
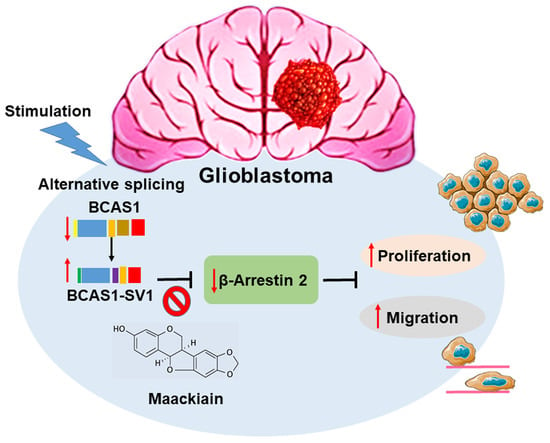
A Novel Splice Variant of BCAS1 Inhibits β-Arrestin 2 to Promote the Proliferation and Migration of Glioblastoma Cells, and This Effect Was Blocked by Maackiain

 , and
, and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents, Cell Lines, and Primary Cells
2.2. Total RNA Extraction and Reverse Transcriptase-Polymerase Chain Reaction
2.3. Western Bolt Analysis
2.4. Recombinant Plasmid Construction and Transfection
2.5. Cell Proliferation Assay
2.6. Cell Migration Assay
2.7. Cell Invasion Assay
2.8. Transient Transfection of Small RNA Interference
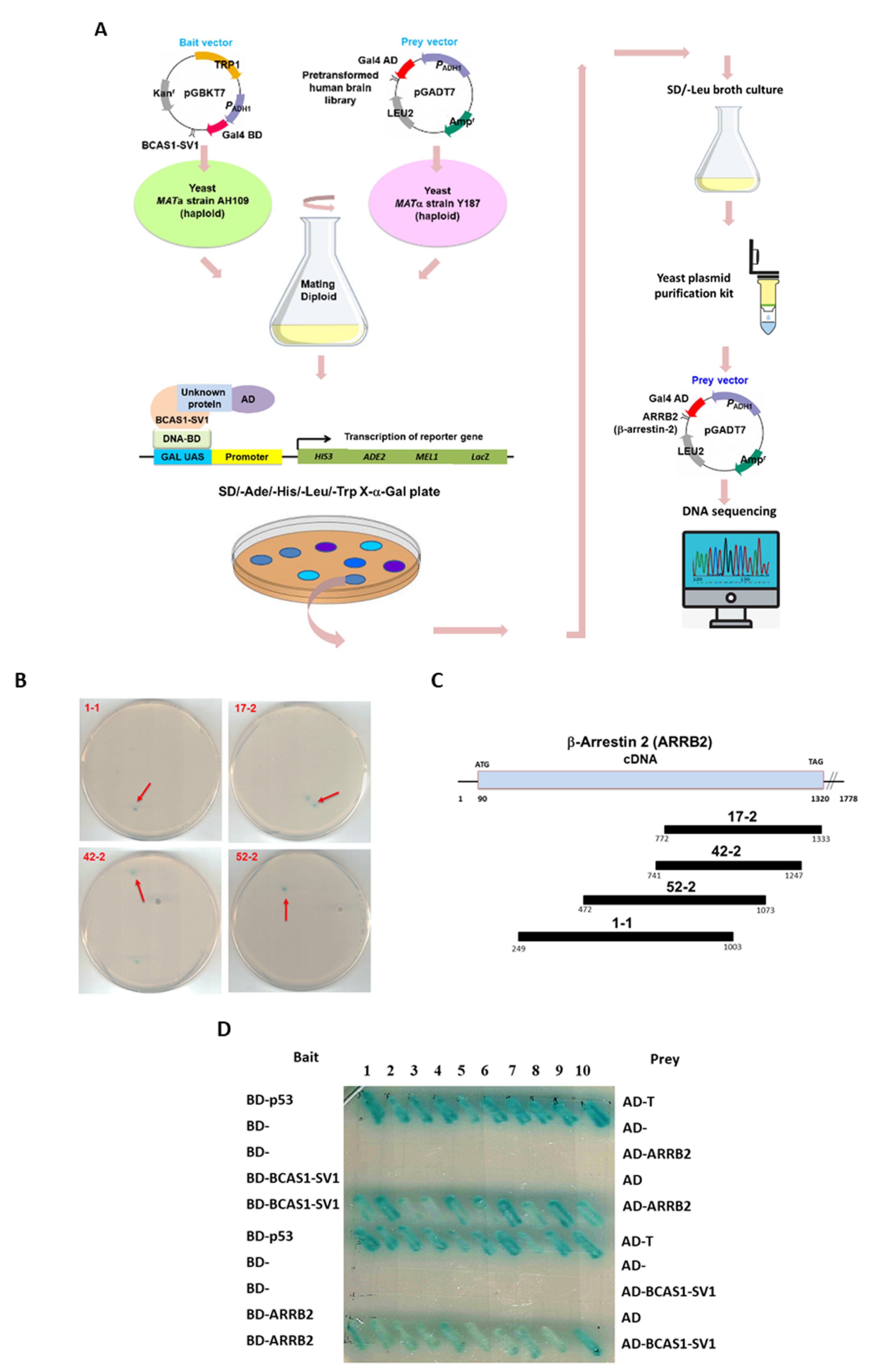
2.9. Yeast Two-Hybrid Library Screening
2.10. Yeast Two-Hybrid Assay
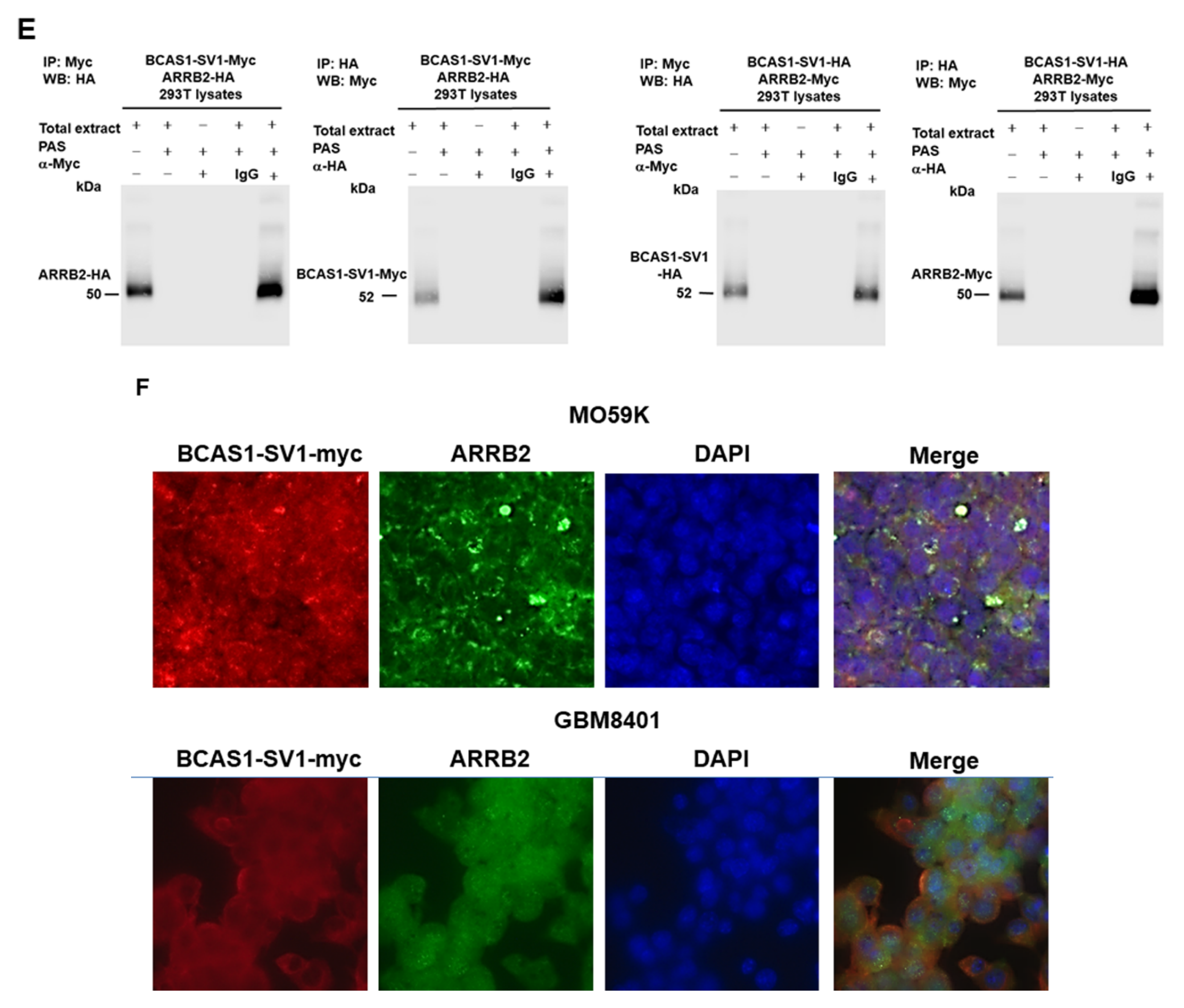
2.11. Co-Immunoprecipitation Analysis
2.12. Immunofluorescence Analysis
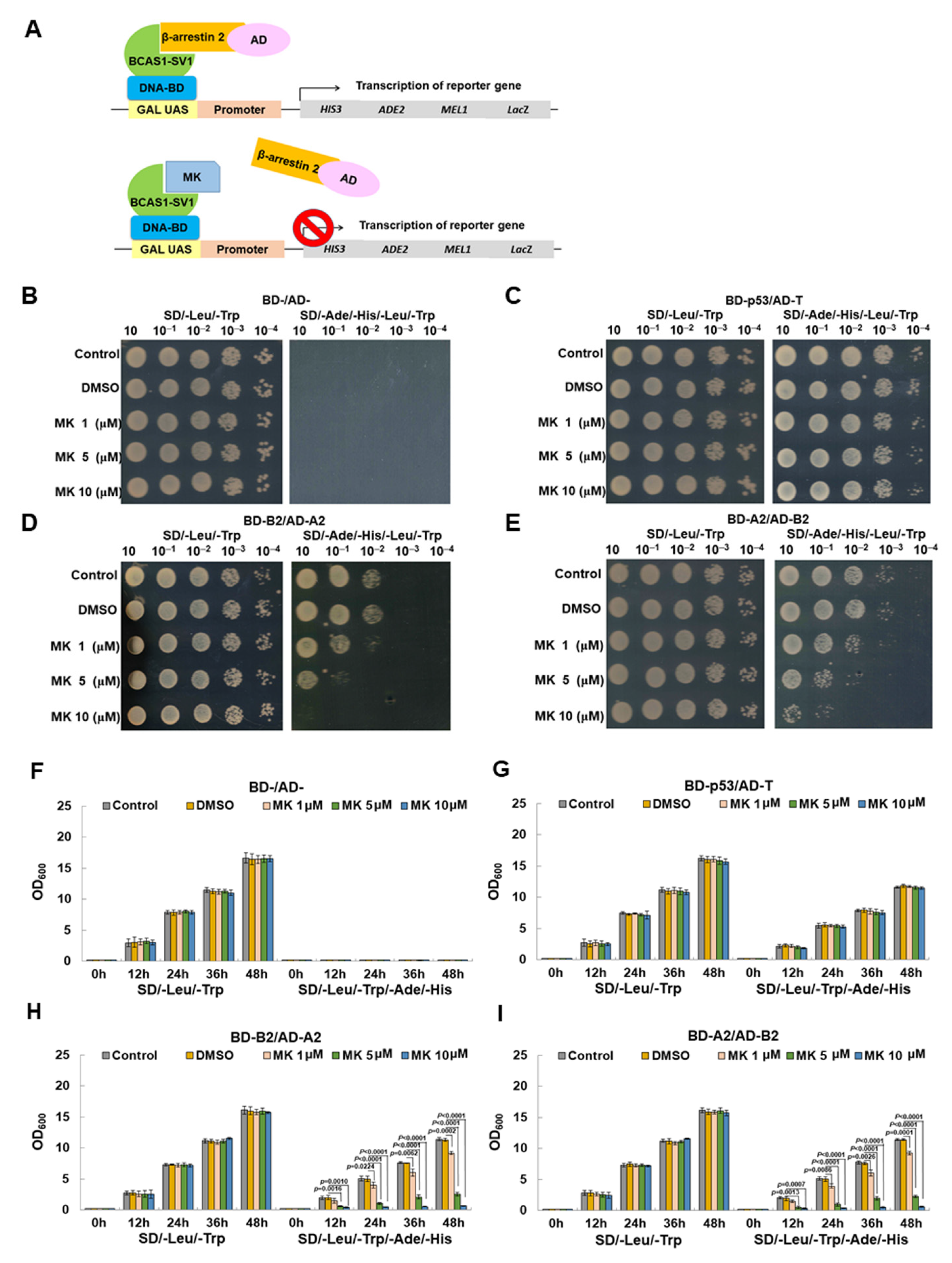
2.13. Maackiain Treatment, Yeast Two-Hybrid-Based Spotting Assay, and Optical Density Measurement
2.14. Cytotoxicity Analysis of Maackiain in GBM Cells
2.15. Tumor Xenograft Mouse Model
2.16. Intracranial Implantation of U87-Luc GBM Cells in Mice
2.17. In Vivo Imaging of Intracranial Tumors
2.18. Statistical Analysis
3. Results
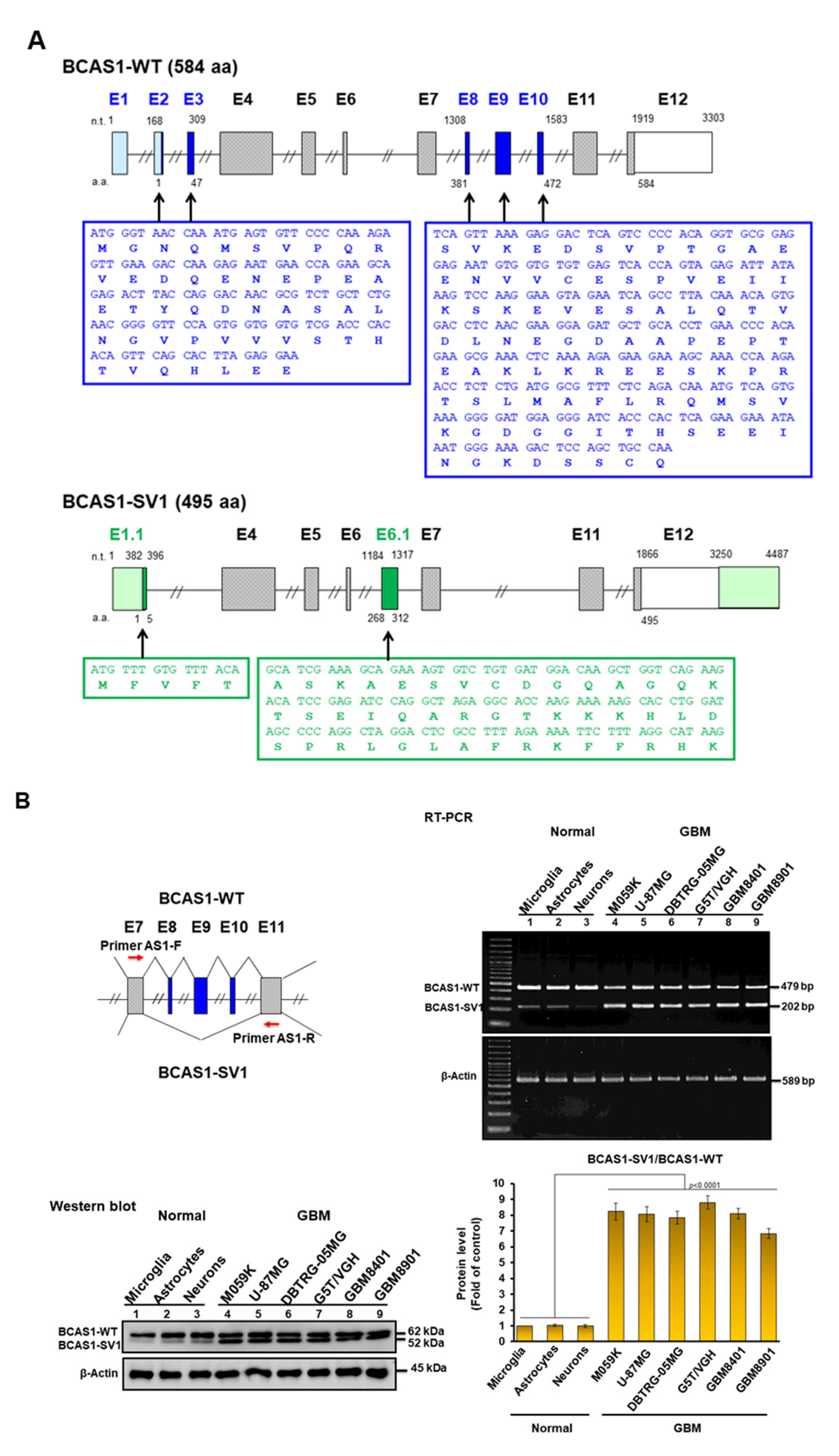
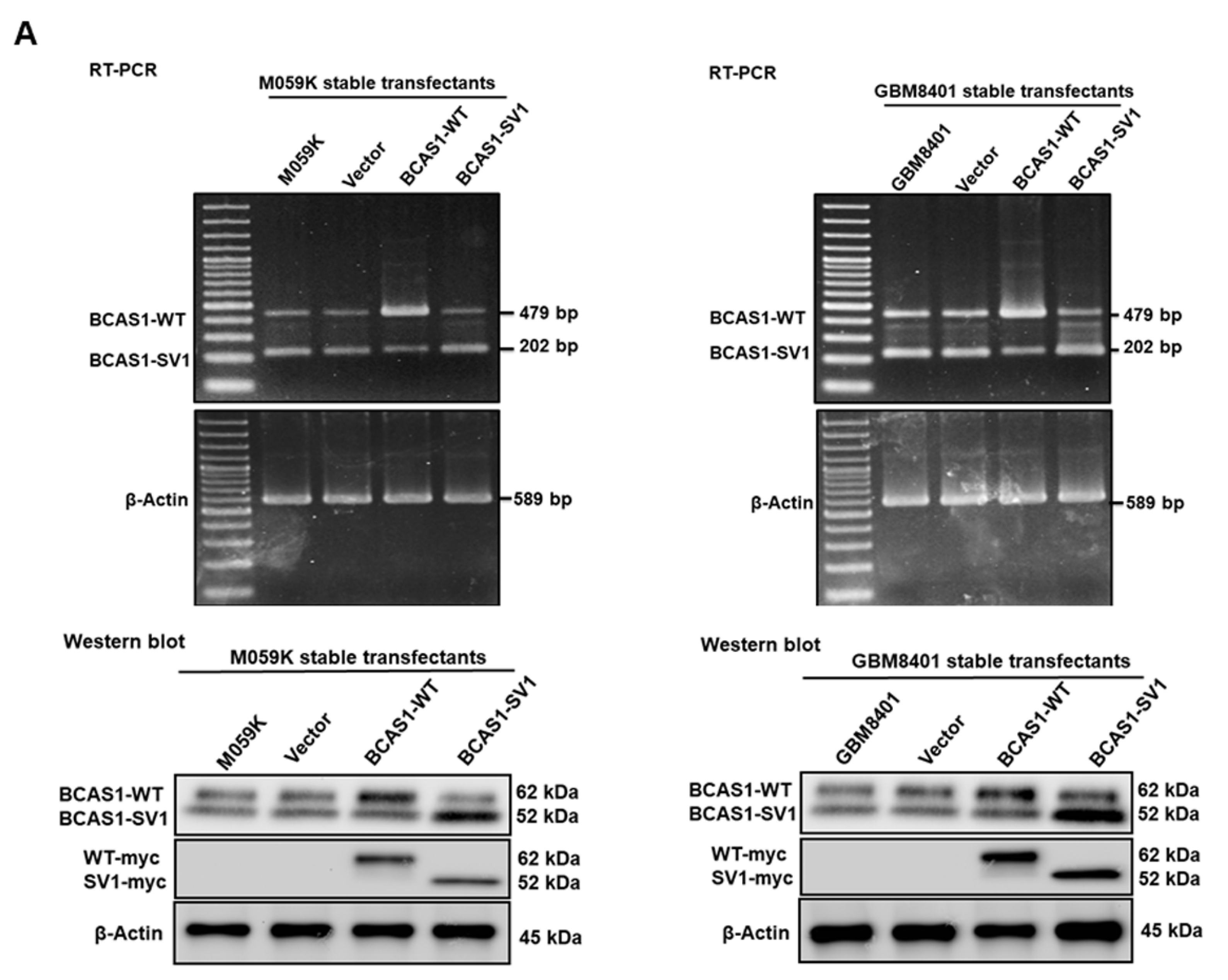
3.1. Identification of a Novel BCAS1 Alternative Splicing Variant
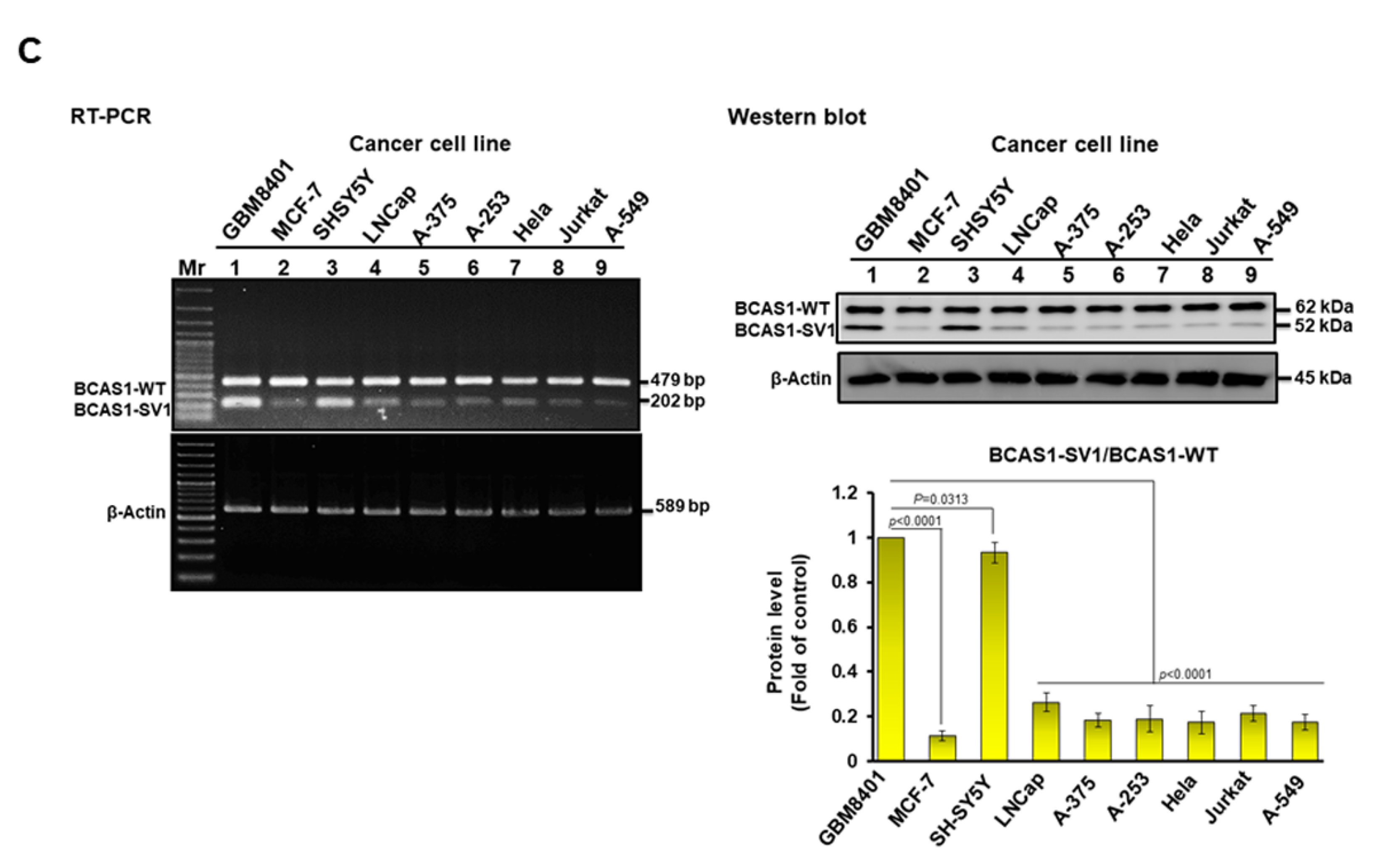
3.2. BCAS1-SV1 Is Highly Expressed in GBM but Not in Normal Brain Cells
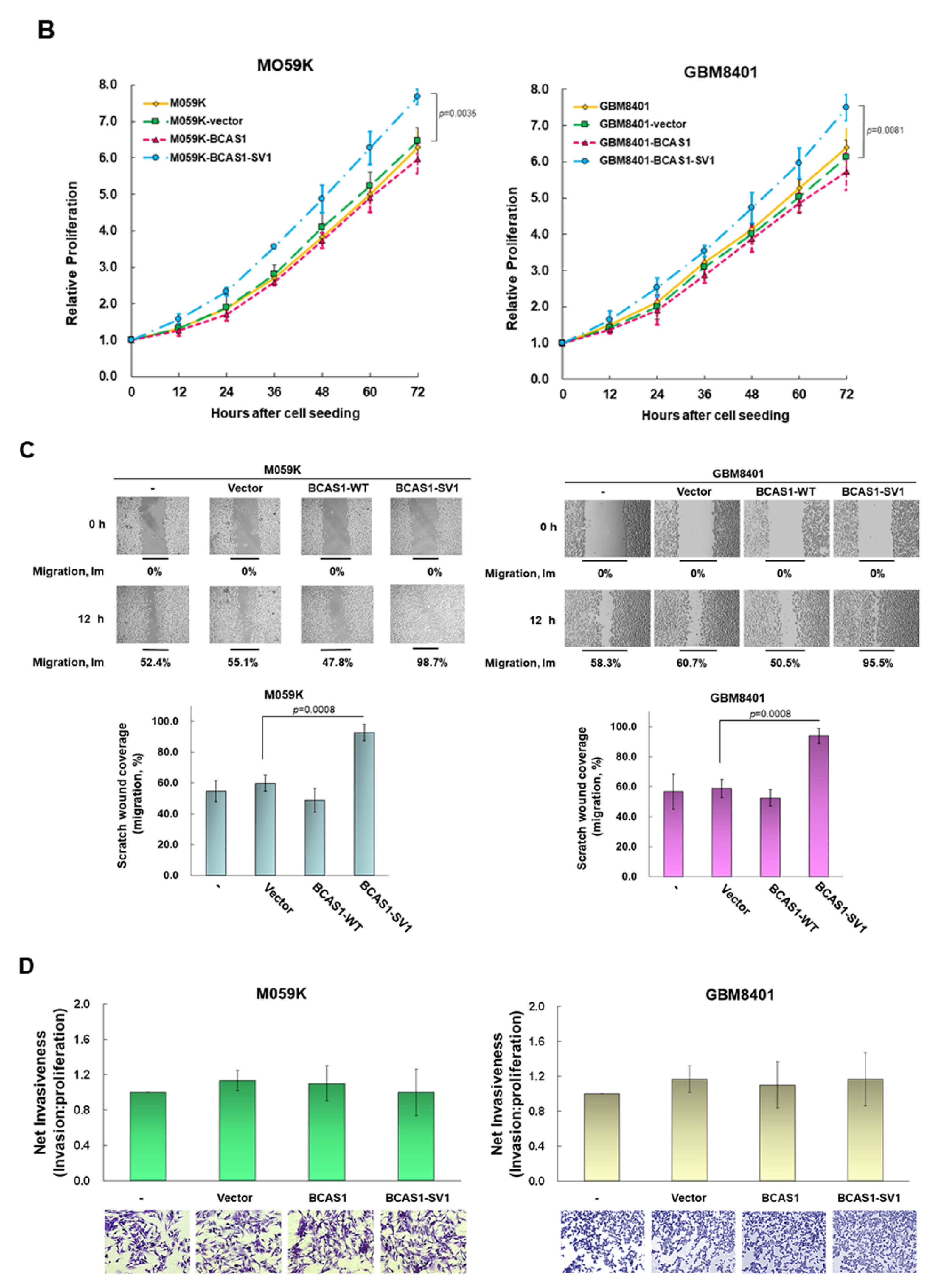
3.3. BCAS1-SV1 Has a Greater Capability to Promote the Proliferation and Migration of GBM Cells Than BCAS1
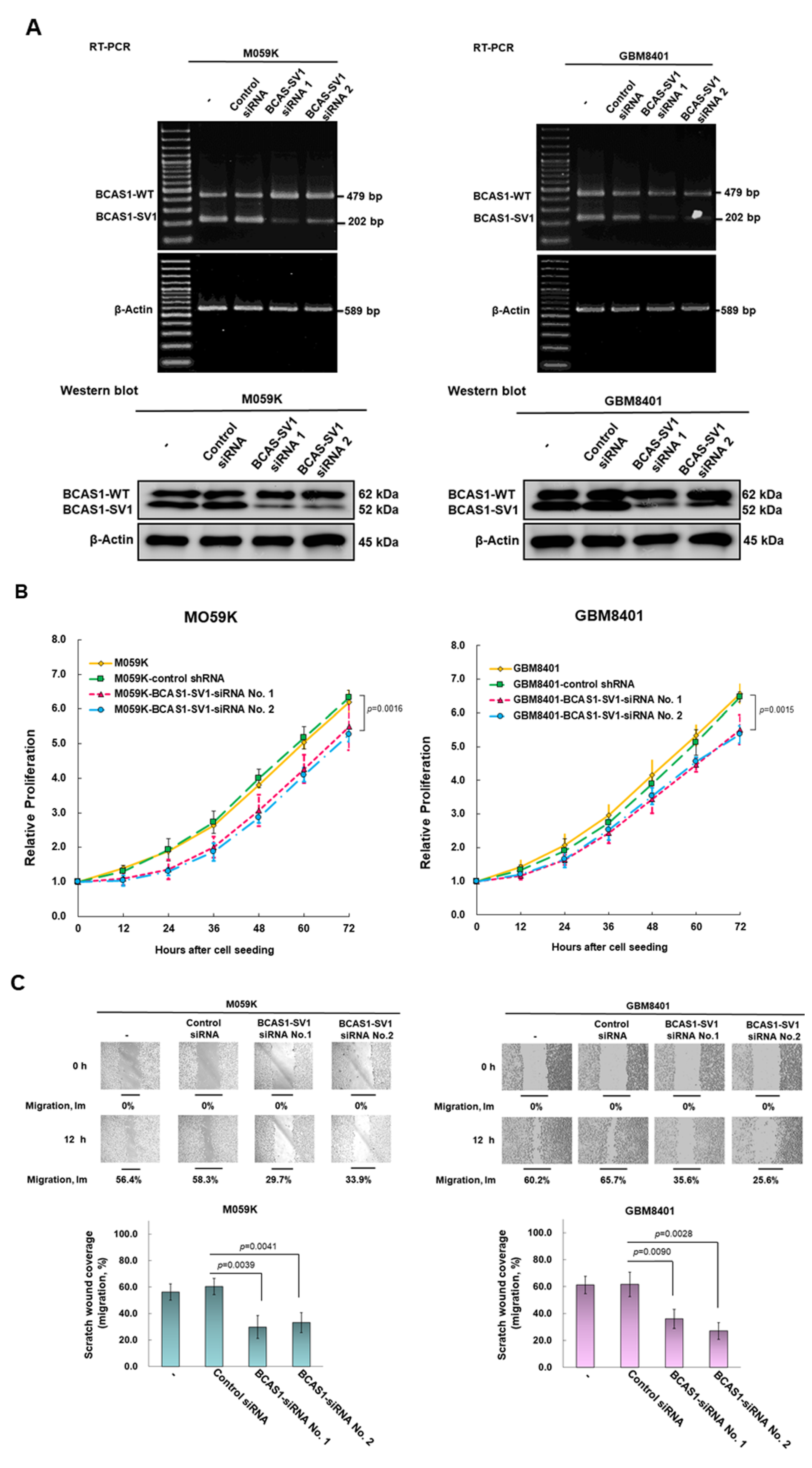
3.4. Blocking of Expression of BCAS1-SV1 Inhibits the Proliferation and Migration of GBM Cells
3.5. β-Arrestin 2 Is a Specific Interaction Partner of BCAS1-SV1
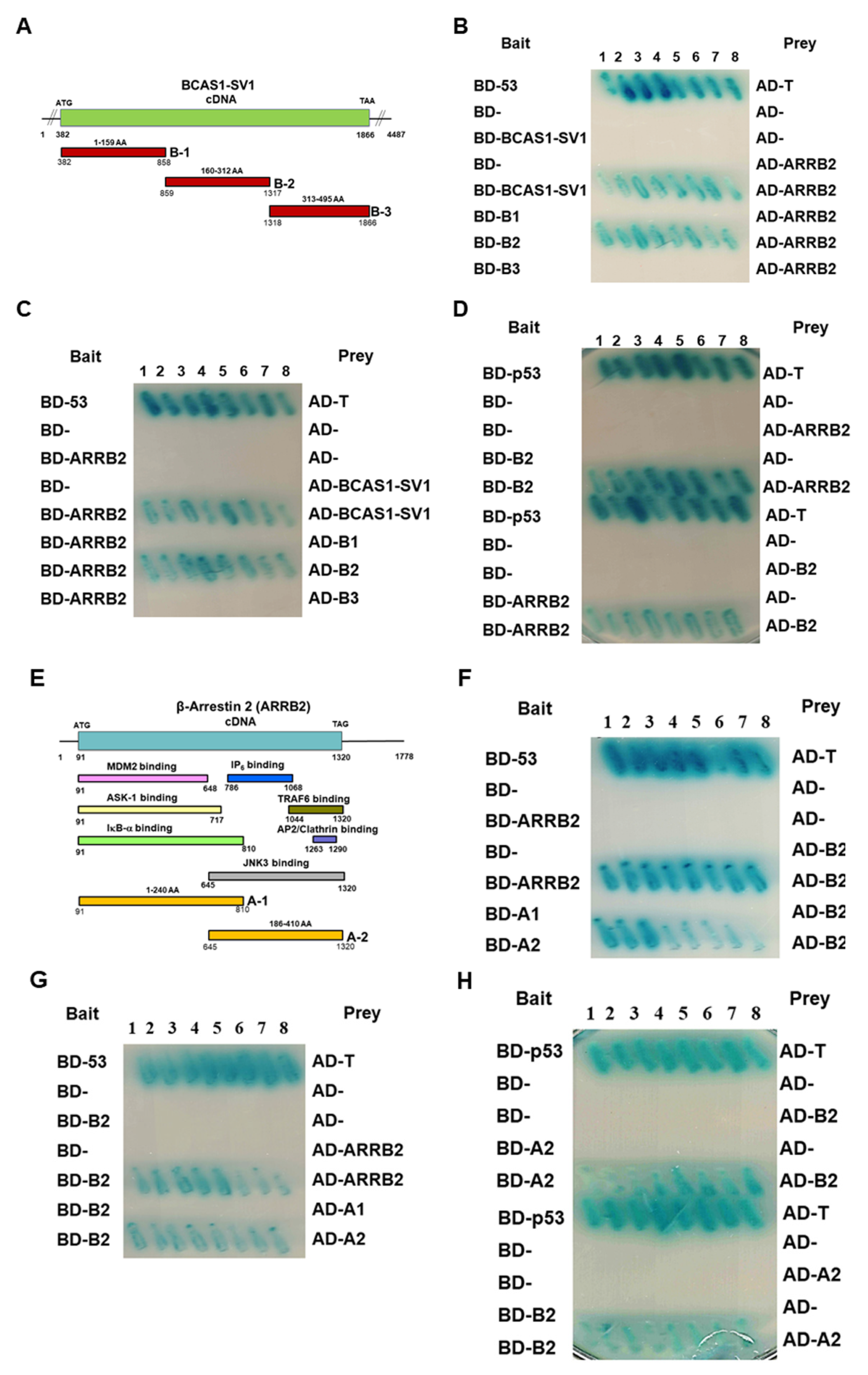
3.6. BCAS1-SV1 Interacts with the C-terminus of β-Arrestin through an Intermediate Region Containing Exon 6.1
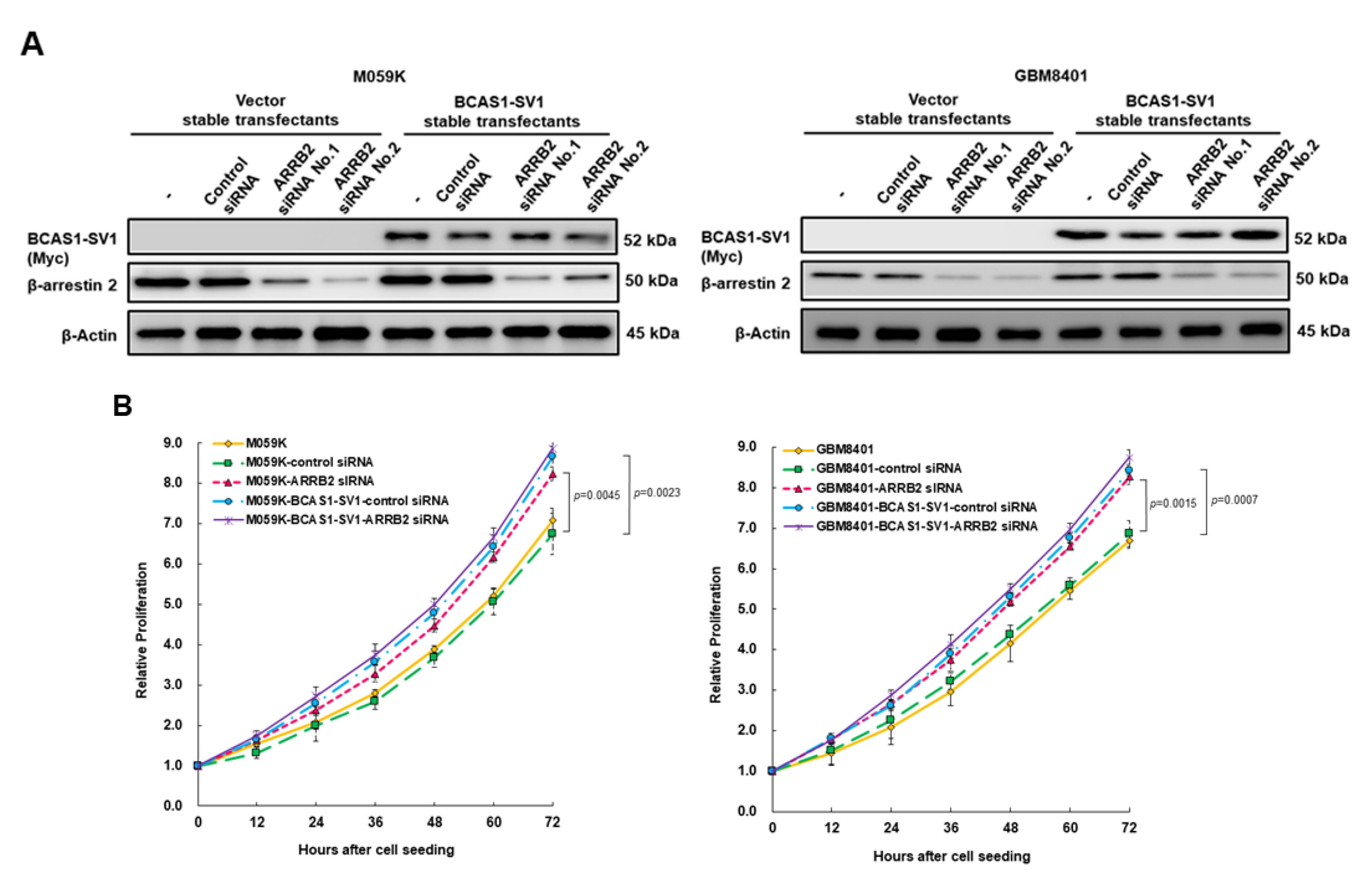
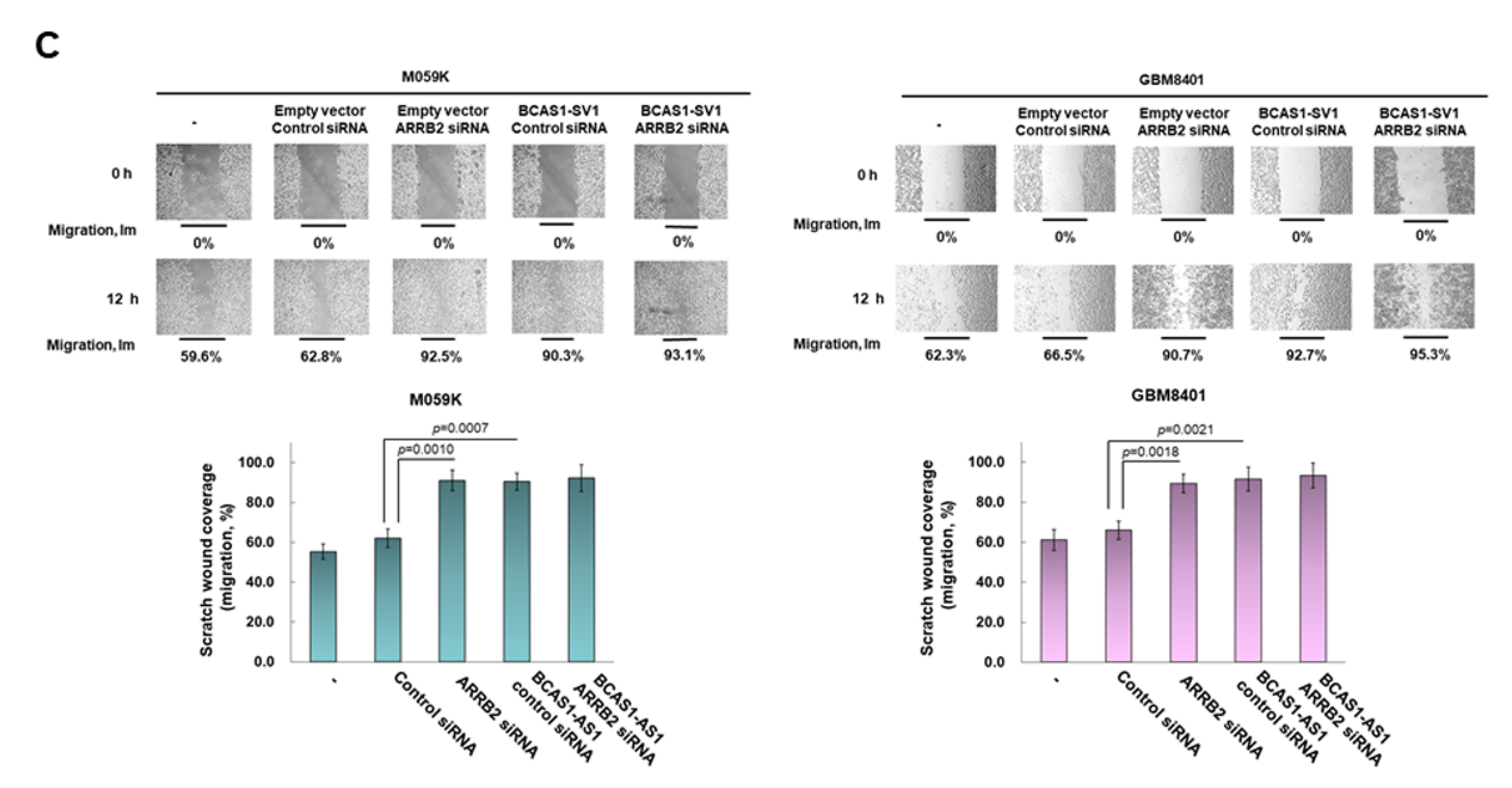
3.7. Downregulation of β-Arrestin 2 Increases Proliferation and Migration of GBM Cells and Abolishes the Effect of BCAS1-SV1
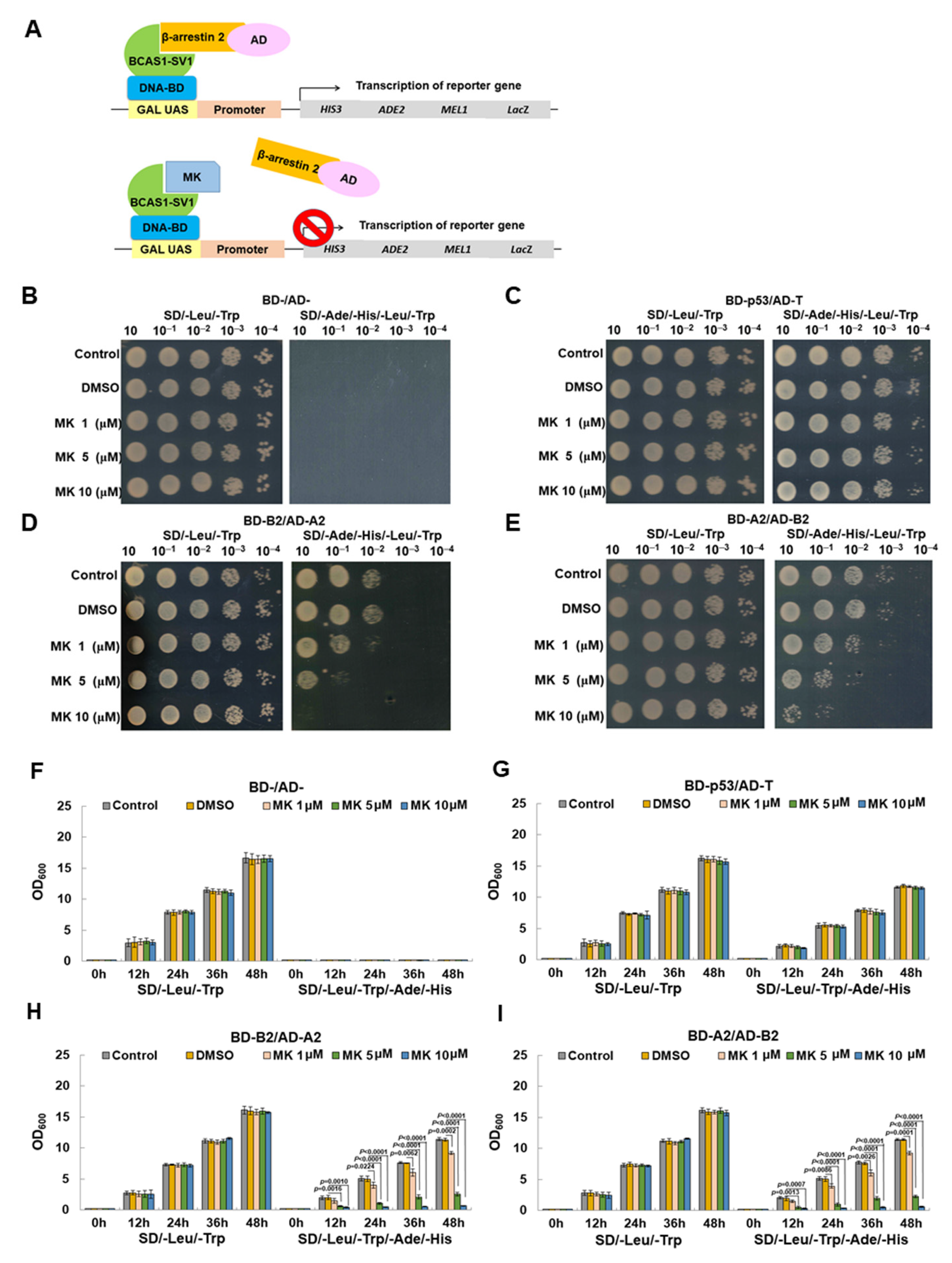
3.8. Maackiain Directly Weakens the Interaction of BCAS1-SV1 with β-Arrestin 2 in a Yeast Two-Hybrid-Based Growth Assay
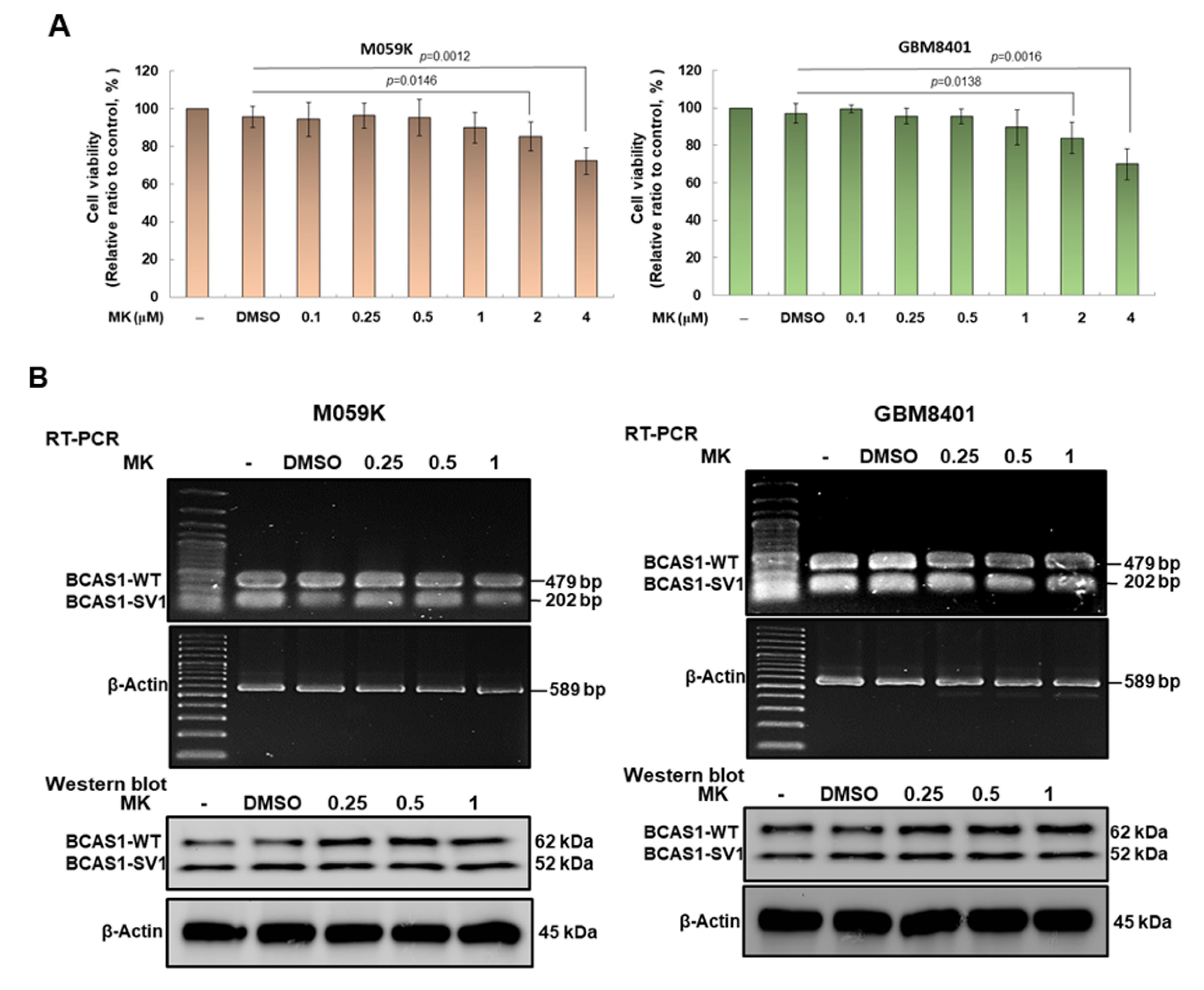
3.9. Maackiain Can Lessen the Proliferation and Migration of GBM Cells In Vitro
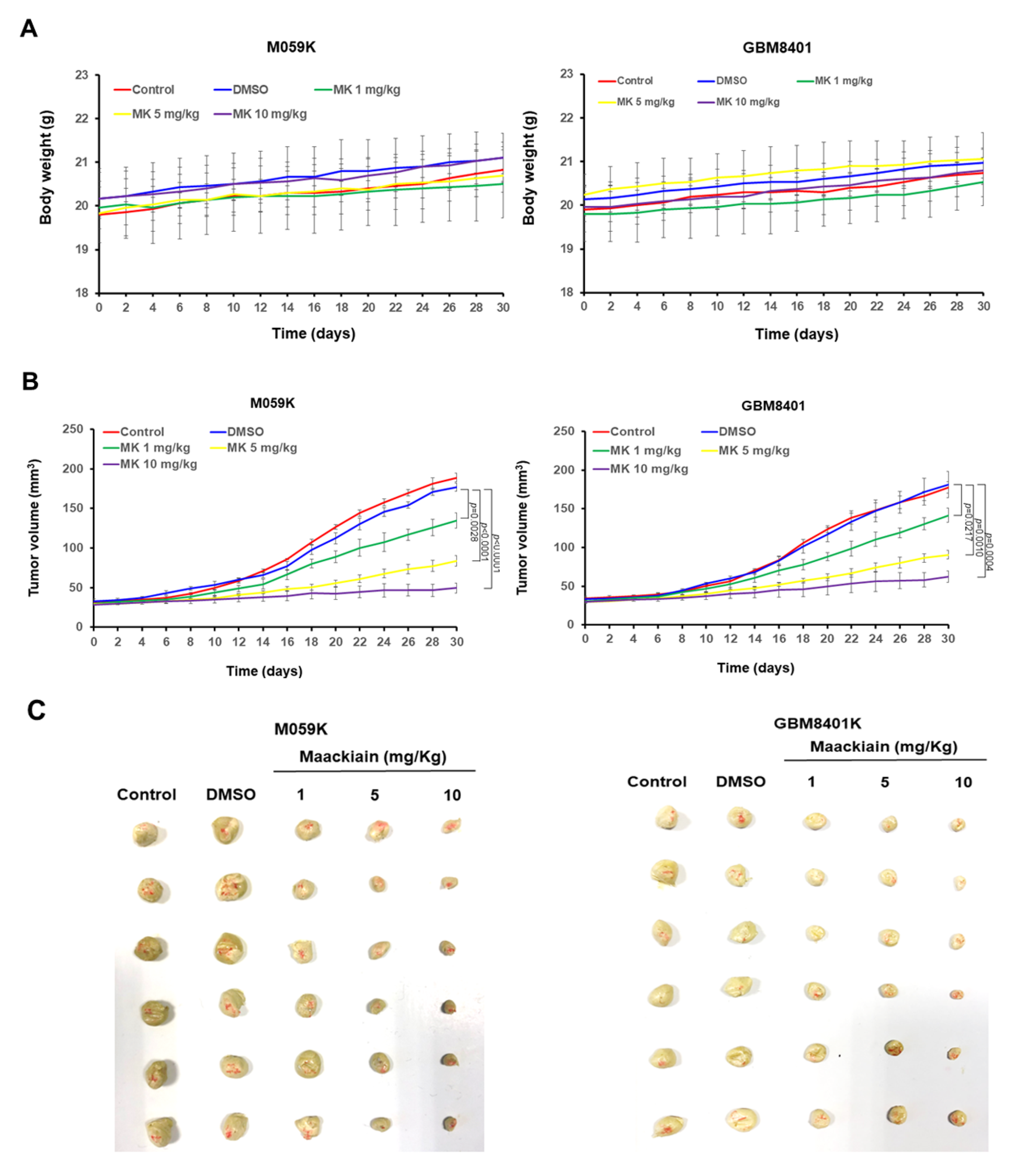
3.10. Maackiain Treatment Reduces Tumor Size in an Immunodeficient Mouse Model
3.11. Maackiain Treatment Diminishes the Size of Orthotopic Xenografted GBM Tumors of Immunodeficient Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 39. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Braunschweig, U.; Gueroussov, S.; Plocik, A.M.; Graveley, B.R.; Blencowe, B.J. Dynamic integration of splicing within gene regulatory pathways. Cell 2013, 152, 1252–1269. [Google Scholar] [CrossRef] [PubMed]
- Kornblihtt, A.R.; Schor, I.E.; Allo, M.; Dujardin, G.; Petrillo, E.; Munoz, M.J. Alternative splicing: A pivotal step between eukaryotic transcription and translation. Nat. Rev. Mol. Cell Biol. 2013, 14, 153–165. [Google Scholar] [CrossRef]
- Luco, R.F.; Allo, M.; Schor, I.E.; Kornblihtt, A.R.; Misteli, T. Epigenetics in alternative pre-mRNA splicing. Cell 2011, 144, 16–26. [Google Scholar] [CrossRef]
- Lokody, I. Alternative splicing: Aberrant splicing promotes colon tumour growth. Nat. Rev. Cancer 2014, 14, 382–383. [Google Scholar] [CrossRef]
- Sahin, I.; George, A.; Seyhan, A.A. Therapeutic targeting of alternative RNA splicing in gastrointestinal malignancies and other cancers. Int. J. Mol. Sci. 2021, 22, 11790. [Google Scholar] [CrossRef]
- Fu, R.H.; Liu, S.P.; Huang, S.J.; Chen, H.J.; Chen, P.R.; Lin, Y.H.; Ho, Y.C.; Chang, W.L.; Tsai, C.H.; Shyu, W.C.; et al. Aberrant alternative splicing events in Parkinson’s disease. Cell Transplant. 2013, 22, 653–661. [Google Scholar] [CrossRef]
- Zhu, H.; Carpenter, R.L.; Han, W.; Lo, H.W. The GLI1 splice variant TGLI1 promotes glioblastoma angiogenesis and growth. Cancer Lett. 2014, 343, 51–61. [Google Scholar] [CrossRef]
