
The Role of PTEN in Epithelial–Mesenchymal Transition



Abstract
Simple Summary
Abstract
1. Introduction
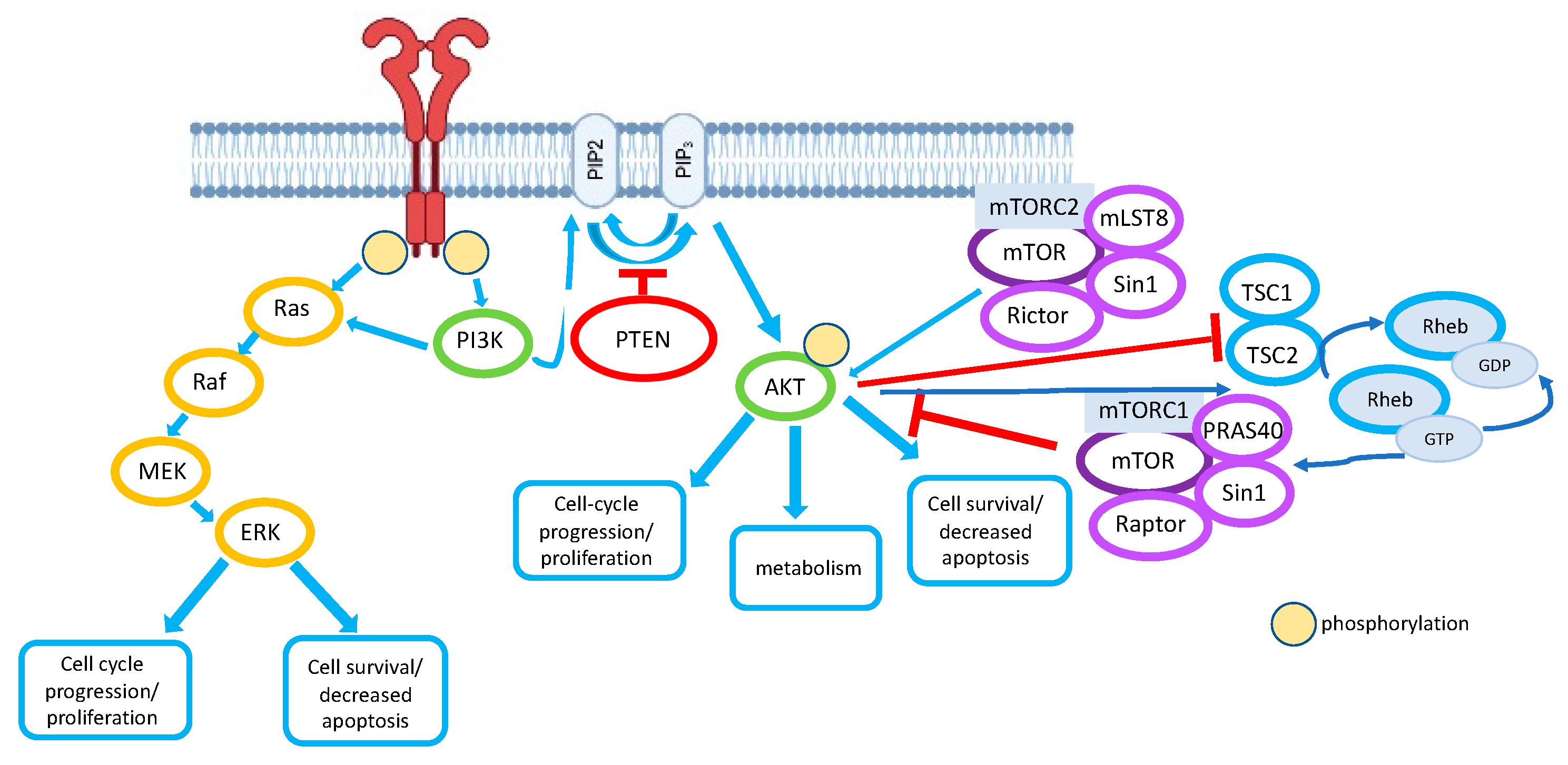
2. PI3K Pathway and PTEN
2.1. The PI3K Pathway
2.2. PTEN
3. PTEN and EMT
3.1. PTEN and EMT Transcription Factors
3.2. PTEN and Tumor Microenviroment
3.3. TGF-β and PTEN
3.4. NOTCH and PTEN
3.5. NF-κB and PTEN
3.6. Integrins and PTEN
3.7. PTEN and Some Scaffolding Proteins
4. miRNA and PTEN
5. lncRNA and PTEN
6. Conclusions
- -
- Does complete ablation of PTEN or loss of its enzymatic function activate EMT on its own, or does this process require additional signaling?
