
Novel Necroptosis-Related Gene Signature for Predicting Early Diagnosis and Prognosis and Immunotherapy of Gastric Cancer

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
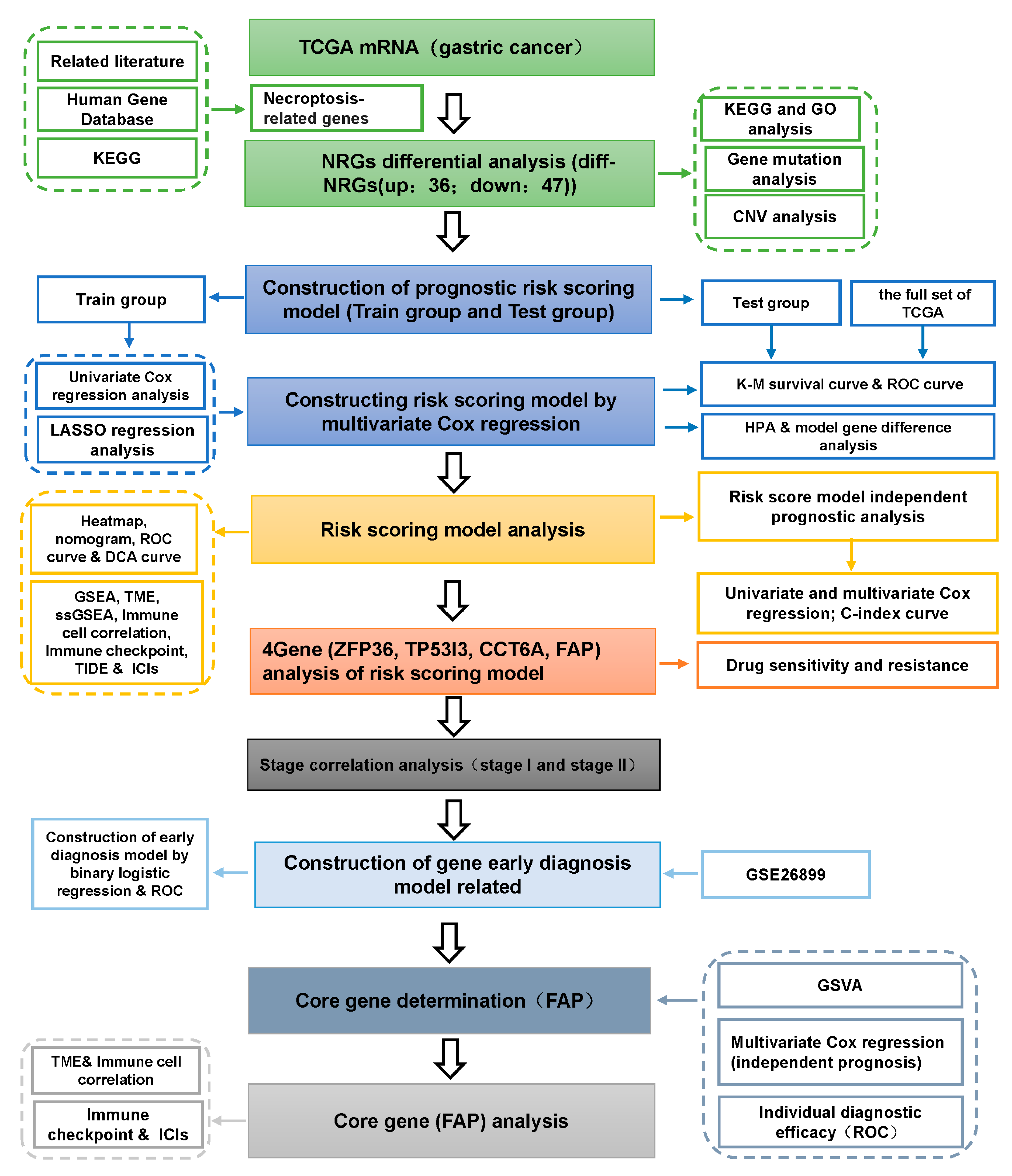
2. Materials and methods
2.1. Acquisition and Preprocessing of the Datasets
2.2. Acquisition of Human Necroptosis-Related Gene Set
2.3. Gene Annotation and NRGs Differential Analysis
2.4. Gene Mutation, CNV and Biological Function Analysis of DE-NRGs
2.5. Construction and Validation of DE-NRGs Prognostic Risk Scoring Model
2.6. Independent Prognosis of Risk Score and Correlation Analysis of Clinical Characteristics
2.7. Nomogram Construction and Analysis
2.8. GSEA Enrichment Analysis of Risk Scores
2.9. Correlation Analysis between Risk Score and TME
2.10. Correlation Analysis of Risk Score with Immune Cell Infiltration and Immune Checkpoints
2.11. Correlation Analysis of Risk Score with Immunotherapy Response and ICIs
2.12. Drug Susceptibility and Resistance Analysis of Genes in Risk Scoring Models
2.13. Correlation Analysis of Gene Expression and Pathological Stage in Risk Scoring Model and Construction of Early Diagnosis Model
2.14. Determination of Core Genes
2.15. Correlation Analysis of Core Gene Expression and TME
2.16. Correlation Analysis of Core Gene Expression and Immune Cells and Immune Cell Infiltration
2.17. Analysis of the Correlation between Core Gene Expression and Immune Checkpoints and ICIs
3. Results
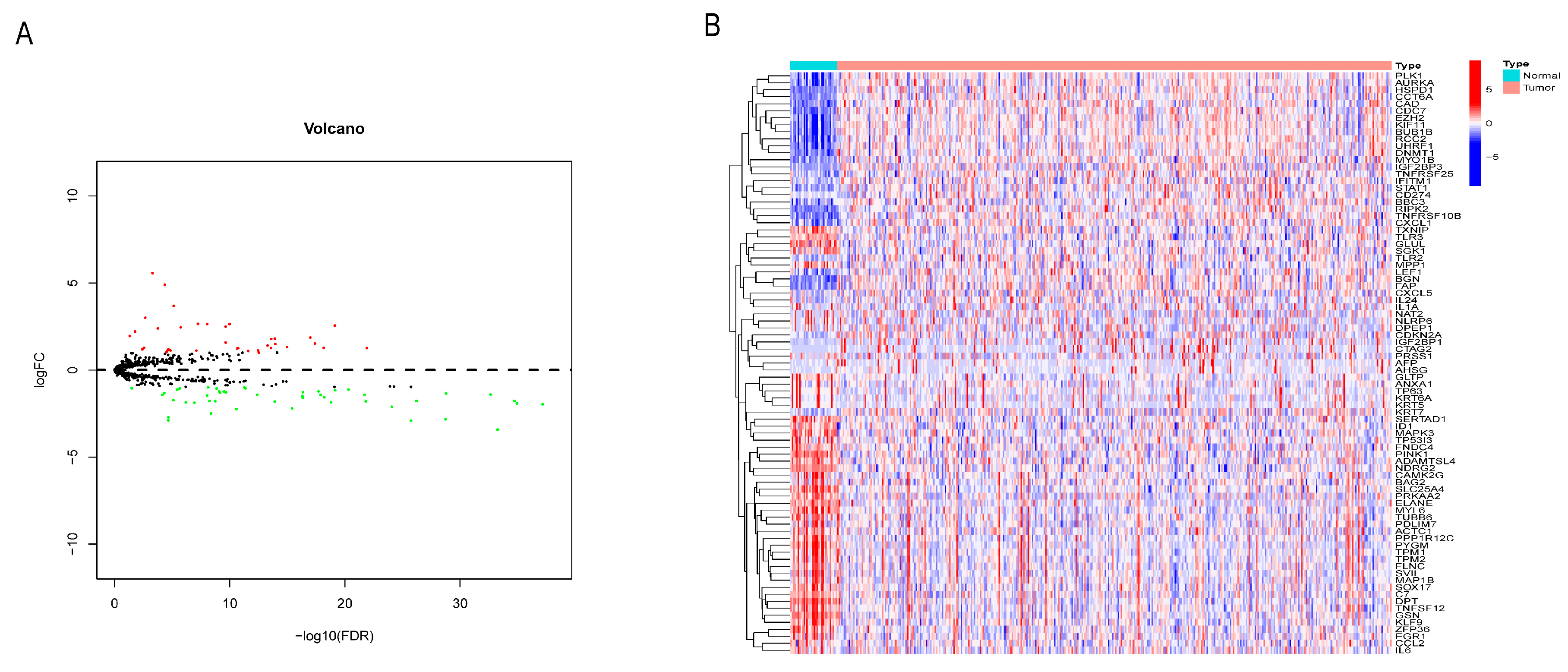
3.1. Identification of NRG Differentially Expressed in GC
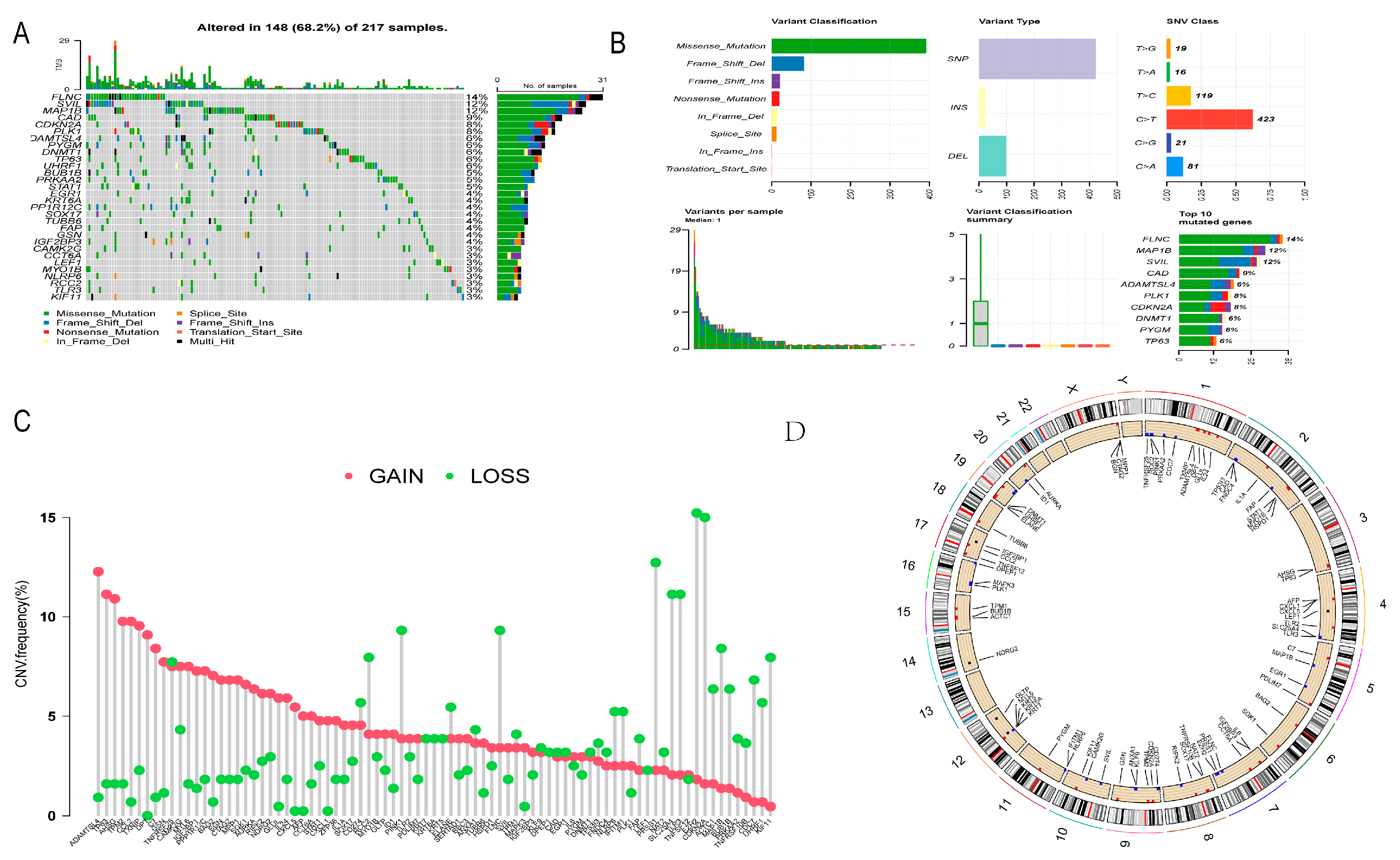
3.2. Gene Mutation and CNV Analysis of DE-NRGs
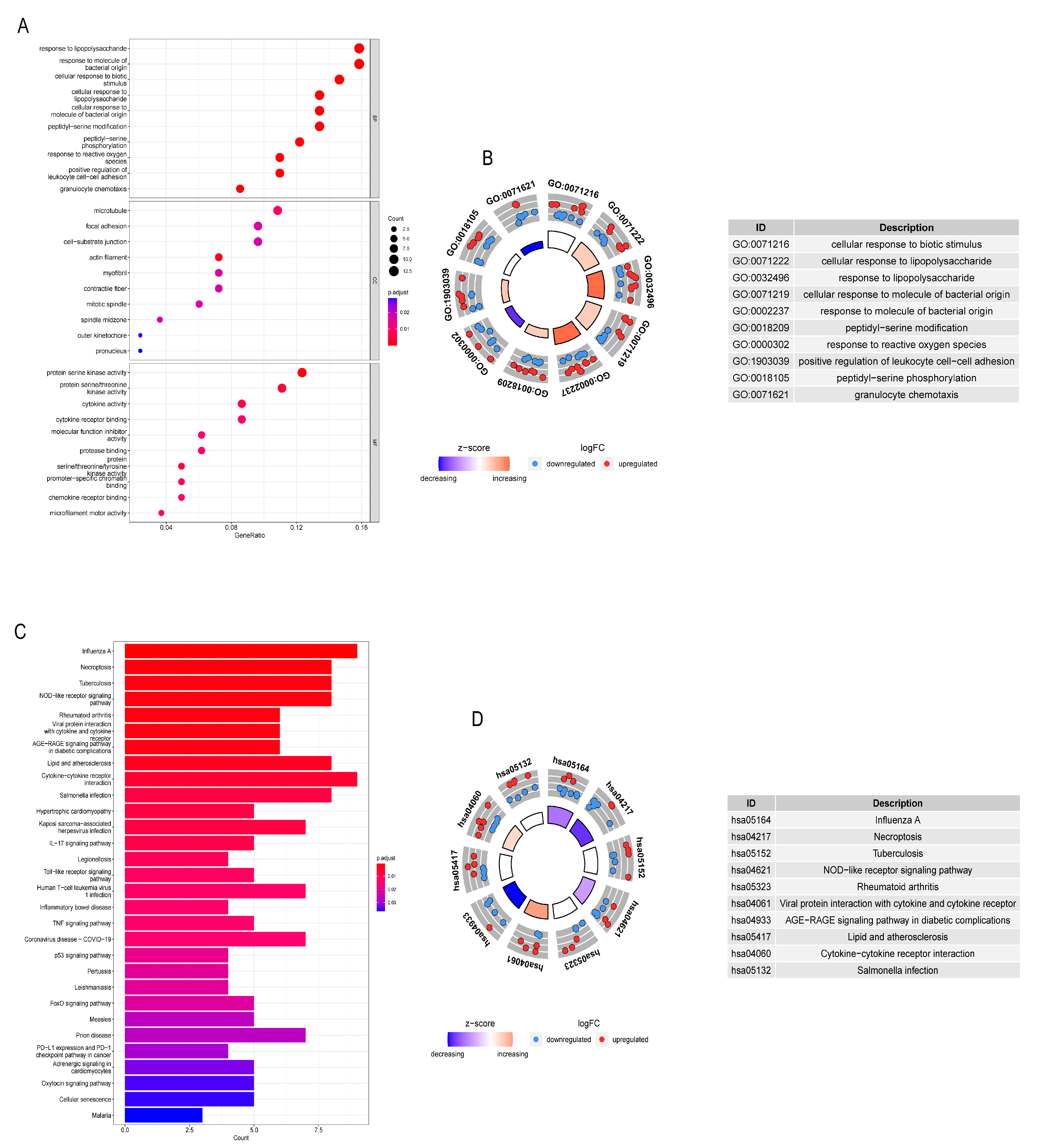
3.3. GO and KEGG Analysis of DE-NRGs
3.4. Building a Prognostic Risk Scoring Model
3.5. Validation of Risk Score
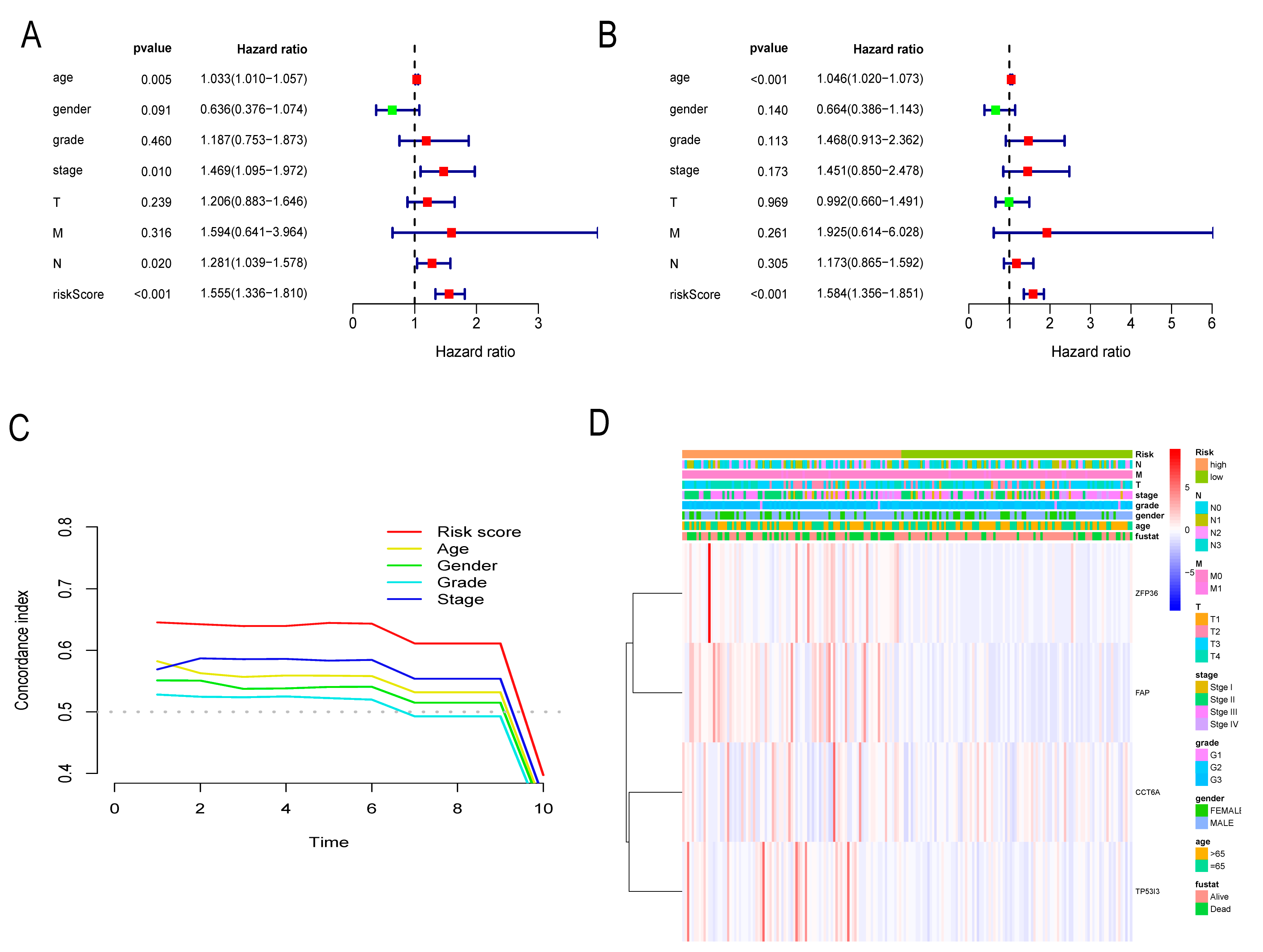