- Ferrarese, R.; Harsh, G.R.; Yadav, A.K.; Bug, E.; Maticzka, D.; Reichardt, W.; Dombrowski, S.M.; Miller, T.E.; Masilamani, A.P.; Dai, F.; et al. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J. Clin. Investig. 2014, 124, 2861–2876. [Google Scholar] [CrossRef]
- Siddaway, R.; Milos, S.; Vadivel, A.K.A.; Dobson, T.H.W.; Swaminathan, J.; Ryall, S.; Pajovic, S.; Patel, P.G.; Nazarian, J.; Becher, O.; et al. Splicing is an alternate oncogenic pathway activation mechanism in glioma. Nat. Commun. 2022, 13, 588. [Google Scholar] [CrossRef]
- Lo, H.W.; Zhu, H.; Cao, X.; Aldrich, A.; Ali-Osman, F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009, 69, 6790–6798. [Google Scholar] [CrossRef]
- Camacho-Vanegas, O.; Narla, G.; Teixeira, M.S.; DiFeo, A.; Misra, A.; Singh, G.; Chan, A.M.; Friedman, S.L.; Feuerstein, B.G.; Martignetti, J.A. Functional inactivation of the KLF6 tumor suppressor gene by loss of heterozygosity and increased alternative splicing in glioblastoma. Int. J. Cancer 2007, 121, 1390–1395. [Google Scholar] [CrossRef]
- Yu, Y.; Jiang, X.; Schoch, B.S.; Carroll, R.S.; Black, P.M.; Johnson, M.D. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007, 67, 130–138. [Google Scholar] [CrossRef]
- van Bodegraven, E.J.; van Asperen, J.V.; Sluijs, J.A.; van Deursen, C.B.J.; van Strien, M.E.; Stassen, O.; Robe, P.A.J.; Hol, E.M. GFAP alternative splicing regulates glioma cell-ECM interaction in a DUSP4-dependent manner. FASEB J. 2019, 33, 12941–12959. [Google Scholar] [CrossRef]
- Xia, X.; Li, X.; Li, F.; Wu, X.; Zhang, M.; Zhou, H.; Huang, N.; Yang, X.; Xiao, F.; Liu, D.; et al. A novel tumor suppressor protein encoded by circular AKT3 RNA inhibits glioblastoma tumorigenicity by competing with active phosphoinositide-dependent Kinase-1. Mol. Cancer 2019, 18, 131. [Google Scholar] [CrossRef]
- Ngo, T.; Corrales, A.; Bourne, T.; Elmojahid, S.; Lam, K.S.; Diaz, E. Alternative Splicing of MXD3 and Its Regulation of MXD3 Levels in Glioblastoma. Front. Mol. Biosci. 2019, 6, 5. [Google Scholar] [CrossRef]
- Cheng, W.Y.; Shen, C.C.; Chiao, M.T.; Liang, Y.J.; Mao, T.F.; Liu, B.S.; Chen, J.P. High expression of a novel splicing variant of VEGF, L-VEGF144 in glioblastoma multiforme is associated with a poorer prognosis in bevacizumab treatment. J. Neuro-Oncol. 2018, 140, 37–47. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, M.; Shi, B.; Zhang, Q.; Jiang, H.; Sun, Y.; Liu, J.; Zhou, K.; Yao, M.; Gu, J.; et al. Identification of an exon 4-deletion variant of epidermal growth factor receptor with increased metastasis-promoting capacity. Neoplasia 2011, 13, 461–471. [Google Scholar] [CrossRef]
- Jin, W.; McCutcheon, I.E.; Fuller, G.N.; Huang, E.S.; Cote, G.J. Fibroblast growth factor receptor-1 alpha-exon exclusion and polypyrimidine tract-binding protein in glioblastoma multiforme tumors. Cancer Res. 2000, 60, 1221–1224. [Google Scholar]
- Aldave, G.; Gonzalez-Huarriz, M.; Rubio, A.; Romero, J.P.; Ravi, D.; Minana, B.; Cuadrado-Tejedor, M.; Garcia-Osta, A.; Verhaak, R.; Xipell, E.; et al. The aberrant splicing of BAF45d links splicing regulation and transcription in glioblastoma. Neuro-Oncology 2018, 20, 930–941. [Google Scholar] [CrossRef]
- Fontana, L.; Rovina, D.; Novielli, C.; Maffioli, E.; Tedeschi, G.; Magnani, I.; Larizza, L. Suggestive evidence on the involvement of polypyrimidine-tract binding protein in regulating alternative splicing of MAP/microtubule affinity-regulating kinase 4 in glioma. Cancer Lett. 2015, 359, 87–96. [Google Scholar] [CrossRef]
- Desai, S.; Ding, M.; Wang, B.; Lu, Z.; Zhao, Q.; Shaw, K.; Yung, W.K.; Weinstein, J.N.; Tan, M.; Yao, J. Tissue-specific isoform switch and DNA hypomethylation of the pyruvate kinase PKM gene in human cancers. Oncotarget 2014, 5, 8202–8210. [Google Scholar] [CrossRef]
- Izaguirre, D.I.; Zhu, W.; Hai, T.; Cheung, H.C.; Krahe, R.; Cote, G.J. PTBP1-dependent regulation of USP5 alternative RNA splicing plays a role in glioblastoma tumorigenesis. Mol. Carcinog. 2012, 51, 895–906. [Google Scholar] [CrossRef]