- -
- Is PTEN required for the reverse process of EMT, mesenchymal-to-epithelial transition?
- -
- Can PTEN be a therapeutic target for the treatment of metastatic cancer?
Author Contributions
Funding
Conflicts of Interest
References
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta 2018, 1871, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial-mesenchymal transition-activating transcription factors-multifunctional regulators in cancer. World J. Stem Cells 2013, 5, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Chaffer, C.L.; San Juan, B.P.; Lim, E.; Weinberg, R.A. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016, 35, 645–654. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Inoue, A.; Seidel, M.G.; Wu, W.; Kamizono, S.; Ferrando, A.A.; Bronson, R.T.; Iwasaki, H.; Akashi, K.; Morimoto, A.; Hitzler, J.K.; et al. Slug, a highly conserved zinc finger transcriptional repressor, protects hematopoietic progenitor cells from radiation-induced apoptosis in vivo. Cancer Cell 2002, 2, 279–288. [Google Scholar] [CrossRef]
- Olmeda, D.; Moreno-Bueno, G.; Flores, J.M.; Fabra, A.; Portillo, F.; Cano, A. SNAI1 Is Required for Tumor Growth and Lymph Node Metastasis of Human Breast Carcinoma MDA-MB-231 Cells. Cancer Res. 2007, 67, 11721–11731. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2018, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer Stem Cells: The Promise and the Potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomolecules 2019, 9, 153. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-Surface Receptors Transactivation Mediated by G Protein-Coupled Receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef]
- Fruman, D.A.; Meyers, R.E.; Cantley, L.C. PHOSPHOINOSITIDE KINASES. Annu. Rev. Biochem. 1998, 67, 481–507. [Google Scholar] [CrossRef]
- Dbouk, H.A.; Backer, J.M. A beta version of life: p110β takes center stage. Oncotarget 2010, 1, 729–733. [Google Scholar] [CrossRef][Green Version]
- Fox, M.; Mott, H.R.; Owen, D. Class IA PI3K regulatory subunits: p110-independent roles and structures. Biochem. Soc. Trans. 2020, 48, 1397–1417. [Google Scholar] [CrossRef]
- Koch, P.A.; Dornan, G.L.; Hessenberger, M.; Haucke, V. The molecular mechanisms mediating class II PI 3-kinase function in cell physiology. FEBS J. 2021, 288, 7025–7042. [Google Scholar] [CrossRef] [PubMed]
- Vara, J.Á.F.; Casado, E.; De Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Guo, H.; German, P.; Bai, S.; Barnes, S.; Guo, W.; Qi, X.; Lou, H.; Liang, J.; Jonasch, E.; Mills, G.B.; et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J. Genet. Genom. 2015, 42, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.F.; Jakubowicz, T.; Hemmings, B.A. Molecular cloning of a second form of rac protein kinase. Cell Regul. 1991, 2, 1001–1009. [Google Scholar] [CrossRef][Green Version]
- Jones, P.F.; Jakubowicz, T.; Pitossi, F.J.; Maurer, F.A.; Hemmings, B. Molecular cloning and identification of a serine/threonine protein kinase of the second-messenger subfamily. Proc. Natl. Acad. Sci. USA 1991, 88, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Masure, S.; Haefner, B.; Wesselink, J.-J.; Hoefnagel, E.; Mortier, E.; Verhasselt, P.; Tuytelaars, A.; Gordon, R.; Richardson, A. Molecular cloning, expression and characterization of the human serine/threonine kinase Akt-3. JBIC J. Biol. Inorg. Chem. 1999, 265, 353–360. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell. Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Coffer, P.J.; Jin, J.; Woodgett, J. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998, 335, 1–13. [Google Scholar] [CrossRef]
- Scheid, M.P.; Woodgett, J.R. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003, 546, 108–112. [Google Scholar] [CrossRef]