3.6. Independent Prognostic Assessment of Risk Score
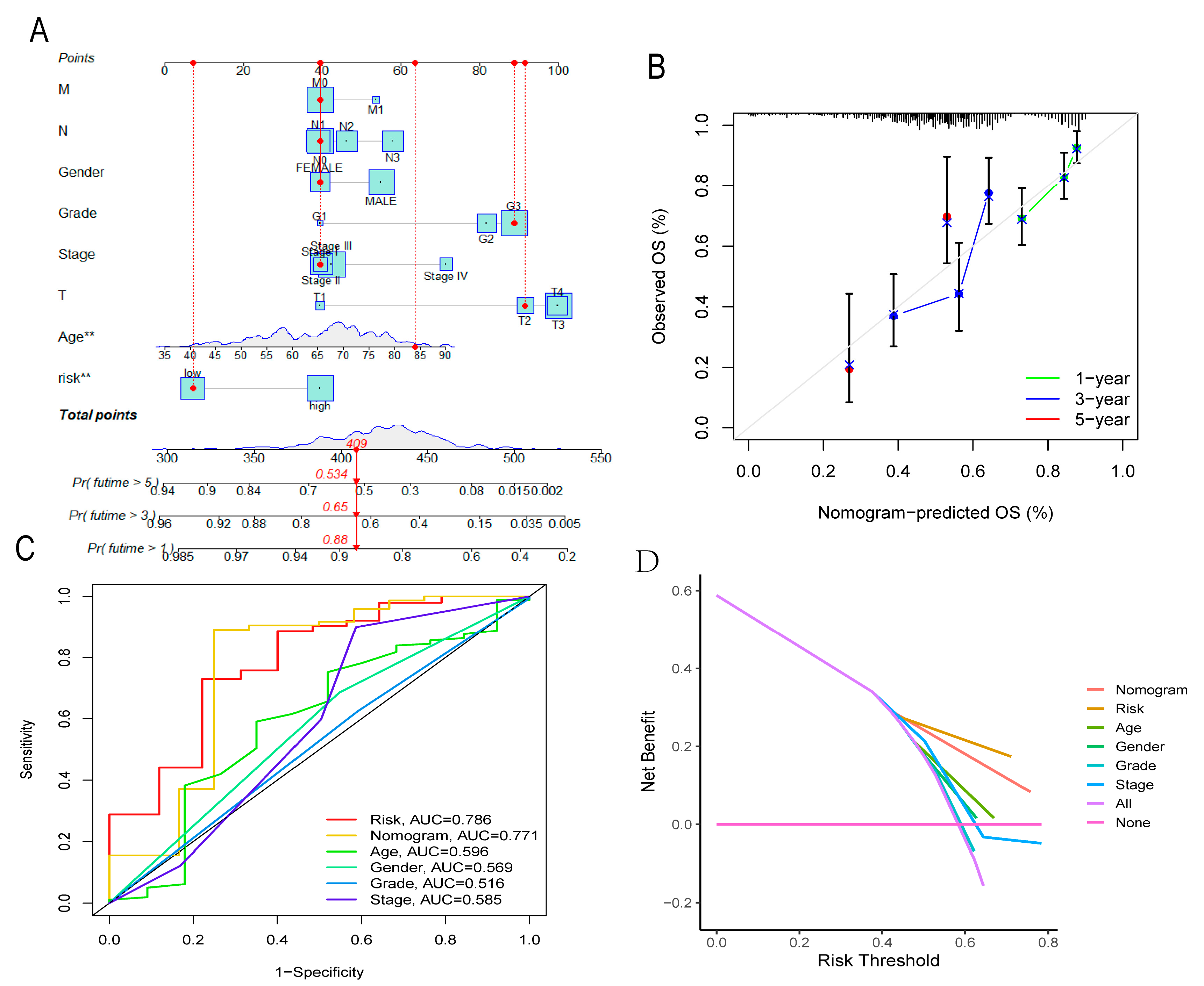
3.7. Nomogram
3.8. GSEA
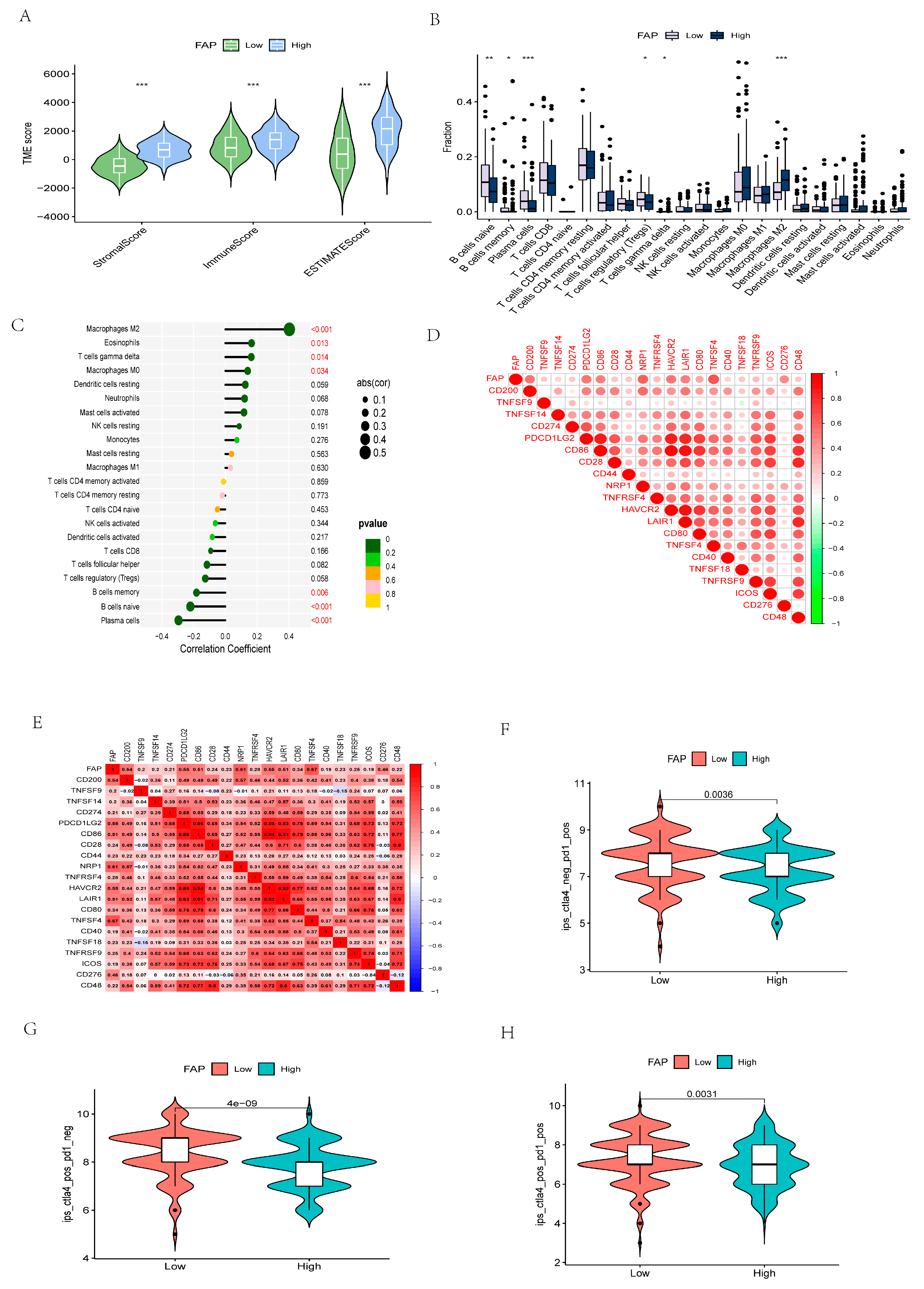
3.9. Identification of Different Risk Groups Associated with Immunity
3.10. The Importance of Risk Scores in Immunotherapy Response and ICIs
3.11. Identification of 4-Gene Drug Susceptibility and Resistance
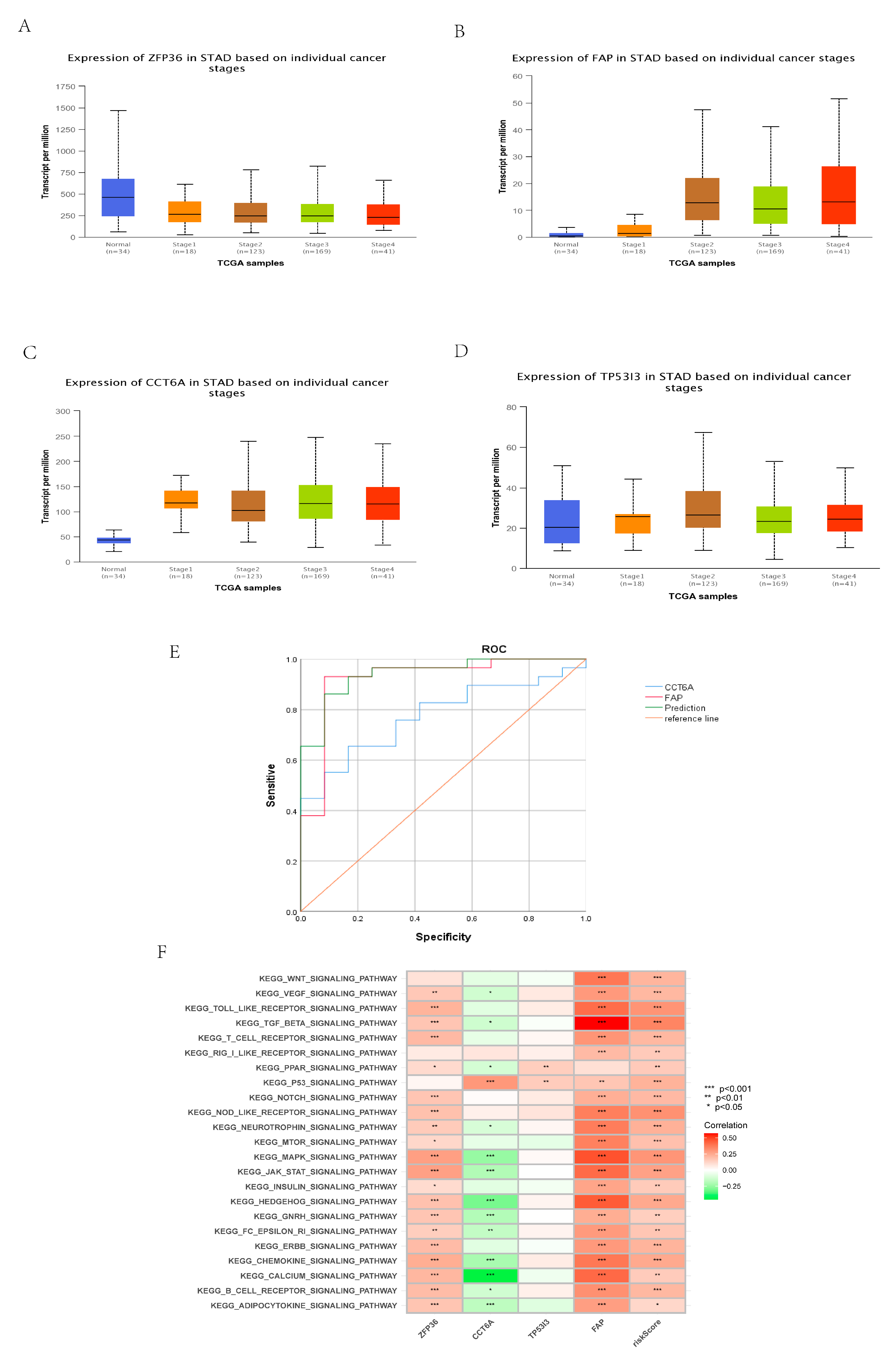
3.12. Construction and Identification of Early Diagnostic Models
3.13. Identification of Core Gene
3.14. Comprehensive Analysis of Core Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GC | gastric cancer |
| NRGs | necroptosis-related genes |
| ICIs | immune checkpoint inhibitors |
| PRRs | pattern recognition receptors |
| RIPK1 | serine-threonine kinase 1 |
| RIPK3 | serine-threonine kinase 3 |
| MLKL | mixed lineage kinase domain-like protein |
| ZBP1 | Z-DNA-binding protein 1 |
| TLR | Toll-like receptor |
| TME | immune microenvironment |
| TCGA | the Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| DE-NRG | differentially expressed necroptosis-related genes |
| GO | Gene Ontology |
| CNV | copy number variation |
| CI | confidence interval |
| LASSO | absolute minimum shrinkage and selection operator |
| OS | overall survival |
| K-M | Kaplan-Meier |
| ROC | receiver operating characteristic |
| PCA | principal component analysis |
| HPA | Human Protein Atlas |
| C-index | concordance index |
| DCA | decision curve analysis |
| ssGSEA | single-sample enrichment analysis |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| CAF | cancer associated fibroblast |
| TCIA | the Cancer Immunome Atlas |
References
