
CAR-NK Cells: A Chimeric Hope or a Promising Therapy?

 ,
,
Abstract
Simple Summary
Abstract
1. NK Cells’ Biology: A Plenitude of Receptors and a Balance between Immunological Self-Tolerance and Immune Surveillance
2. Anti-Leukemic Effect of Allogenic NK Cells in Haploidentical Stem Cell Transplantation
3. CAR-T Immunotherapy
4. Immunotherapy with CAR-NK Cells
5. CAR-NK Cells’ Sources
5.1. NK-92 Cell Line
5.2. NK Cells Derived from the Umbilical Cord
5.3. NK Cells Derived from Peripheral Blood
5.4. NK Cells Derived from iPSCs
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Caligiuri, M.A. Human natural killer cells. Blood J. Am. Soc. Hematol. 2008, 112, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Vacca, P.; Del Zotto, G.; Munari, E.; Mingari, M.C.; Moretta, L. Human NK cells: Surface receptors, inhibitory checkpoints, and translational applications. Cell. Mol. Immunol. 2019, 16, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Pahl, J.; Cerwenka, A. Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 2017, 222, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H. Missing self recognition and self tolerance of natural killer (NK) cells. Semin. Immunol. 2006, 18, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Lanier, L.L. Natural Killer Cell Licensing During Viral Infection. In Crossroads between Innate and Adaptive Immunity III; Springer: New York, NY, USA, 2011; pp. 37–44. [Google Scholar] [CrossRef]
- Passweg, J.R.; Baldomero, H.; Chabannon, C.; Basak, G.W.; de la Cámara, R.; Corbacioglu, S.; Dolstra, H.; Duarte, R.; Glass, B.; Greco, R.; et al. Hematopoietic cell transplantation and cellular therapy survey of the EBMT: Monitoring of activities and trends over 30 years. Bone Marrow Transpl. 2021, 56, 1651–1664. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of Donor Natural Killer Cell Alloreactivity in Mismatched Hematopoietic Transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef]
- Nguyen, S.; Béziat, V.; Roos-Weil, D.; Vieillard, V. Role of natural killer cells in hematopoietic stem cell transplanta-tion: Myth or reality? J. Innate Immun. 2011, 3, 383–394. [Google Scholar] [CrossRef]
- Nguyen, S.; Achour, A.; Souchet, L.; Vigouroux, S.; Chevallier, P.; Furst, S.; Sirvent, A.; Bay, J.-O.; Socié, G.; Ceballos, C.; et al. Clinical impact of NK-cell reconstitution after reduced intensity conditioned unrelated cord blood transplantation in patients with acute myeloid leukemia: Analysis of a prospective phase II multicenter trial on behalf of the Société Française de Greffe de Moelle Osseuse et Thérapie Cellulaire and Eurocord. Bone Marrow Transpl. 2017, 52, 1428–1435. [Google Scholar] [CrossRef]