- Scheid, M.P.; Woodgett, J.R. PKB/AKT: Functional insights from genetic models. Nat. Rev. Mol. Cell Biol. 2001, 2, 760–768. [Google Scholar] [CrossRef]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Delcommenne, M.; Tan, C.; Gray, V.; Rue, L.; Woodgett, J.; Dedhar, S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, K.M.; Anderson, N.G. The protein kinase B/Akt signalling pathway in human malignancy. Cell. Signal. 2002, 14, 381–395. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Ebner, M.; Lučić, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P 3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell 2017, 65, 416–431. [Google Scholar] [CrossRef]
- Girardi, C.; James, P.; Zanin, S.; Pinna, L.A.; Ruzzene, M. Differential phosphorylation of Akt1 and Akt2 by protein kinase CK2 may account for isoform specific functions. Biochim. Biophys. Acta 2014, 1843, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Di Maira, G.; Salvi, M.; Arrigoni, G.; Marin, O.; Sarno, S.; Brustolon, F.A.; Pinna, L.; Ruzzene, M. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ. 2005, 12, 668–677. [Google Scholar] [CrossRef]
- Liu, P.; Begley, M.J.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef]
- Liu, R.; Chen, Y.; Liu, G.; Li, C.; Song, Y.; Cao, Z.; Li, W.; Hu, J.; Lu, C.; Liu, Y. PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis. 2020, 11, 797. [Google Scholar] [CrossRef]
- Zhang, X.; Tang, N.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Burgering, B.M.T.; Medema, R.H. Decisions on life and death: FOXO Forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukoc. Biol. 2003, 73, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; de Mattos, S.F.; van der Horst, A.; Klompmaker, R.; Kops, G.J.P.L.; Lam, E.W.-F.; Burgering, B.M.T.; Medema, R.H. Cell Cycle Inhibition by FoxO Forkhead Transcription Factors Involves Downregulation of Cyclin D. Mol. Cell. Biol. 2002, 22, 7842–7852. [Google Scholar] [CrossRef]
- Ciechomska, I.; Pyrzynska, B.; Kazmierczak, P.; Kaminska, B. Inhibition of Akt kinase signalling and activation of Forkhead are indispensable for upregulation of FasL expression in apoptosis of glioma cells. Oncogene 2003, 22, 7617–7627. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted Roles of GSK-3 in Cancer and Autophagy-Related Diseases. Oxidative Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and -independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Paluch, B.E.; Wang, X.; Jiang, X. PTEN at a glance. J. Cell Sci. 2012, 125, 4687–4692. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Trifault, B.; Chastre, E.; Scott, M.G.H. Posttranslational Regulation and Conformational Plasticity of PTEN. Cold Spring Harb. Perspect. Med. 2020, 10, a036095. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.L. Phosphatase and tensin homologue deleted on chromosome 10: Extending its PTENtacles. Int. J. Biochem. Cell Biol. 2009, 41, 757–761. [Google Scholar] [CrossRef]
- Molinari, F.; Frattini, M. Functions and Regulation of the PTEN Gene in Colorectal Cancer. Front. Oncol. 2014, 3, 326. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, T.; Thangada, S.; Wu, M.-T.; Kontos, C.D.; Wu, D.; Wu, H.; Hla, T. PTEN as an effector in the signaling of antimigratory G protein-coupled receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Lima-Fernandes, E.; Misticone, S.; Boularan, C.; Paradis, J.S.; Enslen, H.; Roux, P.P.; Bouvier, M.; Baillie, G.S.; Marullo, S.; Scott, M. A biosensor to monitor dynamic regulation and function of tumour suppressor PTEN in living cells. Nat. Commun. 2014, 5, 4431. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Y.; Wang, Y.; Chan, A.M. Multifaceted Regulation of PTEN Subcellular Distributions and Biological Functions. Cancers 2019, 11, 1247. [Google Scholar] [CrossRef]