- Cao, W.; Chen, H.D.; Yu, Y.W.; Li, N.; Chen, W.Q. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Coburn, N.; Cosby, R.; Klein, L.; Knight, G.; Malthaner, R.; Mamazza, J.; Mercer, C.D.; Ringash, J. Staging and surgical approaches in gastric cancer: A systematic review. Cancer Treat. Rev. 2018, 63, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.T.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Tirino, G.; Pompella, L.; Petrillo, A.; Laterza, M.M.; Pappalardo, A.; Caterino, M.; Orditura, M.; Ciardiello, F.; Galizia, G.; De Vita, F. What’s New in Gastric Cancer: The Therapeutic Implications of Molecular Classifications and Future Perspectives. Int. J. Mol. Sci. 2018, 19, 2659. [Google Scholar] [CrossRef]
- Wang, N.; Liu, D. Identification and Validation a Necroptosis-related Prognostic Signature and Associated Regulatory Axis in Stomach Adenocarcinoma. Onco Targets Ther. 2021, 14, 5373–5383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Kodali, S.; Chen, M.; Wang, J. Maintenance of Germinal Center B Cells by Caspase-9 through Promotion of Apoptosis and Inhibition of Necroptosis. J. Immunol. 2020, 205, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, H.; Zhou, X.; Fang, D.; Ou, X.; Ye, J.; Peng, J.; Xu, J. Necroptosis-Related lncRNAs: Predicting Prognosis and the Dis-tinction between the Cold and Hot Tumors in Gastric Cancer. J. Oncol. 2021, 2021, 6718443. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Ly, N.N.G.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune sup-pression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.V.; Pierotti, C.L.; Lowes, K.N.; Au, A.E.; Zhang, Y.; Etemadi, N.; Fitzgibbon, C.; Kersten, W.J.A.; Samson, A.L.; van Delft, M.F.; et al. The Lck inhibitor, AMG-47a, blocks necroptosis and implicates RIPK1 in signalling downstream of MLKL. Cell Death Dis. 2022, 13, 291. [Google Scholar] [CrossRef]