- Nguyen, S.; Dhedin, N.; Vernant, J.-P.; Kuentz, M.; Al Jijakli, A.; Rouas-Freiss, N.; Carosella, E.D.; Boudifa, A.; Debré, P.; Vieillard, V. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: Immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood 2005, 105, 4135–4142. [Google Scholar] [CrossRef]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef]
- Cooley, S.; Parham, P.; Miller, J.S. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood 2018, 131, 1053–1062. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated with High Serum Interleu-kin-15 Levels. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Turtle, C.J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable Molecular Remissions in Chronic Lymphocytic Leukemia Treated with CD19-Specific Chimeric Antigen Recep-tor-Modified T Cells After Failure of Ibrutinib. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2017, 35, 3010–3020. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (Transcend NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Locke, F.L.; Ghobadi, A.; Jacobson, C.A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): A single-arm, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 31–42. [Google Scholar] [CrossRef]
- Locke, F.L.; Miklos, D.B.; Jacobson, C.; Perales, M.-A.; Kersten, M.J.; Oluwole, O.O.; Ghobadi, A.; Rapoport, A.P.; McGuirk, J.P.; Pagel, J.M.; et al. Primary Analysis of ZUMA-7: A Phase 3 Randomized Trial of Axicabtagene Ciloleucel (Axi-Cel) Versus Standard-of-Care Therapy in Patients with Relapsed/Refractory Large B-Cell Lymphoma. Blood 2021, 138, 2. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, Ö.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.; Locke, F.L.; Ghobadi, A.; Miklos, D.B.; Lekakis, L.J.; Oluwole, O.O.; Lin, Y.; Braunschweig, I.; Hill, B.T.; Timmerman, J.M.; et al. Long-Term Survival and Gradual Recovery of B Cells in Patients with Refractory Large B Cell Lymphoma Treated with Axicabtagene Ciloleucel (Axi-Cel). Blood 2020, 136, 40–42. [Google Scholar] [CrossRef]
- Pfefferle, A.; Huntington, N.D. You Have Got a Fast CAR: Chimeric Antigen Receptor NK Cells in Cancer Therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.; Mufti, G.; Poirot, L. ‘Off-the-shelf’allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Vasic, D.; Lee, J.B.; Leung, Y.; Khatri, I.; Na, Y.; Abate-Daga, D.; Zhang, L. Allogeneic double-negative CAR-T cells inhibit tumor growth without off-tumor toxicities. Sci. Immunol. 2022, 7, eabl3642. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. In Hematology 2014: The American Society of Hematology Education Program Book; American Society of Hematology: Washington, DC, USA, 2020; pp. 570–578. [Google Scholar]
- Myers, J.A.; Miller, J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021, 18, 85–100. [Google Scholar] [CrossRef]