- Maddika, S.; Kavela, S.; Rani, N.; Palicharla, V.R.; Pokorny, J.L.; Sarkaria, J.N.; Chen, J. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011, 13, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Deb, S.; Paul, I.; Chatterjee, A.; Mandal, T.; Chatterjee, U.; Ghosh, M.K. The Chaperone-assisted E3 Ligase C Terminus of Hsc70-interacting Protein (CHIP) Targets PTEN for Proteasomal Degradation. J. Biol. Chem. 2012, 287, 15996–16006. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.-S.; Jeong, M.-H.; Lee, H.-W.; Han, H.-J.; Ko, A.; Hewitt, S.M.; Kim, J.-H.; Chun, K.-H.; Chung, J.-Y.; Lee, C.; et al. PI3K/AKT activation induces PTEN ubiquitination and destabilization accelerating tumourigenesis. Nat. Commun. 2015, 6, 7769. [Google Scholar] [CrossRef]
- Van Themsche, C.; Leblanc, V.; Parent, S.; Asselin, E. X-linked Inhibitor of Apoptosis Protein (XIAP) Regulates PTEN Ubiquitination, Content, and Compartmentalization. J. Biol. Chem. 2009, 284, 20462–20466. [Google Scholar] [CrossRef]
- Wang, X.; Trotman, L.C.; Koppie, T.; Alimonti, A.; Chen, Z.; Gao, Z.; Wang, J.; Erdjument-Bromage, H.; Tempst, P.; Cordon-Cardo, C.; et al. NEDD4-1 Is a Proto-Oncogenic Ubiquitin Ligase for PTEN. Cell 2007, 128, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Wang, X.; Alimonti, A.; Chen, Z.; Teruya-Feldstein, J.; Yang, H.; Pavletich, N.P.; Carver, B.S.; Cordon-Cardo, C.; Erdjument-Bromage, H.; et al. Ubiquitination Regulates PTEN Nuclear Import and Tumor Suppression. Cell 2007, 128, 141–156. [Google Scholar] [CrossRef]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef]
- Minaguchi, T.; Waite, K.A.; Eng, C. Nuclear Localization of PTEN Is Regulated by Ca2+ through a Tyrosil Phosphorylation–Independent Conformational Modification in Major Vault Protein. Cancer Res. 2006, 66, 11677–11682. [Google Scholar] [CrossRef] [PubMed]
- Kavela, S.; Shinde, S.R.; Ratheesh, R.; Viswakalyan, K.; Bashyam, M.D.; Gowrishankar, S.; Vamsy, M.; Pattnaik, S.; Rao, S.; Sastry, R.A.; et al. PNUTS Functions as a Proto-Oncogene by Sequestering PTEN. Cancer Res. 2013, 73, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Cruise, E.S.; Dowling, R.J.O.; Stambolic, V. PTEN Nuclear Functions. Cold Spring Harb. Perspect. Med. 2019, 10, a036079. [Google Scholar] [CrossRef]
- Déléris, P.; Gayral, S.; Breton-Douillon, M. Nuclear Ptdlns(3,4,5)P3 signaling: An ongoing story. J. Cell. Biochem. 2006, 98, 469–485. [Google Scholar] [CrossRef]
- Neri, L.M.; Borgatti, P.; Capitani, S.; Martelli, A.M. The nuclear phosphoinositide 3-kinase/AKT pathway: A new second messenger system. Biochim. Biophys. Acta. 2002, 1584, 73–80. [Google Scholar] [CrossRef]
- Tang, Y.; Eng, C. PTEN Autoregulates Its Expression by Stabilization of p53 in a Phosphatase-Independent Manner. Cancer Res. 2006, 66, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Stambolic, V.; MacPherson, D.; Sas, D.; Lin, Y.; Snow, B.; Jang, Y.; Benchimol, S.; Mak, T.W. Regulation of PTEN Transcription by p53. Mol. Cell 2001, 8, 317–325. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, O.; Daks, A.; Petrova, V.; Petukhov, A.; Lezina, L.; Shuvalov, O.; Davidovich, P.; Kriger, D.; Lomert, E.; Tentler, D.; et al. Novel isatin-derived molecules activate p53 via interference with Mdm2 to promote apoptosis. Cell Cycle 2018, 17, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 affects genotoxic stress response via the Mdm2 axis. Oncotarget 2015, 6, 25843–25855. [Google Scholar] [CrossRef] [PubMed]
