H. Prostate-specific antigen dynamics predict individual responses to intermittent androgen deprivation. Nat. Commun. 2020, 11, 1750. [Google Scholar] [CrossRef] [Green Version]
- West, J.; You, L.; Zhang, J.; Gatenby, R.A.; Brown, J.S.; Newton, P.K.; Anderson, A.R.A. Towards Multidrug Adaptive Therapy. Cancer Res. 2020, 80, 1578–1589. [Google Scholar] [CrossRef]
- Hirata, Y.; Bruchovsky, N.; Aihara, K. Development of a mathematical model that predicts the outcome of hormone therapy for prostate cancer. J. Theor. Biol. 2010, 264, 517–527. [Google Scholar] [CrossRef]
- Nakanishi, A.; Hirata, Y. Practically scheduling hormone therapy for prostate cancer using a mathematical model. J. Theor. Biol. 2019, 478, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Jain, H.V.; Clinton, S.K.; Bhinder, A.; Friedman, A. Mathematical modeling of prostate cancer progression in response to androgen ablation therapy. Proc. Natl. Acad. Sci. USA 2011, 108, 19701–19706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, H.V.; Sorribes, I.C.; Handelman, S.K.; Barnaby, J.; Jackson, T.L. Standing Variations Modeling Captures Inter-Individual Heterogeneity in a Deterministic Model of Prostate Cancer Response to Combination Therapy. Cancers 2021, 13, 1872. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, G.; Scott, M.A.; Tew, K.; Hughes, T.J.R.; Zhang, Y.J.; Liu, L.; Vilanova, G.; Gomez, H. Tissue-scale, personalized modeling and simulation of prostate cancer growth. Proc. Natl. Acad. Sci. USA 2016, 113, E7663–E7671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzo, G.; Pérez-García, V.M.; Mariño, A.; Pérez-Romasanta, L.A.; Reali, A.; Gomez, H. Mechanistic modelling of prostate-specific antigen dynamics shows potential for personalized prediction of radiation therapy outcome. J. R Soc. Interface 2019, 16, 20190195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerasuolo, M.; Paris, D.; Iannotti, F.A.; Melck, D.; Verde, R.; Mazzarella, E.; Motta, A.; Ligresti, A. Neuroendocrine Transdifferentiation in Human Prostate Cancer Cells: An Integrated Approach. Cancer Res. 2015, 75, 2975–2986. [Google Scholar] [CrossRef] [Green Version]