- Geller, M.A.; Miller, J.S. Use of allogeneic NK cells for cancer immunotherapy. Immunotherapy 2011, 3, 1445–1459. [Google Scholar] [CrossRef]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Front. Immunol. 2021, 11, 61163. [Google Scholar] [CrossRef]
- Allan, D.S.; Chakraborty, M.; Waller, G.C.; Hochman, M.J.; Poolcharoen, A.; Reger, R.N.; Childs, R.W. Systematic improvements in lentiviral transduction of primary human natural killer cells undergoing ex vivo expansion. Mol. Ther.—Methods Clin. Dev. 2021, 20, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C. NK Cells in the Tumor Microenvironment. In Tumor Microenvironment; Springer: Cham, Swizerland, 2020; Volume 1273, pp. 69–90. [Google Scholar] [CrossRef]
- Kaweme, N.M.; Zhou, F. Optimizing NK Cell-Based Immunotherapy in Myeloid Leukemia: Abrogating an Immunosuppressive Microenvironment. Front. Immunol. 2021, 12, 2348. [Google Scholar] [CrossRef] [PubMed]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Louis, C.U.; Savoldo, B.; Dakhova, O.; Durett, A.; Grilley, B.; Liu, H.; Wu, M.F.; Mei, Z.; Gee, A.; et al. CAR T Cells Administered in Combination with Lymphodepletion and PD-1 Inhibition to Patients with Neuroblastoma. Mol. Ther. 2017, 25, 2214–2224. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wu, L.; Yin, L.; Shi, H.; Gu, Y.; Xing, N. Combined treatment with anti-PSMA CAR NK-92 cell and an-ti-PD-L1 monoclonal antibody enhances the antitumour efficacy against castration-resistant prostate cancer. Clin. Transl. Med. 2022, 12, e901. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W.; et al. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083. [Google Scholar]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Tran-fection of chimeric an-ti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K.; et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef]
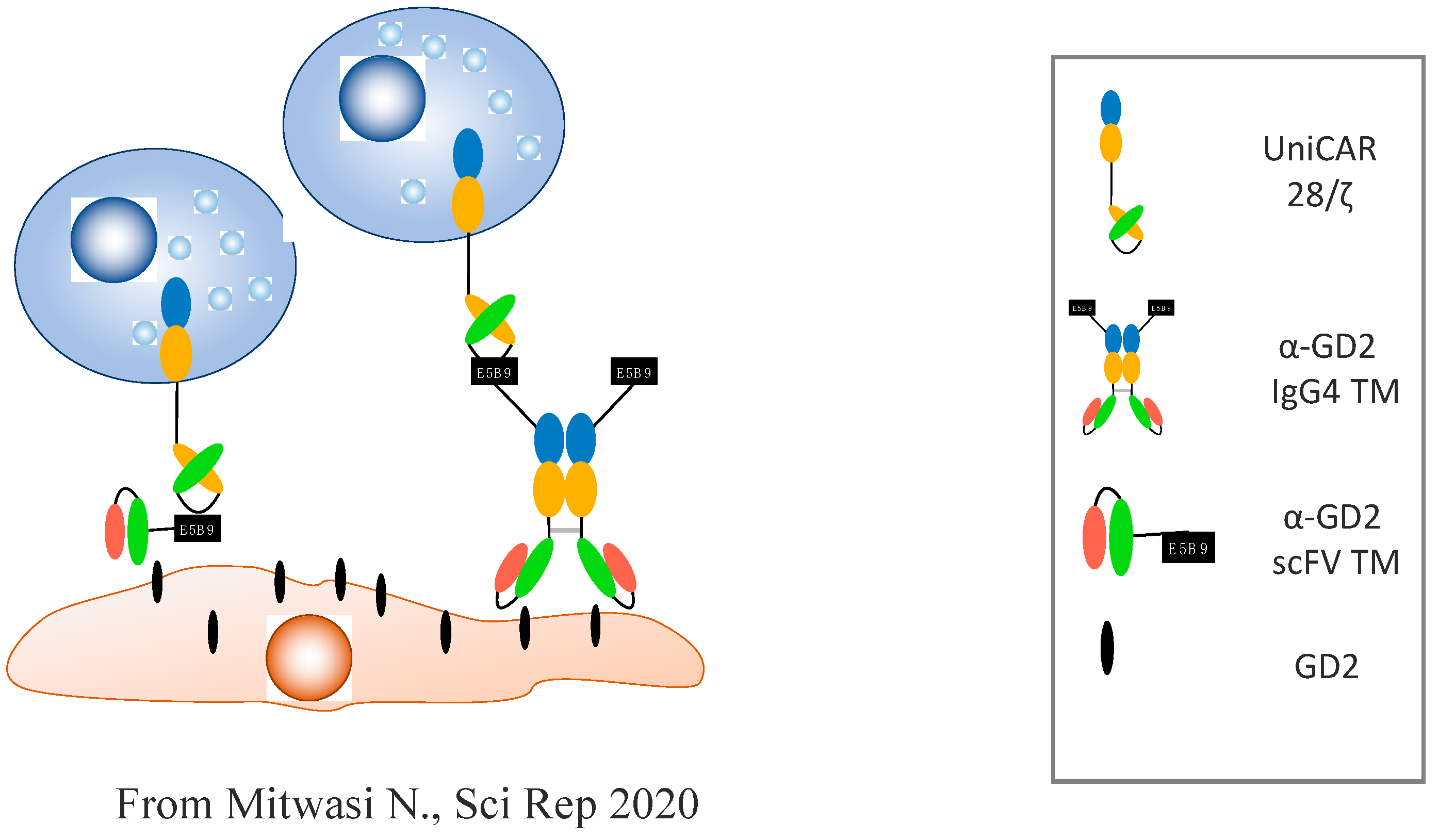
- Mitwasi, N.; Feldmann, A.; Arndt, C.; Koristka, S.; Berndt, N.; Jureczek, J.; Loureiro, L.R.; Bergmann, R.; Máthé, D.; Hegedüs, N. “UniCAR”-modified off-the-shelf NK-92 cells for targeting of GD2-expressing tumour cells. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Béziat, V.; Nguyen, S.; Lapusan, S.; Hervier, B.; Dhedin, N.; Bories, D.; Uzunov, M.; Boudifa, A.; Trebeden-Negre, H.; Norol, F.; et al. Fully functional NK cells after unrelated cord blood transplantation. Leukemia 2009, 23, 721–728. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
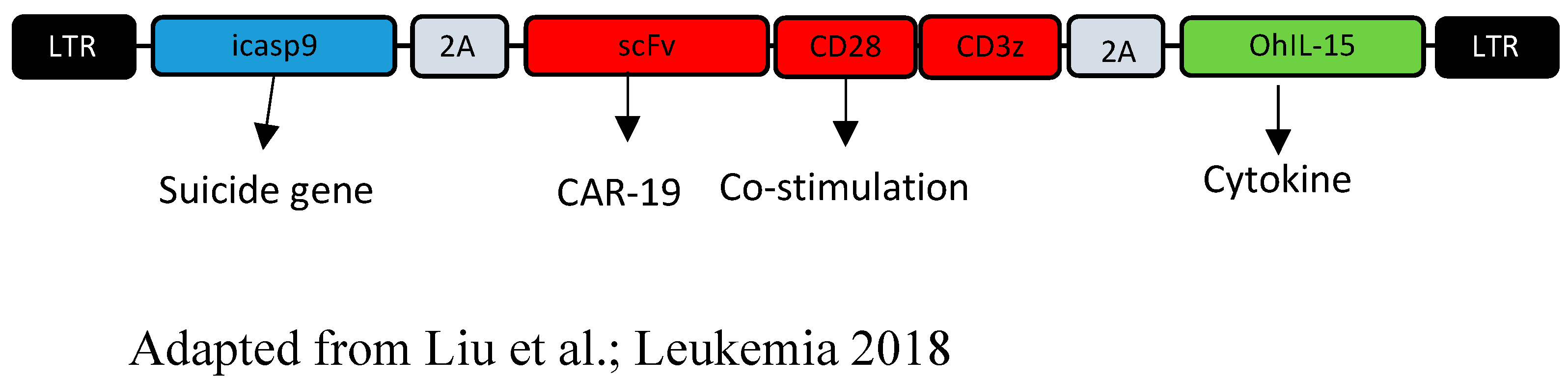