- Chibaya, L.; Karim, B.; Zhang, H.; Jones, S.N. Mdm2 phosphorylation by Akt regulates the p53 response to oxidative stress to promote cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2021, 118, e2003193118. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. The PTEN, Mdm2, p53 tumor suppressor–oncoprotein network. Trends Biochem. Sci. 2002, 27, 462–467. [Google Scholar] [CrossRef]
- Chung, J.-H.; Eng, C. Nuclear-Cytoplasmic Partitioning of Phosphatase and Tensin Homologue Deleted on Chromosome 10 (PTEN) Differentially Regulates the Cell Cycle and Apoptosis. Cancer Res. 2005, 65, 8096–8100. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Mao, Y.; Sullivan, K.F. Centromeres and Kinetochores: From Epigenetics to Mitotic Checkpoint Signaling. Cell 2003, 112, 407–421. [Google Scholar] [CrossRef]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guessous, F.; Kwon, S.; Kumar, M.; Ibidapo, O.; Fuller, L.; Johnson, E.; Lal, B.; Hussaini, I.; Bao, Y.; et al. PTEN Has Tumor-Promoting Properties in the Setting of Gain-of-Function p53 Mutations. Cancer Res. 2008, 68, 1723–1731. [Google Scholar] [CrossRef]
- Liu, Y.; Siles, L.; Lu, X.; Dean, K.C.; Cuatrecasas, M.; Postigo, A.; Dean, D.C. Mitotic polarization of transcription factors during asymmetric division establishes fate of forming cancer cells. Nat. Commun. 2018, 9, 2424. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Wan, G.; Yang, Y.; Liu, Y.; Yang, X.; Zheng, Y.; Jiang, L.; Zhang, P.; Liu, D.; Zhao, W.; et al. SLFN5 influences proliferation and apoptosis by upregulating PTEN transcription via ZEB1 and inhibits the purine metabolic pathway in breast cancer. Am. J. Cancer Res. 2020, 10, 2832–2850. [Google Scholar] [PubMed]
- Karreth, F.A.; Tay, Y.; Perna, D.; Ala, U.; Tan, S.M.; Rust, A.G.; DeNicola, G.; Webster, K.A.; Weiss, D.; Perez-Mancera, P.A.; et al. In Vivo Identification of Tumor-Suppressive PTEN ceRNAs in an Oncogenic BRAF-Induced Mouse Model of Melanoma. Cell 2011, 147, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Escrivà, M.; Peiró, S.; Herranz, N.; Villagrasa, P.; Dave, N.; Montserrat-Sentís, B.; Murray, S.A.; Francí, C.; Gridley, T.; Virtanen, I.; et al. Repression of PTEN Phosphatase by Snail1 Transcriptional Factor during Gamma Radiation-Induced Apoptosis. Mol. Cell. Biol. 2008, 28, 1528–1540. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Abramo, K.; Leikina, E.; Vary, C.; Liaw, L.; Wu, W.-S. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate 2015, 75, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Tafani, M.; Sansone, L.; Limana, F.; Arcangeli, T.; De Santis, E.; Polese, M.; Fini, M.; Russo, M.A. The Interplay of Reactive Oxygen Species, Hypoxia, Inflammation, and Sirtuins in Cancer Initiation and Progression. Oxidative Med. Cell. Longev. 2015, 2016, 3907147. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Idelchik, M.d.P.S.; Begley, U.; Begley, T.J.; Melendez, J.A. Mitochondrial ROS control of cancer. Proc. Semin. Cancer Biol. 2017, 47, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate hypoxic signaling. Curr. Opin. Cell Biol. 2009, 21, 894–899. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Dada, L.A.; Wu, M.; Kelly, A.; Trejo, H.; Zhou, Q.; Varga, J.; Sznajder, J.I. Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L1120–L1130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Park, J.; Han, S.-J.; Yang, S.Y.; Yoon, H.J.; Park, I.; Woo, H.A.; Lee, S.-R. Redox regulation of tumor suppressor PTEN in cell signaling. Redox Biol. 2020, 34, 101553. [Google Scholar] [CrossRef]
- Lefler, J.E.; Seward, C.; Ostrowski, M.C. PTEN in cancer associated fibroblasts. Adv. Cancer Res. 2022, 154, 203–226. [Google Scholar] [CrossRef]