- Turner, L.; Burbanks, A.; Cerasuolo, M. PCa dynamics with neuroendocrine differentiation and distributed delay. Math. Biosci. Eng. 2021, 18, 8577–8602. [Google Scholar] [CrossRef]
- Coletti, R.; Leonardelli, L.; Parolo, S.; Marchetti, L. A QSP model of prostate cancer immunotherapy to identify effective combination therapies. Sci. Rep. 2020, 10, 9063. [Google Scholar] [CrossRef]
- Valle, P.A.; Coria, L.N.; Carballo, K.D. Chemoimmunotherapy for the treatment of prostate cancer: Insights from mathematical modelling. Appl. Math. Model 2021, 90, 682–702. [Google Scholar] [CrossRef]
- Siewe, N.; Friedman, A. Combination therapy for mCRPC with immune checkpoint inhibitors, ADT and vaccine: A mathematical model. Raza A, editor. PLoS ONE 2022, 17, e0262453. [Google Scholar] [CrossRef]
- Zhang, J.; Cunningham, J.; Brown, J.; Gatenby, R. Evolution-based mathematical models significantly prolong response to abiraterone in metastatic castrate-resistant prostate cancer and identify strategies to further improve outcomes. eLife 2022, 11, e76284. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Nagy, J.; Eikenberry, S. Introduction to Mathematical Oncology; Chapman and Hall/CRC: Boca Raton, FL, USA, 2018. [Google Scholar]
- Pasetto, S.; Enderling, H.; Gatenby, R.A.; Brady-Nicholls, R. Intermittent Hormone Therapy Models Analysis and Bayesian Model Comparison for Prostate Cancer. Bull. Math. Biol. 2022, 84, 2. [Google Scholar] [CrossRef] [PubMed]
- Droop, M.R. Vitamin B 12 and Marine Ecology. IV. The Kinetics of Uptake, Growth and Inhibition in Monochrysis lutheri. J. Mar. Biol. Assoc. UK 1968, 48, 689–733. [Google Scholar] [CrossRef]
- Droop, M.R. The nutrient status of algal cells in continuous culture. J. Mar. Biol. Assoc. UK 1974, 54, 825–855. [Google Scholar] [CrossRef]