- Lefave, C.V.; Squatrito, M.; Vorlova, S.; Rocco, G.L.; Brennan, C.W.; Holland, E.C.; Pan, Y.X.; Cartegni, L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011, 30, 4084–4097. [Google Scholar] [CrossRef]
- Cheung, H.C.; Baggerly, K.A.; Tsavachidis, S.; Bachinski, L.L.; Neubauer, V.L.; Nixon, T.J.; Aldape, K.D.; Cote, G.J.; Krahe, R. Global analysis of aberrant pre-mRNA splicing in glioblastoma using exon expression arrays. BMC Genom. 2008, 9, 216. [Google Scholar] [CrossRef]
- Collins, C.; Rommens, J.M.; Kowbel, D.; Godfrey, T.; Tanner, M.; Hwang, S.I.; Polikoff, D.; Nonet, G.; Cochran, J.; Myambo, K.; et al. Positional cloning of ZNF217 and NABC1: Genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc. Natl. Acad. Sci. USA 1998, 95, 8703–8708. [Google Scholar] [CrossRef]
- Loukopoulos, P.; Shibata, T.; Katoh, H.; Kokubu, A.; Sakamoto, M.; Yamazaki, K.; Kosuge, T.; Kanai, Y.; Hosoda, F.; Imoto, I.; et al. Genome-wide array-based comparative genomic hybridization analysis of pancreatic adenocarcinoma: Identification of genetic indicators that predict patient outcome. Cancer Sci. 2007, 98, 392–400. [Google Scholar] [CrossRef]
- van Dekken, H.; Vissers, K.; Tilanus, H.W.; Kuo, W.L.; Tanke, H.J.; Rosenberg, C.; Ijszenga, M.; Szuhai, K. Genomic array and expression analysis of frequent high-level amplifications in adenocarcinomas of the gastro-esophageal junction. Cancer Genet. Cytogenet. 2006, 166, 157–162. [Google Scholar] [CrossRef]
- Fenne, I.S.; Helland, T.; Flageng, M.H.; Dankel, S.N.; Mellgren, G.; Sagen, J.V. Downregulation of steroid receptor coactivator-2 modulates estrogen-responsive genes and stimulates proliferation of mcf-7 breast cancer cells. PLoS ONE 2013, 8, e70096. [Google Scholar] [CrossRef]
- Burmester, J.K.; Suarez, B.K.; Lin, J.H.; Jin, C.H.; Miller, R.D.; Zhang, K.Q.; Salzman, S.A.; Reding, D.J.; Catalona, W.J. Analysis of candidate genes for prostate cancer. Hum. Hered. 2004, 57, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Taylor, L.; Varkey, M.; Hoxie, N.; Mohammed, Y.; Goo, Y.A.; Peterman, S.; Moghekar, A.; Yuan, Y.; Glaros, T.; et al. Reinspection of a clinical proteomics tumor analysis consortium (CPTAC) dataset with cloud computing reveals abundant post-translational modifications and protein sequence variants. Cancers 2021, 13, 5034. [Google Scholar] [CrossRef] [PubMed]
- Beardsley, D.I.; Kowbel, D.; Lataxes, T.A.; Mannino, J.M.; Xin, H.; Kim, W.J.; Collins, C.; Brown, K.D. Characterization of the novel amplified in breast cancer-1 (NABC1) gene product. Exp. Cell Res. 2003, 290, 402–413. [Google Scholar] [CrossRef]
- Ewing, R.M.; Chu, P.; Elisma, F.; Li, H.; Taylor, P.; Climie, S.; McBroom-Cerajewski, L.; Robinson, M.D.; O’Connor, L.; Li, M.; et al. Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol. Syst. Biol. 2007, 3, 89. [Google Scholar] [CrossRef] [PubMed]
- Fard, M.K.; van der Meer, F.; Sanchez, P.; Cantuti-Castelvetri, L.; Mandad, S.; Jakel, S.; Fornasiero, E.F.; Schmitt, S.; Ehrlich, M.; Starost, L.; et al. BCAS1 expression defines a population of early myelinating oligodendrocytes in multiple sclerosis lesions. Sci. Transl. Med. 2017, 9, eaam7816. [Google Scholar] [CrossRef] [PubMed]
- Kaji, S.; Maki, T.; Ueda, J.; Ishimoto, T.; Inoue, Y.; Yasuda, K.; Sawamura, M.; Hikawa, R.; Ayaki, T.; Yamakado, H.; et al. BCAS1-positive immature oligodendrocytes are affected by the alpha-synuclein-induced pathology of multiple system atrophy. Acta Neuropathol. Commun. 2020, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Lohrberg, M.; Winkler, A.; Franz, J.; van der Meer, F.; Ruhwedel, T.; Sirmpilatze, N.; Dadarwal, R.; Handwerker, R.; Esser, D.; Wiegand, K.; et al. Lack of astrocytes hinders parenchymal oligodendrocyte precursor cells from reaching a myelinating state in osmolyte-induced demyelination. Acta Neuropathol. Commun. 2020, 8, 224. [Google Scholar] [CrossRef]
- Ishimoto, T.; Ninomiya, K.; Inoue, R.; Koike, M.; Uchiyama, Y.; Mori, H. Mice lacking BCAS1, a novel myelin-associated protein, display hypomyelination, schizophrenia-like abnormal behaviors, and upregulation of inflammatory genes in the brain. Glia 2017, 65, 727–739. [Google Scholar] [CrossRef]