- Wallace, J.A.; Li, F.; Leone, G.; Ostrowski, M.C. Pten in the Breast Tumor Microenvironment: Modeling Tumor–Stroma Coevolution. Cancer Res. 2011, 71, 1203–1207. [Google Scholar] [CrossRef]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.-L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming Growth Factor Beta Family: Insight into the Role of Growth Factors in Regulation of Fracture Healing Biology and Potential Clinical Applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef]
- Li, D.-M.; Sun, H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res. 1997, 57, 2124–2129. [Google Scholar] [PubMed]
- Kimbrough-Allah, M.N.; Millena, A.C.; Khan, S.A. Differential role of PTEN in transforming growth factor β (TGF-β) effects on proliferation and migration in prostate cancer cells. Prostate 2018, 78, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Quach, K.T.; Cabrera, B.L.; Cabral, J.A.; Beck, S.E.; Carethers, J.M. RAS/ERK modulates TGFbeta-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis 2007, 28, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.Y.; Cabral, J.A.; Chang, J.; Carethers, J.M. TGFβ modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol. Ther. 2008, 7, 1694–1699. [Google Scholar] [CrossRef]
- Eritja, N.; Felip, I.; Dosil, M.A.; Vigezzi, L.; Mirantes, C.; Yeramian, A.; Navaridas, R.; Santacana, M.; Llobet-Navas, D.; Yoshimura, A.; et al. A Smad3-PTEN regulatory loop controls proliferation and apoptotic responses to TGF-β in mouse endometrium. Cell Death Differ. 2017, 24, 1443–1458. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhen, Y.; Zhou, S.-F.; Lu, N. Posttranslational regulation of phosphatase and tensin homolog (PTEN) and its functional impact on cancer behaviors. Drug Des. Dev. Ther. 2014, 8, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, F.; Ramaswamy, S.; Nakamura, N.; Sellers, W.R. Phosphorylation of the PTEN Tail Regulates Protein Stability and Function. Mol. Cell. Biol. 2000, 20, 5010–5018. [Google Scholar] [CrossRef]
- Aoyama, D.; Hashimoto, N.; Sakamoto, K.; Kohnoh, T.; Kusunose, M.; Kimura, M.; Ogata, R.; Imaizumi, K.; Kawabe, T.; Hasegawa, Y. Involvement of TGFβ-Induced Phosphorylation of the PTEN C-Terminus on TGFβ-Induced Acquisition of Malignant Phenotypes in Lung Cancer Cells. PLoS ONE 2013, 8, e81133. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions Between β-Catenin and Transforming Growth Factor-β Signaling Pathways Mediate Epithelial-Mesenchymal Transition and Are Dependent on the Transcriptional Co-activator cAMP-response Element-binding Protein (CREB)-binding Protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef]
- Lam, A.P.; Herazo-Maya, J.D.; Sennello, J.A.; Flozak, A.S.; Russell, S.; Mutlu, G.M.; Budinger, G.R.S.; DasGupta, R.; Varga, J.; Kaminski, N.; et al. Wnt Coreceptor Lrp5 Is a Driver of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 185–195. [Google Scholar] [CrossRef]
- Tian, X.; Liu, Z.; Niu, B.; Zhang, J.; Tan, T.K.; Lee, S.R.; Zhao, Y.; Harris, D.C.H.; Zheng, G. E-Cadherin/β-Catenin Complex and the Epithelial Barrier. J. Biomed. Biotechnol. 2011, 2011, 567305. [Google Scholar] [CrossRef]
- Masszi, A.; Fan, L.; Rosivall, L.; McCulloch, C.A.; Rotstein, O.D.; Mucsi, I.; Kapus, A. Integrity of cell-cell contacts is a critical regulator of TGF-β1-induced epithelial-to-myofibroblast transition: Role for β-catenin. Am. J. Pathol. 2004, 165, 1955–1967. [Google Scholar] [CrossRef]