- Packer, A.; Li, Y.; Andersen, T.; Hu, Q.; Kuang, Y.; Sommerfeld, M. Growth and neutral lipid synthesis in green microalgae: A mathematical model. Bioresour. Technol. 2011, 102, 111–117. [Google Scholar] [CrossRef]
- Feldman, B.; Feldman, D. The development of androgen independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- Louie, K.S.; Seigneurin, A.; Cathcart, P.; Sasieni, P. Do prostate cancer risk models improve the predictive accuracy of PSA screening? A meta-analysis. Ann. Oncol. 2015, 26, 848–864. [Google Scholar] [CrossRef]
- Carlson, G.D.; Calvanese, C.B.; Kahane, H.; Epstein, J.I. Accuracy of Biopsy Gleason Scores from a Large Uropathology Laboratory: Use of a Diagnostic Protocol to Minimize Observer Variability. Urology 1998, 51, 525–529. [Google Scholar] [CrossRef]
- Eikenberry, S.E.; Nagy, J.D.; Kuang, Y. The evolutionary impact of androgen levels on prostate cancer in a multi-scale mathematical model. Biol. Direct 2010, 5, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Ideta, A.M.; Tanaka, G.; Takeuchi, T.; Aihara, K. A Mathematical model of intermittent androgen suppression for prostate cancer. J. Nonlinear Sci. 2008, 18, 593–614. [Google Scholar] [CrossRef]
- Nishiyama, T. Serum testosterone levels after medical or surgical androgen deprivation: A comprehensive review of the literature. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 38.e17–38.e28. [Google Scholar] [CrossRef] [PubMed]
- Berges, R.R.; Vukanovic, J.; Epstein, J.I.; CarMichel, M.; Cisek, L.; Johnson, D.E.; Veltri, R.W.; Walsh, P.C.; Isaacs, J.T. Implication of cell kinetic changes during the progression of human prostatic cancer. Clin. Cancer Res. 1995, 1, 473–480. [Google Scholar] [PubMed]


{kind=link}
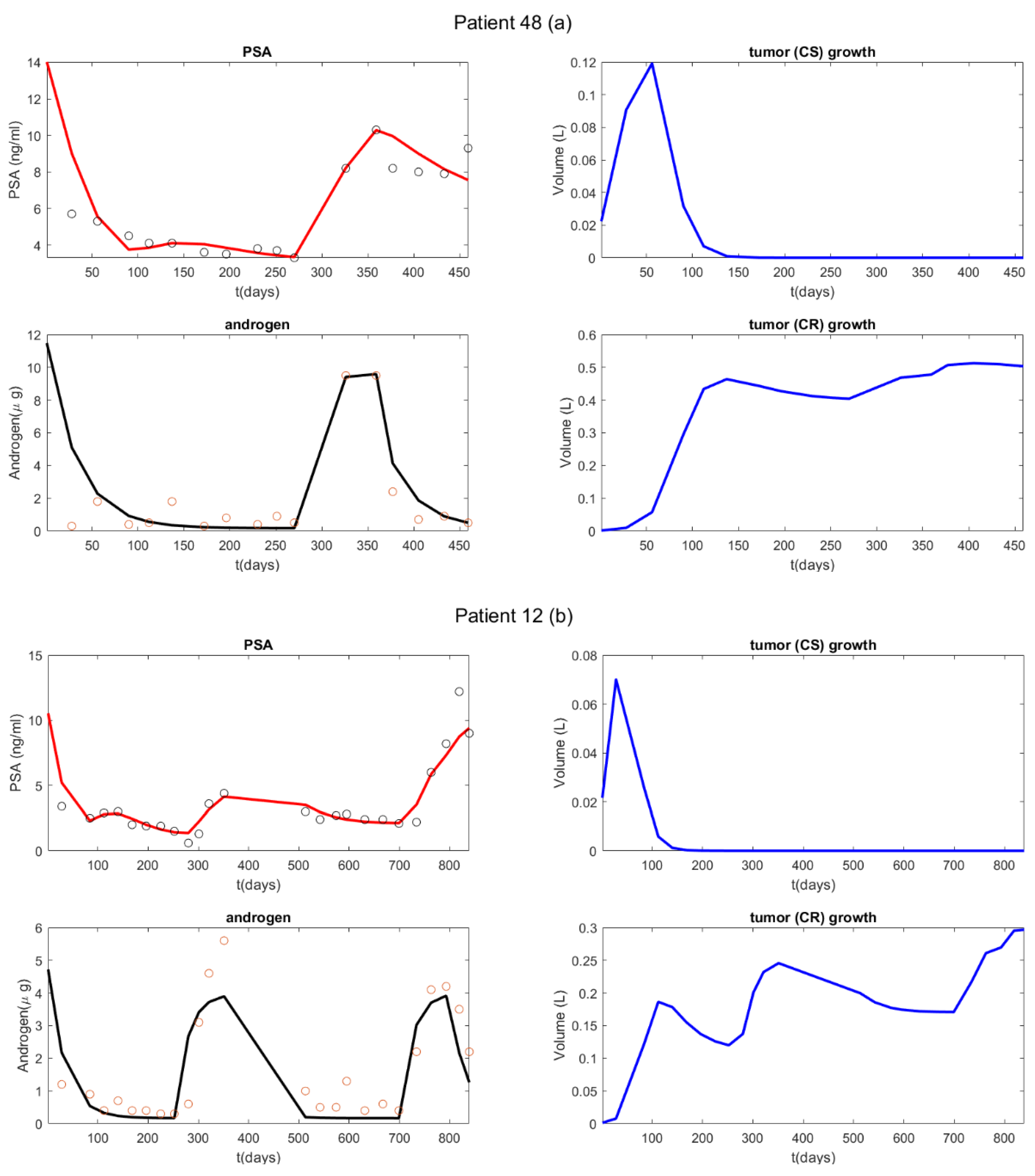{kind=link}
{kind=link}
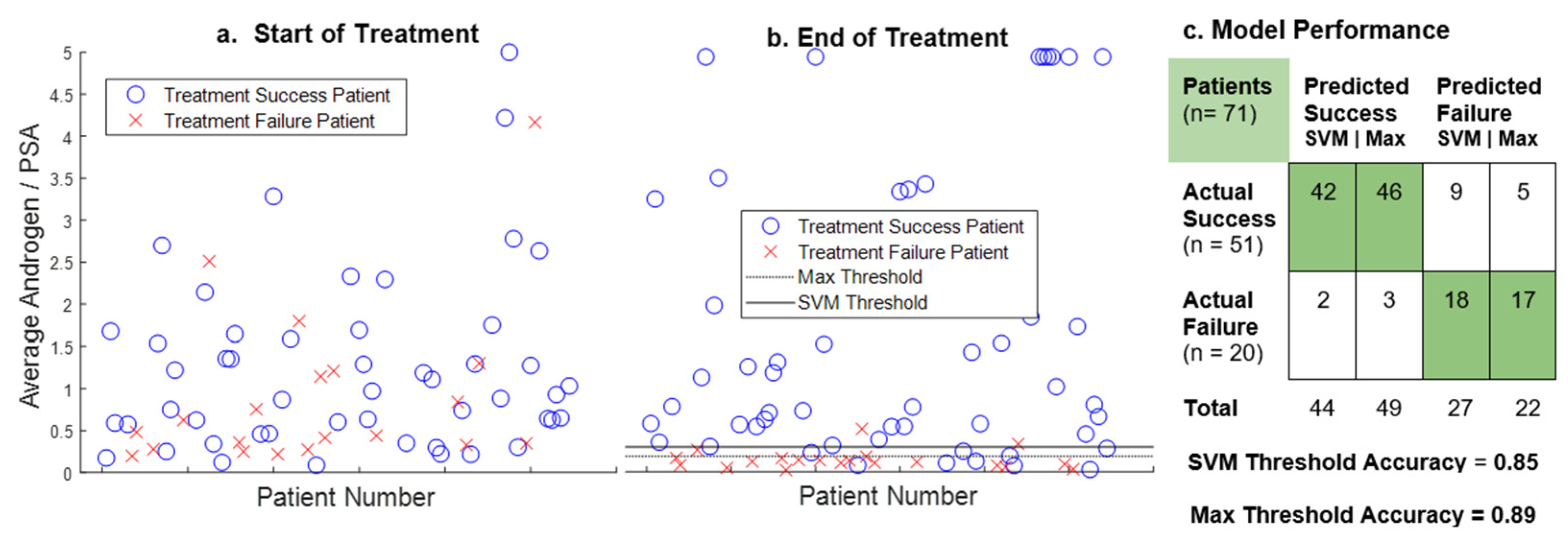{kind=link}
| Parameter | Description | Range | Unit |