- Lauridsen, J.B.; Johansen, J.L.; Rekling, J.C.; Thirstrup, K.; Moerk, A.; Sager, T.N. Regulation of the Bcas1 and Baiap3 transcripts in the subthalamic nucleus in mice recovering from MPTP toxicity. Neurosci. Res. 2011, 70, 269–276. [Google Scholar] [CrossRef]
- Bae, W.Y.; Choi, J.S.; Nam, S.; Jeong, J.W. beta-arrestin 2 stimulates degradation of HIF-1alpha and modulates tumor progression of glioblastoma. Cell Death Differ. 2021, 28, 3092–3104. [Google Scholar] [CrossRef]
- Cory, G. Scratch-wound assay. Methods Mol. Biol. 2011, 769, 25–30. [Google Scholar]
- Marrero, L.; Wyczechowska, D.; Musto, A.E.; Wilk, A.; Vashistha, H.; Zapata, A.; Walker, C.; Velasco-Gonzalez, C.; Parsons, C.; Wieland, S.; et al. Therapeutic efficacy of aldoxorubicin in an intracranial xenograft mouse model of human glioblastoma. Neoplasia 2014, 16, 874–882. [Google Scholar] [CrossRef]
- Wong, J.H.; Alfatah, M.; Sin, M.F.; Sim, H.M.; Verma, C.S.; Lane, D.P.; Arumugam, P. A yeast two-hybrid system for the screening and characterization of small-molecule inhibitors of protein-protein interactions identifies a novel putative Mdm2-binding site in p53. BMC Biol. 2017, 15, 108. [Google Scholar] [CrossRef]
- Guo, J.; Li, J.; Wei, H.; Liang, Z. Maackiain protects the kidneys of type 2 diabetic rats via modulating the Nrf2/HO-1 and TLR4/NF-kappaB/caspase-3 pathways. Drug Des. Dev. Ther. 2021, 15, 4339–4358. [Google Scholar] [CrossRef]
- Fabro, F.; Lamfers, M.L.M.; Leenstra, S. Advancements, challenges, and future directions in tackling glioblastoma resistance to small kinase inhibitors. Cancers 2022, 14, 600. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhang, Y.; Xiong, F.; Zhang, S.; Gong, Z.; Yan, Q.; He, Y.; Wei, F.; Zhang, W.; Zhou, M.; et al. The role of alternative splicing in human cancer progression. Am. J. Cancer Res. 2021, 11, 4642–4667. [Google Scholar]
- Chompunud Na Ayudhya, C.; Amponnawarat, A.; Ali, H. Substance P serves as a balanced agonist for MRGPRX2 and a single tyrosine residue is required for beta-arrestin recruitment and receptor internalization. Int. J. Mol. Sci. 2021, 22, 5318. [Google Scholar] [CrossRef]
- Mohamed, R.; Ahmad, E.A.; Omran, B.H.F.; Sakr, A.T.; Ibrahim, I.; Mahmoud, M.F.; El-Naggar, M.E. Mitigation of dexamethasone-induced nephrotoxicity by modulating the activity of adrenergic receptors: Implication of Wnt/beta-arrestin2/beta-catenin pathway. Life Sci. 2022, 293, 120304. [Google Scholar] [CrossRef]
- Raghuwanshi, S.K.; Nasser, M.W.; Chen, X.; Strieter, R.M.; Richardson, R.M. Depletion of beta-arrestin-2 promotes tumor growth and angiogenesis in a murine model of lung cancer. J. Immunol. 2008, 180, 5699–5706. [Google Scholar] [CrossRef]
- Sun, W.Y.; Hu, S.S.; Wu, J.J.; Huang, Q.; Ma, Y.; Wang, Q.T.; Chen, J.Y.; Wei, W. Down-regulation of beta-arrestin2 promotes tumour invasion and indicates poor prognosis of hepatocellular carcinoma. Sci. Rep. 2016, 6, 35609. [Google Scholar] [CrossRef]
- Cao, Y.J.; Zhou, Y.J.; He, X.Z.; Zhou, C.X.; Cui, L.; Zhuang, Q.F.; Xu, R.F. Overexpression of beta-arrestin2 induces G1-phase cell cycle arrest and suppresses tumorigenicity in renal cell carcinoma. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 1729–1737. [Google Scholar] [PubMed]
- Bedini, A.; Baiula, M.; Vincelli, G.; Formaggio, F.; Lombardi, S.; Caprini, M.; Spampinato, S. Nociceptin/orphanin FQ antagonizes lipopolysaccharide-stimulated proliferation, migration and inflammatory signaling in human glioblastoma U87 cells. Biochem. Pharmacol. 2017, 140, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Lei, Z.; Yan, Z.; Ji, X.; Chang, X.; Cai, Z.; Lu, L.; Qi, Y.; Yin, X.; Han, X.; et al. Exploiting D2 receptor beta-arrestin2-biased signalling to suppress tumour growth of pituitary adenomas. Br. J. Pharmacol. 2021, 178, 3570–3586. [Google Scholar] [CrossRef] [PubMed]