- Long, F.; Lin, H.; Zhang, X.; Zhang, J.; Xiao, H.; Wang, T. Atractylenolide-I Suppresses Tumorigenesis of Breast Cancer by Inhibiting Toll-Like Receptor 4-Mediated Nuclear Factor-kappaB Signaling Pathway. Front. Pharmacol. 2020, 11, 598939. [Google Scholar] [CrossRef]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, G.; Liang, L.; Liu, H.; Pan, J.; Cheng, H.; Wang, H.; Qu, A.; Wang, Y. RIPK3-Mediated Necroptosis and Neutrophil Infiltration Are Associated with Poor Prognosis in Patients with Alcoholic Cirrhosis. J. Immunol. Res. 2018, 2018, 1509851. [Google Scholar] [CrossRef]
- Sun, W.; Yu, W.; Shen, L.; Huang, T. MLKL is a potential prognostic marker in gastric cancer. Oncol. Lett. 2019, 18, 3830–3836. [Google Scholar] [CrossRef]
- Bolandi, N.; Derakhshani, A.; Hemmat, N.; Baghbanzadeh, A.; Asadzadeh, Z.; Nour, M.A.; Brunetti, O.; Bernardini, R.; Silvestris, N.; Baradaran, B. The Positive and Negative Immunoregulatory Role of B7 Family: Promising Novel Targets in Gas-tric Cancer Treatment. Int. J. Mol. Sci. 2021, 22, 10719. [Google Scholar] [CrossRef]
- Chang, X.; Ge, X.; Zhang, Y.; Xue, X. The current management and biomarkers of immunotherapy in advanced gastric cancer. Medicine 2022, 101, e29304. [Google Scholar] [CrossRef]
- Zeng, Y.; Zhang, X.; Li, F.; Wang, Y.; Wei, M. AFF3 is a novel prognostic biomarker and a potential target for immunotherapy in gastric cancer. J. Clin. Lab. Anal. 2022, 36, e24437. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Wen, X.; He, X.; Jiao, F.; Wang, C.; Sun, Y.; Ren, X.; Li, Q. Fibroblast Activation Protein-alpha-Positive Fibroblasts Promote Gas-tric Cancer Progression and Resistance to Immune Checkpoint Blockade. Oncol. Res. 2017, 25, 629–640. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Chan, Y.T.; Zhang, C.; Guo, W.; Chen, F.; Zhong, Z.; Li, S.; Feng, Y. ID1 overexpression increases gefitinib sensitivity in non-small cell lung cancer by activating RIP3/MLKL-dependent necroptosis. Cancer Lett. 2020, 475, 109–118. [Google Scholar] [CrossRef]