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Ciurea, S.O.; Schafer, J.R.; Bassett, R.; Denman, C.J.; Cao, K.; Willis, D.; Rondon, G.; Chen, J.; Soebbing, D.; Kaur, I.; et al. Phase 1 clinical trial using mbIL21 ex vivo–expanded donor-derived NK cells after haploidentical transplantation. Blood 2017, 130, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; DeFor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Björklund, A.T.; Carlsten, M.; Sohlberg, E.; Liu, L.L.; Clancy, T.; Karimi, M.; Cooley, S.; Miller, J.S.; Klimkowska, M.; Schaffer, M.; et al. Complete Remission with Reduction of High-Risk Clones following Haploidentical NK-Cell Therapy against MDS and AML. Clin. Cancer Res. 2018, 24, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Trebeden-Negre, H.; Vieillard, V.; Rosenzwajg, M.; Garderet, L.; Cherai, M.; Nguyen-Quoc, S.; Tanguy, M.; Norol, F. Polyvalent immunoglobulins, platelet lysate and lenalidomide: Cocktail for polyfunctional NK cells expansion for multiple myeloma. Bone Marrow Transplant. 2017, 52, 480–483. [Google Scholar] [CrossRef][Green Version]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-modified memory-like NK cells exhibit potent responses to NK-resistant lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Romee, R.; Schneider, S.E.; Leong, J.W.; Chase, J.M.; Keppel, C.R.; Sullivan, R.P.; Cooper, M.A.; Fehniger, T.A. Cytokine activation induces human memory-like NK cells. Blood J. Am. Soc. Hematol. 2012, 120, 4751–4760. [Google Scholar] [CrossRef]
- Berrien-Elliott, M.; Wagner, J.A.; Fehniger, T.A. Human Cytokine-Induced Memory-Like Natural Killer Cells. J. Innate Immun. 2015, 7, 563–571. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef]
- Likhite, S.; Elmas, E.; Yamamoto, K.; Schwartz, M.; Sorathia, K.; Pereira, M.; Devine, R.; Lyberger, J.; Behbehani, G.; Chakravarti, N. CRISPR-Targeted CAR Gene Insertion Using Cas9/RNP and AAV6 Enhances Anti-AML Activity of Primary NK Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Yamanaka, S. Pluripotent Stem Cell-Based Cell Therapy—Promise and Challenges. Cell Stem Cell 2020, 27, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.-J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, J.P.; Mahmood, S.; Zhu, H.; Gaidarova, M.S.; Blum, R.; Bjordahl, R.; Cichocki, F.; Chu, H.-Y.; Bonello, G.; Lee, T.; et al. FT596: Translation of First-of-Kind Multi-Antigen Targeted Off-the-Shelf CAR-NK Cell with Engineered Persistence for the Treatment of B Cell Malignancies. Blood 2019, 134, 301. [Google Scholar] [CrossRef]
- Bachanova, V.; Cayci, Z.; Lewis, D.; Maakaron, J.E.; Janakiram, M.; Bartz, A.; Payne, S.; Wong, C.; Cooley, S.; Valamehr, B. Initial clinical activity of FT596, a first-in-class, multi-antigen targeted, off-the-shelf, iPSC-derived CD19 CAR NK cell therapy in relapsed/refractory B-cell lymphoma. Blood 2020, 136, 8. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autologous CAR-T | Allogeneic CAR-NK | ||
|---|---|---|---|