- Blondel-Tepaz, E.; Leverve, M.; Sokrat, B.; Paradis, J.S.; Kosic, M.; Saha, K.; Auffray, C.; Lima-Fernandes, E.; Zamborlini, A.; Poupon, A.; et al. The RanBP2/RanGAP1-SUMO complex gates beta-arrestin2 nuclear entry to regulate the Mdm2-p53 signaling axis. Oncogene 2021, 40, 2243–2257. [Google Scholar] [CrossRef]
- Rosano, L.; Cianfrocca, R.; Masi, S.; Spinella, F.; Di Castro, V.; Biroccio, A.; Salvati, E.; Nicotra, M.R.; Natali, P.G.; Bagnato, A. Beta-arrestin links endothelin A receptor to beta-catenin signaling to induce ovarian cancer cell invasion and metastasis. Proc. Natl. Acad. Sci. USA 2009, 106, 2806–2811. [Google Scholar] [CrossRef]
- Zajac, M.; Law, J.; Cvetkovic, D.D.; Pampillo, M.; McColl, L.; Pape, C.; Di Guglielmo, G.M.; Postovit, L.M.; Babwah, A.V.; Bhattacharya, M. GPR54 (KISS1R) transactivates EGFR to promote breast cancer cell invasiveness. PLoS ONE 2011, 6, e21599. [Google Scholar] [CrossRef]
- Bonnans, C.; Flaceliere, M.; Grillet, F.; Dantec, C.; Desvignes, J.P.; Pannequin, J.; Severac, D.; Dubois, E.; Bibeau, F.; Escriou, V.; et al. Essential requirement for beta-arrestin2 in mouse intestinal tumors with elevated Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 3047–3052. [Google Scholar] [CrossRef]
- Cao, C.; Zhang, Y.; Cheng, J.; Wu, F.; Niu, X.; Hu, X.; Duan, X.; Fu, X.; Zhang, J.; Zhang, X.; et al. Beta-arrestin2 inhibits the apoptosis and facilitates the proliferation of fibroblast-like synoviocytes in diffuse-type tenosynovial giant cell tumor. Cancer Genom. Proteom. 2021, 18 (Suppl. S3), 461–470. [Google Scholar] [CrossRef]
- Liang, H.; Lin, Z.; Ye, Y.; Luo, R.; Zeng, L. ARRB2 promotes colorectal cancer growth through triggering WTAP. Acta Biochim. Biophys. Sin. 2021, 53, 85–93. [Google Scholar] [CrossRef]
- Czogalla, B.; Partenheimer, A.; Jeschke, U.; von Schonfeldt, V.; Mayr, D.; Mahner, S.; Burges, A.; Simoni, M.; Melli, B.; Benevelli, R.; et al. Beta-arrestin 2 is a prognostic factor for survival of ovarian cancer patients upregulating cell proliferation. Front. Endocrinol. 2020, 11, 554733. [Google Scholar] [CrossRef]
- Ge, L.; Shenoy, S.K.; Lefkowitz, R.J.; DeFea, K. Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both beta-arrestin-1 and -2. J. Biol. Chem. 2004, 279, 55419–55424. [Google Scholar] [CrossRef]
- Luttrell, L.M.; Lefkowitz, R.J. The role of beta-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 2002, 115 Pt 3, 455–465. [Google Scholar] [CrossRef]
- Vucenik, I.; Druzijanic, A.; Druzijanic, N. Inositol hexaphosphate (IP6) and colon cancer: From concepts and first experiments to clinical application. Molecules 2020, 25, 5931. [Google Scholar] [CrossRef]
- Chen, C.; Yang, F.; Liu, C.; Cui, L.; Fu, M.; Song, Y. Inositol hexaphosphate hydrolysate competitively binds to AKT to inhibit the proliferation of colon carcinoma. Oncol. Rep. 2017, 38, 2901–2910. [Google Scholar] [CrossRef]
- Schroterova, L.; Jezkova, A.; Rudolf, E.; Caltova, K.; Kralova, V.; Hanusova, V. Inositol hexaphosphate limits the migration and the invasiveness of colorectal carcinoma cells in vitro. Int. J. Oncol. 2018, 53, 1625–1632. [Google Scholar]
- Wang, Z.; Liu, Y.; Huang, S.; Fang, M. TRAF6 interacts with and ubiquitinates PIK3CA to enhance PI3K activation. FEBS Lett. 2018, 592, 1882–1892. [Google Scholar] [CrossRef]
- Shi, J.; Liu, Z.; Xu, Q. Tumor necrosis factor receptor-associated factor 6 contributes to malignant behavior of human cancers through promoting AKT ubiquitination and phosphorylation. Cancer Sci. 2019, 110, 1909–1920. [Google Scholar] [CrossRef]
- Peng, Z.; Shuangzhu, Y.; Yongjie, J.; Xinjun, Z.; Ying, L. TNF receptor-associated factor 6 regulates proliferation, apoptosis, and invasion of glioma cells. Mol. Cell. Biochem. 2013, 377, 87–96. [Google Scholar] [CrossRef]
- Yang, M.; Jin, M.; Li, K.; Liu, H.; Yang, X.; Zhang, X.; Zhang, B.; Gong, A.; Bie, Q. TRAF6 promotes gastric cancer cell self-renewal, proliferation, and migration. Stem Cells Int. 2020, 2020, 3296192. [Google Scholar] [CrossRef]
- Rong, Y.; Wang, D.; Wu, W.; Jin, D.; Kuang, T.; Ni, X.; Zhang, L.; Lou, W. TRAF6 is over-expressed in pancreatic cancer and promotes the tumorigenicity of pancreatic cancer cells. Med. Oncol. 2014, 31, 260. [Google Scholar] [CrossRef]