- Tang, X.; Li, Y.; Liu, L.; Guo, R.; Zhang, P.; Zhang, Y.; Zhang, Y.; Zhao, J.; Su, J.; Sun, L.; et al. Sirtuin 3 induces apoptosis and necroptosis by regulating mutant p53 expression in small-cell lung cancer. Oncol. Rep. 2020, 43, 591–600. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR con-trols RIPK3-dependent necroptosis in intestinal inflammation and cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]
- Yang, D.; Liang, Y.; Zhao, S.; Ding, Y.; Zhuang, Q.; Shi, Q.; Ai, T.; Wu, S.Q.; Han, J. ZBP1 mediates interferon-induced necroptosis. Cell. Mol. Immunol. 2020, 17, 356–368. [Google Scholar] [CrossRef]
- Yang, W.; Liu, S.; Li, Y.; Wang, Y.; Deng, Y.; Sun, W.; Huang, H.; Xie, J.; He, A.; Chen, H.; et al. Pyridoxine induces mono-cyte-macrophages death as specific treatment of acute myeloid leukemia. Cancer Lett. 2020, 492, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.G.; Wang, Y.; Zhu, R.J.; Tang, K.; Tang, X.B.; Su, X.M. EMS1/DLL4-Notch Signaling Axis Augments Cell Cy-cle-Mediated Tumorigenesis and Progress in Human Adrenocortical Carcinoma. Front. Oncol. 2021, 11, 771579. [Google Scholar] [CrossRef]
- Li, T.; Wang, H.; Xu, J.; Li, C.; Zhang, Y.; Wang, G.; Liu, Y.; Cai, S.; Fang, W.; Li, J.; et al. TGFBR2 mutation predicts resistance to immune checkpoint inhibitors in patients with non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 17588359211038477. [Google Scholar] [CrossRef]
- Li, K.; Zhang, A.; Li, X.; Zhang, H.; Zhao, L. Advances in clinical immunotherapy for gastric cancer. Biochim. Biophys. Acta 2021, 1876, 188615. [Google Scholar] [CrossRef]
- Malapelle, U.; Parente, P.; Pepe, F.; De Luca, C.; Pisapia, P.; Sgariglia, R.; Nacchio, M.; Gragnano, G.; Russo, G.; Conticelli, F.; et al. Evaluation of Micro Satellite Instability and Mismatch Repair Status in Different Solid Tumors: A Multicenter Analysis in a Real World Setting. Cells 2021, 10, 1878. [Google Scholar] [CrossRef]