|---|---|---|---|
| max proliferation Rate | 0.001–0.09 | [day]−1 | |
| q1 | to proliferate | 0.41–1.73 | [nmol][day]−1 |
| q2 | to proliferate | 0.01–0.41 | [nmole][day]−1 |
| d | density death rate | 0.001–0.30 | [L]−1[day]−1 |
| c | maximum mutation rate | 0.00015–0.00015 | [day]−1 |
| K | half-saturation constant for mutation | 1–1 | [nmole][day]−1 |
| androgen production by testes | 0.008–0.8 | [nmol][day]−1 | |
| androgen production rate by adrenal gland | 0.005–0.005 | [nmol][day]−1 | |
| homeostasis serum androgen level | * | [nmol] | |
| androgen degradation rate | 0.03–0.15 | [day]−1 | |
| b | baseline PSA production rate | 0.0001–0.1 | g][nmol]−1[day]−1 |
| 0.001–1 | g][nmol]−1[L]−1[day]−1 | ||
| maximum PSA production rate by x2 | 0.001–1 | g][nmol]−1[L]−1[day]−1 | |
| PSA clearance rate | 0.0001–0.1 | [day]−1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meade, W.; Weber, A.; Phan, T.; Hampston, E.; Resa, L.F.; Nagy, J.; Kuang, Y. High Accuracy Indicators of Androgen Suppression Therapy Failure for Prostate Cancer—A Modeling Study. Cancers 2022, 14, 4033. https://doi.org/10.3390/cancers14164033
Meade W, Weber A, Phan T, Hampston E, Resa LF, Nagy J, Kuang Y. High Accuracy Indicators of Androgen Suppression Therapy Failure for Prostate Cancer—A Modeling Study. Cancers. 2022; 14(16):4033. https://doi.org/10.3390/cancers14164033
Chicago/Turabian StyleMeade, William, Allison Weber, Tin Phan, Emily Hampston, Laura Figueroa Resa, John Nagy, and Yang Kuang. 2022. "High Accuracy Indicators of Androgen Suppression Therapy Failure for Prostate Cancer—A Modeling Study" Cancers 14, no. 16: 4033. https://doi.org/10.3390/cancers14164033
APA StyleMeade, W., Weber, A., Phan, T., Hampston, E., Resa, L. F., Nagy, J., & Kuang, Y. (2022). High Accuracy Indicators of Androgen Suppression Therapy Failure for Prostate Cancer—A Modeling Study. Cancers, 14(16), 4033. https://doi.org/10.3390/cancers14164033