- He, Z.; Huang, C.; Lin, G.; Ye, Y. siRNA-induced TRAF6 knockdown promotes the apoptosis and inhibits the invasion of human lung cancer SPC-A1 cells. Oncol. Rep. 2016, 35, 1933–1940. [Google Scholar] [CrossRef]
- Meng, Q.; Zhang, W.; Xu, X.; Li, J.; Mu, H.; Liu, X.; Qin, L.; Zhu, X.; Zheng, M. The effects of TRAF6 on proliferation, apoptosis and invasion in osteosarcoma are regulated by miR-124. Int. J. Mol. Med. 2018, 41, 2968–2976. [Google Scholar] [CrossRef]
- Zhu, G.; Cheng, Z.; Huang, Y.; Zheng, W.; Yang, S.; Lin, C.; Ye, J. TRAF6 promotes the progression and growth of colorectal cancer through nuclear shuttle regulation NF-kB/c-jun signaling pathway. Life Sci. 2019, 235, 116831. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Zeng, W.; Zhang, J.; Cai, L.; Wu, Z.; Su, J.; Xiao, Y.; Liu, N.; Tang, L.; et al. TRAF6 activates fibroblasts to cancer-associated fibroblasts through FGF19 in tumor microenvironment to benefit the malignant phenotype of melanoma cells. J. Investig. Dermatol. 2020, 140, 2268–2279.e11. [Google Scholar] [CrossRef]
- Pignatelli, J.; Jones, M.C.; LaLonde, D.P.; Turner, C.E. Beta2-adaptin binds actopaxin and regulates cell spreading, migration and matrix degradation. PLoS ONE 2012, 7, e46228. [Google Scholar] [CrossRef]
- Tsygankova, O.M.; Keen, J.H. A unique role for clathrin light chain A in cell spreading and migration. J. Cell Sci. 2019, 132, jcs22403. [Google Scholar] [CrossRef]
- Zang, Y.; Zhu, J.; Li, Q.; Tu, J.; Li, X.; Hu, R.; Yang, D. miR-137-3p Modulates the progression of prostate cancer by regulating the JNK3/EZH2 axis. OncoTargets Ther. 2020, 13, 7921–7932. [Google Scholar] [CrossRef]
- Witherow, D.S.; Garrison, T.R.; Miller, W.E.; Lefkowitz, R.J. beta-Arrestin inhibits NF-kappaB activity by means of its interaction with the NF-kappaB inhibitor IkappaBalpha. Proc. Natl. Acad. Sci. USA 2004, 101, 8603–8607. [Google Scholar] [CrossRef]
- Sun, Y.; Cheng, Z.; Ma, L.; Pei, G. Beta-arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J. Biol. Chem. 2002, 277, 49212–49219. [Google Scholar] [CrossRef]
- Huh, J.W.; Lee, J.H.; Jeon, E.; Ryu, H.W.; Oh, S.R.; Ahn, K.S.; Jun, H.S.; Ha, U.H. Maackiain, a compound derived from Sophora flavescens, increases IL-1beta production by amplifying nigericin-mediated inflammasome activation. FEBS Open Bio 2020, 10, 1482–1491. [Google Scholar] [CrossRef]
- Yoo, H.; Kang, M.; Pyo, S.; Chae, H.S.; Ryu, K.H.; Kim, J.; Chin, Y.W. SKI3301, a purified herbal extract from Sophora tonkinensis, inhibited airway inflammation and bronchospasm in allergic asthma animal models in vivo. J. Ethnopharmacol. 2017, 206, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, H.; Nariai, Y.; Kato, S.; Nakano, T.; Kanayama, T.; Kashiwada, Y.; Nemoto, H.; Kawazoe, K.; Takaishi, Y.; Kitamura, Y.; et al. Maackiain is a novel antiallergic compound that suppresses transcriptional upregulation of the histamine H1 receptor and interleukin-4 genes. Pharmacol. Res. Perspect. 2015, 3, e00166. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Zhu, Y.; Jie, J.; Li, D.; Song, L.; Luo, J. Maackiain protects against sepsis via activating AMPK/Nrf2/HO-1 pathway. Int. Immunopharmacol. 2022, 108, 108710. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, S.G.; Savova, M.S.; Marchev, A.S.; Ferrante, C.; Orlando, G.; Wabitsch, M.; Georgiev, M.I. Anti-adipogenic activity of maackiain and ononin is mediated via inhibition of PPARgamma in human adipocytes. Biomed. Pharmacother. 2022, 149, 112908. [Google Scholar] [CrossRef]
- Li, P.; Chai, W.C.; Wang, Z.Y.; Tang, K.J.; Chen, J.Y.; Venter, H.; Semple, S.J.; Xiang, L. Bioactivity-guided isolation of compounds from Sophora flavescens with antibacterial activity against Acinetobacter baumannii. Nat. Prod. Res. 2021, 1–9. [Google Scholar] [CrossRef]
- Lee, H.W.; Ryu, H.W.; Kang, M.G.; Park, D.; Oh, S.R.; Kim, H. Potent selective monoamine oxidase B inhibition by maackiain, a pterocarpan from the roots of Sophora flavescens. Bioorg. Med. Chem. Lett. 2016, 26, 4714–4719. [Google Scholar] [CrossRef]