- Kang, Y.-K.; Reck, M.; Nghiem, P.; Feng, Y.; Plautz, G.; Kim, H.R.; Owonikoko, T.K.; Boku, N.; Chen, L.-T.; Lei, M.; et al. Assessment of hyperprogression versus the natural course of disease development with nivolumab with or without ipilimumab versus placebo in phase III, randomized, controlled trials. J. Immunother. Cancer 2022, 10, e004273. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.W.; Chau, I. Current status and future potential of predictive biomarkers for immune checkpoint inhibitors in gastric cancer. ESMO Open 2020, 5, e000791. [Google Scholar] [CrossRef]
- Zheng, S.; Wang, J.; Ding, N.; Chen, W.; Chen, H.; Xue, M.; Chen, F.; Ni, J.; Wang, Z.; Lin, Z.; et al. Prodrug polymeric micelles integrating cancer-associated fibroblasts deactivation and synergistic chemotherapy for gastric cancer. J. Nanobiotechnol. 2021, 19, 381. [Google Scholar] [CrossRef]
- Liu, J.; Huang, C.; Peng, C.; Xu, F.; Li, Y.; Yutaka, Y.; Xiong, B.; Yang, X. Stromal fibroblast activation protein alpha promotes gastric cancer progression via epithelial-mesenchymal transition through Wnt/ beta-catenin pathway. BMC Cancer 2018, 18, 1099. [Google Scholar] [CrossRef]
- Gao, L.-M.; Wang, F.; Zheng, Y.; Fu, Z.-Z.; Zheng, L.; Chen, L.-L. Roles of Fibroblast Activation Protein and Hepatocyte Growth Factor Expressions in Angiogenesis and Metastasis of Gastric Cancer. Pathol. Oncol. Res. 2019, 25, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cheng, X.; Rong, R.; Gao, Y.; Tang, X.; Chen, Y. High expression of fibroblast activation protein (FAP) predicts poor outcome in high-grade serous ovarian cancer. BMC Cancer 2020, 20, 1032. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Ruiz, P.; Corvigno, S.; Grootenhuis, N.C.T.; la Fleur, L.; Backman, M.; Strell, C.; Mezheyeuski, A.; Hoelzlwimmer, G.; Klein, C.; Botling, J.; et al. Stromal FAP is an independent poor prognosis marker in non-small cell lung ade-nocarcinoma and associated with p53 mutation. Lung Cancer. 2021, 155, 10–19. [Google Scholar] [CrossRef]
- Qi, J.; Sun, H.; Zhang, Y.; Wang, Z.; Xun, Z.; Li, Z.; Ding, X.; Bao, R.; Hong, L.; Jia, W.; et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat. Commun. 2022, 13, 1742. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TCGA | GEO | ||
|---|---|---|---|
| Variables | Number of Samples | Variables | Number of Samples |
| Gender | Gender | ||
| Male | 159 | Male | 75 |
| Female | 290 | Female | 21 |
| Unknown | 12 | ||
| Age at diagnosis | Age at diagnosis | ||
| ≤65 | 198 | ≤65 | 63 |
| >65 | 242 | >65 | 33 |
| Unknown | 9 | Unknown | 12 |
| Grade | Grade | ||
| G1 | 12 | G1 | Unknown |
| G2 | 159 | G2 | Unknown |
| G3 | 263 | G3 | Unknown |
| G4 | 9 | G4 | Unknown |
| Unknown | 6 | NA | Unknown |
| Stage | Stage | ||
| I | 59 | I | 11 |
| II | 131 | II | 18 |
| III | 184 | III | 27 |
| IV | 44 | IV | 36 |
| Unknown | 31 | NA | 16 |
| T | T | ||
| T1 | 23 | T1 | Unknown |
| T2 | 93 | T2 | Unknown |
| T3 | 200 | T3 | Unknown |
| T4 | 119 | T4 | Unknown |
| Unknown | 14 | NA | Unknown |
| M | M | ||
| M0 | 393 | M0 | Unknown |
| M1 | 30 | M1 | Unknown |
| Unknown | 26 | NA | Unknown |
| N | N | ||
| N0 | 133 | N0 | Unknown |
| N1 | 120 | N1 | Unknown |
| N2 | 85 | N2 | Unknown |
| N3 | 88 | N3 | Unknown |
| Unknown | 23 | NA | Unknown |
| fustat | fustat | ||
| Alive | 277 | Alive | Unknown |
| Dead | 172 | Dead | Unknown |
| Gene | Antibody Number | Patient Id | Type | Staining | Intensity | Quantity |
|---|---|---|---|---|---|---|