| Advantage | Disadvantage | Advantage | Disadvantage |
| Better expansion | Preparation time | “off the shelf” and immediate availability | Short lifespan |
| Better persistence | Higher cost; 1 patient = 1 product | Lower cost; 1 source = many products | Difficult to modify, especially with frozen cells |
| Autologous setting (the patient is their own donor) | Variable cell quality | Multiple allogeneic sources (PBMC, UCB units, NK92 cell line, iPSC) | If derived from immature cells (UCB/iPSC) with a CD56 bright, KIR neg, NKG2A+, and low CD16, meaning proliferative but less cytotoxic and, thus, the need for further manipulation; if derived from tumor cell lines, need to be irradiated |
| Easier modification | Toxicity (CRS, neurotoxicity, persistent cytopenia) | No GVHD, good safety | If derived from PBMCs (CD56dim, CD16+, KIR+), a need for a specific haploidentical donor and promotion of proliferation. The criteria of choice of the donor are to be defined (interest of the mismatch KIR ligand?) |
| T-cell senescence (high tumor burden, consistent antigen stimulation) | Superior quality and product homogeneity | Inhibition by inhibitory receptors and the need of further modification to surpass it (HLA-based donor or addition of a cytokine/cytokine receptor fusion protein) | |
| Distinct manufacturing methods with heterogeneous products dependent on the cells and the collection, the patient | Possible expansion to produce many batches from a single source | Risk of immune rejection | |
| Proven clinical efficacy in B-cell hemopathies and multiple myeloma | Three mechanisms of action: classical cytotoxicity via the CAR and ADCC (with the possibility to target a second antigen, limiting the risk of relapse by antigen loss), cytokine pathways (IL-15) | Preclinical observations | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabbah, M.; Jondreville, L.; Lacan, C.; Norol, F.; Vieillard, V.; Roos-Weil, D.; Nguyen, S. CAR-NK Cells: A Chimeric Hope or a Promising Therapy? Cancers 2022, 14, 3839. https://doi.org/10.3390/cancers14153839
Sabbah M, Jondreville L, Lacan C, Norol F, Vieillard V, Roos-Weil D, Nguyen S. CAR-NK Cells: A Chimeric Hope or a Promising Therapy? Cancers. 2022; 14(15):3839. https://doi.org/10.3390/cancers14153839
Chicago/Turabian StyleSabbah, Mohamad, Ludovic Jondreville, Claire Lacan, Francoise Norol, Vincent Vieillard, Damien Roos-Weil, and Stéphanie Nguyen. 2022. "CAR-NK Cells: A Chimeric Hope or a Promising Therapy?" Cancers 14, no. 15: 3839. https://doi.org/10.3390/cancers14153839
APA StyleSabbah, M., Jondreville, L., Lacan, C., Norol, F., Vieillard, V., Roos-Weil, D., & Nguyen, S. (2022). CAR-NK Cells: A Chimeric Hope or a Promising Therapy? Cancers, 14(15), 3839. https://doi.org/10.3390/cancers14153839