- Tsai, R.T.; Tsai, C.W.; Liu, S.P.; Gao, J.X.; Kuo, Y.H.; Chao, P.M.; Hung, H.S.; Shyu, W.C.; Lin, S.Z.; Fu, R.H. Maackiain ameliorates 6-hydroxydopamine and SNCA pathologies by modulating the PINK1/Parkin pathway in models of Parkinson’s disease in Caenorhabditis elegans and the SH-SY5Y cell line. Int. J. Mol. Sci. 2020, 21, 4455. [Google Scholar] [CrossRef]
- Peng, F.; Wang, L.; Xiong, L.; Tang, H.; Du, J.; Peng, C. Maackiain modulates miR-374a/GADD45A axis to inhibit triple-negative breast cancer initiation and progression. Front. Pharmacol. 2022, 13, 806869. [Google Scholar] [CrossRef]
- Hamada, T.; Akahane, T.; Yokoyama, S.; Higa, N.; Kirishima, M.; Matsuo, K.; Shimokawa, M.; Yoshimoto, K.; Tanimoto, A. An oncogenic splice variant of PDGFRalpha in adult glioblastoma as a therapeutic target for selective CDK4/6 inhibitors. Sci. Rep. 2022, 12, 1275. [Google Scholar] [CrossRef]
- Kim, J.H.; Jeong, K.; Li, J.; Murphy, J.M.; Vukadin, L.; Stone, J.K.; Richard, A.; Tran, J.; Gillespie, G.Y.; Flemington, E.K.; et al. SON drives oncogenic RNA splicing in glioblastoma by regulating PTBP1/PTBP2 switching and RBFOX2 activity. Nat. Commun. 2021, 12, 5551. [Google Scholar] [CrossRef]
- Golan-Gerstl, R.; Cohen, M.; Shilo, A.; Suh, S.S.; Bakacs, A.; Coppola, L.; Karni, R. Splicing factor hnRNP A2/B1 regulates tumor suppressor gene splicing and is an oncogenic driver in glioblastoma. Cancer Res. 2011, 71, 4464–4472. [Google Scholar] [CrossRef]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef]
- Fuentes-Fayos, A.C.; Perez-Gomez, J.M.; ME, G.G.; Jimenez-Vacas, J.M.; Blanco-Acevedo, C.; Sanchez-Sanchez, R.; Solivera, J.; Breunig, J.J.; Gahete, M.D.; Castano, J.P.; et al. SF3B1 inhibition disrupts malignancy and prolongs survival in glioblastoma patients through BCL2L1 splicing and mTOR/ss-catenin pathways imbalances. J. Exp. Clin. Cancer Res. 2022, 41, 39. [Google Scholar] [CrossRef]
- Song, X.; Wan, X.; Huang, T.; Zeng, C.; Sastry, N.; Wu, B.; James, C.D.; Horbinski, C.; Nakano, I.; Zhang, W.; et al. SRSF3-regulated RNA alternative splicing promotes glioblastoma tumorigenicity by affecting multiple cellular processes. Cancer Res. 2019, 79, 5288–5301. [Google Scholar] [CrossRef]
- Fuentes-Fayos, A.C.; Vazquez-Borrego, M.C.; Jimenez-Vacas, J.M.; Bejarano, L.; Pedraza-Arevalo, S.; López, F.L.; Blanco-Acevedo, C.; Sanchez-Sanchez, R.; Reyes, O.; Ventura, S.; et al. Splicing machinery dysregulation drives glioblastoma development/aggressiveness: Oncogenic role of SRSF3. Brain 2020, 143, 3273–3293. [Google Scholar] [CrossRef]
- Sun, S.; Gao, T.; Pang, B.; Su, X.; Guo, C.; Zhang, R.; Pang, Q. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m(6)A-dependent manner. Cell Death Dis. 2022, 13, 73. [Google Scholar] [CrossRef]
- Cun, Y.; An, S.; Zheng, H.; Lan, J.; Chen, W.; Luo, W.; Yao, C.; Li, X.; Huang, X.; Sun, X.; et al. Specific regulation of m(6)A by SRSF7 promotes the progression of glioblastoma. Genom. Proteom. Bioinform. 2021. [Google Scholar] [CrossRef]
- Sun, C.; Zheng, X.; Sun, Y.; Yu, J.; Sheng, M.; Yan, S.; Zhu, Q.; Lan, Q. Identification of IGF2BP3 as an Adverse Prognostic Biomarker of Gliomas. Front. Genet. 2021, 12, 743738. [Google Scholar] [CrossRef]
- Ma, J.; Gao, Z.; Liu, X. DDX46 accelerates the proliferation of glioblastoma by activating the MAPK-p38 signaling. J. B.U.ON. 2021, 26, 2084–2089. [Google Scholar]
- Broggi, G.; Salvatorelli, L.; Barbagallo, D.; Certo, F.; Altieri, R.; Tirro, E.; Massimino, M.; Vigneri, P.; Guadagno, E.; Maugeri, G.; et al. Diagnostic utility of the immunohistochemical expression of serine and arginine rich splicing factor 1 (SRSF1) in the differential diagnosis of adult gliomas. Cancers 2021, 13, 2086. [Google Scholar] [CrossRef]
- Hua, D.; Zhao, Q.; Yu, Y.; Yu, H.; Yu, L.; Zhou, X.; Wang, Q.; Sun, C.; Shi, C.; Luo, W.; et al. Eucalyptal A inhibits glioma by rectifying oncogenic splicing of MYO1B mRNA via suppressing SRSF1 expression. Eur. J. Pharmacol. 2021, 890, 173669. [Google Scholar] [CrossRef]