| ZFP36 | HPA006009 | 2130 | Normal tissue | Medium | Moderate | >75% |
| ZFP36 | HPA006009 | 2195 | Adenocarcinoma | High | Strong | >75% |
| TP53I3 | HPA022012 | 2583 | Normal tissue | Medium | Strong | <25% |
| TP53I3 | HPA022012 | 3270 | Adenocarcinoma | High | Strong | >75% |
| FAP | HPA059739 | 1924 | Normal tissue | High | Strong | 75–25% |
| FAP | HPA059739 | 2378 | Adenocarcinoma | High | Strong | >75% |
| CCT6A | HPA042996 | 3379 | Normal tissue | High | Strong | >75% |
| CCT6A | HPA042996 | 2105 | Adenocarcinoma | High | Strong | >75% |
| GC | Normal | All | Accuracy | |
|---|---|---|---|---|
| 2-mRNA positive | 27 | 2 | 29 | 93.1 |
| 2-mRNA negative | 2 | 10 | 12 | 83.3 |
| All accuracy | 27 | 10 | 41 | 90.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Zhang, B.; Zheng, G.; Zhang, Z.; Wu, J.; Du, K.; Zhang, J. Novel Necroptosis-Related Gene Signature for Predicting Early Diagnosis and Prognosis and Immunotherapy of Gastric Cancer. Cancers 2022, 14, 3891. https://doi.org/10.3390/cancers14163891
Zhou X, Zhang B, Zheng G, Zhang Z, Wu J, Du K, Zhang J. Novel Necroptosis-Related Gene Signature for Predicting Early Diagnosis and Prognosis and Immunotherapy of Gastric Cancer. Cancers. 2022; 14(16):3891. https://doi.org/10.3390/cancers14163891
Chicago/Turabian StyleZhou, Xiaozhu, Baizhuo Zhang, Guoliang Zheng, Zhen Zhang, Jiaoqi Wu, Ke Du, and Jing Zhang. 2022. "Novel Necroptosis-Related Gene Signature for Predicting Early Diagnosis and Prognosis and Immunotherapy of Gastric Cancer" Cancers 14, no. 16: 3891. https://doi.org/10.3390/cancers14163891
APA StyleZhou, X., Zhang, B., Zheng, G., Zhang, Z., Wu, J., Du, K., & Zhang, J. (2022). Novel Necroptosis-Related Gene Signature for Predicting Early Diagnosis and Prognosis and Immunotherapy of Gastric Cancer. Cancers, 14(16), 3891. https://doi.org/10.3390/cancers14163891