- Barbagallo, D.; Caponnetto, A.; Brex, D.; Mirabella, F.; Barbagallo, C.; Lauretta, G.; Morrone, A.; Certo, F.; Broggi, G.; Caltabiano, R.; et al. CircSMARCA5 regulates VEGFA mRNA splicing and Angiogenesis in glioblastoma multiforme through the binding of SRSF1. Cancers 2019, 11, 194. [Google Scholar] [CrossRef]
- Sigala, I.; Tsamis, K.I.; Gousia, A.; Alexiou, G.; Voulgaris, S.; Giannakouros, T.; Kyritsis, A.P.; Nikolakaki, E. Expression of SRPK1 in gliomas and its role in glioma cell lines viability. Tumour Biol. 2016, 37, 8699–8707. [Google Scholar] [CrossRef]
- Uren, P.J.; Vo, D.T.; de Araujo, P.R.; Potschke, R.; Burns, S.C.; Bahrami-Samani, E.; Qiao, M.; de Sousa Abreu, R.; Nakaya, H.I.; Correa, B.R.; et al. RNA-binding protein musashi1 is a central regulator of adhesion pathways in glioblastoma. Mol. Cell. Biol. 2015, 35, 2965–2978. [Google Scholar] [CrossRef]
- Voss, D.M.; Sloan, A.; Spina, R.; Ames, H.M.; Bar, E.E. The alternative splicing factor, MBNL1, inhibits glioblastoma tumor initiation and progression by reducing hypoxia-induced stemness. Cancer Res. 2020, 80, 4681–4692. [Google Scholar] [CrossRef]
- Hu, J.; Ho, A.L.; Yuan, L.; Hu, B.; Hua, S.; Hwang, S.S.; Zhang, J.; Hu, T.; Zheng, H.; Gan, B.; et al. From the Cover: Neutralization of terminal differentiation in gliomagenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14520–14527. [Google Scholar] [CrossRef]
- Pavlyukov, M.S.; Yu, H.; Bastola, S.; Minata, M.; Shender, V.O.; Lee, Y.; Zhang, S.; Wang, J.; Komarova, S.; Wang, J.; et al. Apoptotic cell-derived extracellular vesicles promote malignancy of glioblastoma via intercellular transfer of splicing factors. Cancer Cell 2018, 34, 119–135.e10. [Google Scholar] [CrossRef]
- Li, Y.; Guo, D. Genome-wide profiling of alternative splicing in glioblastoma and their clinical value. BMC Cancer 2021, 21, 958. [Google Scholar] [CrossRef]
- Paz, I.; Kosti, I.; Ares, M., Jr.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 2014, 42, W361–W367. [Google Scholar] [CrossRef]
- Mogilevsky, M.; Shimshon, O.; Kumar, S.; Mogilevsky, A.; Keshet, E.; Yavin, E.; Heyd, F.; Karni, R. Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res. 2018, 46, 11396–11404. [Google Scholar] [CrossRef]
- Raguraman, P.; Balachandran, A.A.; Chen, S.; Diermeier, S.D.; Veedu, R.N. Antisense oligonucleotide-mediated splice switching: Potential therapeutic approach for cancer mitigation. Cancers 2021, 13, 5555. [Google Scholar] [CrossRef] [PubMed]
- Denichenko, P.; Mogilevsky, M.; Clery, A.; Welte, T.; Biran, J.; Shimshon, O.; Barnabas, G.D.; Danan-Gotthold, M.; Kumar, S.; Yavin, E.; et al. Specific inhibition of splicing factor activity by decoy RNA oligonucleotides. Nat. Commun. 2019, 10, 1590. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, Y.-H.; Hung, H.-S.; Tsai, C.-W.; Chiu, S.-C.; Liu, S.-P.; Chiang, Y.-T.; Shyu, W.-C.; Lin, S.-Z.; Fu, R.-H. A Novel Splice Variant of BCAS1 Inhibits β-Arrestin 2 to Promote the Proliferation and Migration of Glioblastoma Cells, and This Effect Was Blocked by Maackiain. Cancers 2022, 14, 3890. https://doi.org/10.3390/cancers14163890
Kuo Y-H, Hung H-S, Tsai C-W, Chiu S-C, Liu S-P, Chiang Y-T, Shyu W-C, Lin S-Z, Fu R-H. A Novel Splice Variant of BCAS1 Inhibits β-Arrestin 2 to Promote the Proliferation and Migration of Glioblastoma Cells, and This Effect Was Blocked by Maackiain. Cancers. 2022; 14(16):3890. https://doi.org/10.3390/cancers14163890
Chicago/Turabian StyleKuo, Yun-Hua, Huey-Shan Hung, Chia-Wen Tsai, Shao-Chih Chiu, Shih-Ping Liu, Yu-Ting Chiang, Woei-Cherng Shyu, Shinn-Zong Lin, and Ru-Huei Fu. 2022. "A Novel Splice Variant of BCAS1 Inhibits β-Arrestin 2 to Promote the Proliferation and Migration of Glioblastoma Cells, and This Effect Was Blocked by Maackiain" Cancers 14, no. 16: 3890. https://doi.org/10.3390/cancers14163890
APA StyleKuo, Y.-H., Hung, H.-S., Tsai, C.-W., Chiu, S.-C., Liu, S.-P., Chiang, Y.-T., Shyu, W.-C., Lin, S.-Z., & Fu, R.-H. (2022). A Novel Splice Variant of BCAS1 Inhibits β-Arrestin 2 to Promote the Proliferation and Migration of Glioblastoma Cells, and This Effect Was Blocked by Maackiain. Cancers, 14(16), 3890. https://doi.org/10.3390/cancers14163890