- Wang, M.M. Notch signaling and Notch signaling modifiers. Int. J. Biochem. Cell Biol. 2011, 43, 1550–1562. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.; Stanners, S.R.; Yong, R.; Tang, O.; Pollock, C.A. Notch mediated epithelial to mesenchymal transformation is associated with increased expression of the Snail transcription factor. Int. J. Biochem. Cell Biol. 2010, 42, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Zhang, L.; He, C.-S.; Xu, F.; Liu, J.-L.; Hu, Z.-H.; Zhao, L.-P.; Tian, Y. Activation of Notch-1 enhances epithelial-mesenchymal transition in gefitinib-acquired resistant lung cancer cells. J. Cell. Biochem. 2011, 113, 1501–1513. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, X.; Luo, J.; Xiao, W.; Ye, X.; Chen, M.; Li, Y.; Zhang, G.-J. Notch3 inhibits epithelial–mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis 2016, 5, e269. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Q.; Liang, Y.-K.; Wu, Y.; Chen, M.; Chen, W.-L.; Li, R.-H.; Zeng, Y.-Z.; Huang, W.-H.; Wu, J.-D.; Zeng, D.; et al. Notch3 inhibits cell proliferation and tumorigenesis and predicts better prognosis in breast cancer through transactivating PTEN. Cell Death Dis. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.-I.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Huang, W.-X.; Zhou, X.; Chen, J.; Li, Z. Numb inhibits epithelial-mesenchymal transition via RBP-Jκ-dependent Notch1/PTEN/FAK signaling pathway in tongue cancer. BMC Cancer 2019, 19, 391. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.; Wyatt, D.; Bocchetta, M.; Li, J.; Filipovic, A.; Green, A.; Peiffer, D.S.; Fuqua, S.; Miele, L.; Albain, K.S.; et al. Notch-1-PTEN-ERK1/2 signaling axis promotes HER2+ breast cancer cell proliferation and stem cell survival. Oncogene 2018, 37, 4489–4504. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Pires, B.R.B.; Mencalha, A.L.; Ferreira, G.M.; De Souza, W.F.; Morgado-Díaz, J.A.; Maia, A.M.; Corrêa, S.; Abdelhay, E.S.F.W. NF-kappaB Is Involved in the Regulation of EMT Genes in Breast Cancer Cells. PLoS ONE 2017, 12, e0169622. [Google Scholar] [CrossRef]
- Neil, J.R.; Schiemann, W.P. Altered TAB1:I κB kinase interaction promotes transforming growth factor beta-mediated nuclear factor-kappaB activation during breast cancer progression. Cancer Res. 2008, 68, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Pantuck, A.J.; An, J.; Liu, H.; Rettig, M.B. NF-κB–Dependent Plasticity of the Epithelial to Mesenchymal Transition Induced by Von Hippel-Lindau Inactivation in Renal Cell Carcinomas. Cancer Res. 2010, 70, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Helfand, B.T.; Jang, T.L.; Zhu, L.J.; Chen, L.; Yang, X.J.; Kozlowski, J.; Smith, N.; Kundu, S.D.; Yang, G.; et al. Nuclear Factor-κB-Mediated Transforming Growth Factor-β-Induced Expression of Vimentin Is an Independent Predictor of Biochemical Recurrence after Radical Prostatectomy. Clin. Cancer Res. 2009, 15, 3557–3567. [Google Scholar] [CrossRef]
- Lin, K.; Baritaki, S.; Militello, L.; Malaponte, G.; Bevelacqua, Y.; Bonavida, B. The Role of B-RAF Mutations in Melanoma and the Induction of EMT via Dysregulation of the NF-κB/Snail/RKIP/PTEN Circuit. Genes Cancer 2010, 1, 409–420. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Zhu, Y. A-kinase-interacting protein 1 promotes EMT and metastasis via PI3K/Akt/IKKβ pathway in cervical cancer. Cell Biochem. Funct. 2020, 38, 782–791. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, X.; Wang, G.; Lan, Z.; Liao, W.; Li, J.; Liang, X.; Chen, J.R.; Shah, S.; Shang, X.; et al. Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 2017, 542, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res. 2009, 339, 269–280. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin Signaling. Science 1999, 285, 1028–1033. [Google Scholar] [CrossRef]
- Taherian, A.; Li, X.; Liu, Y.; Haas, T.A. Differences in integrin expression and signaling within human breast cancer cells. BMC Cancer 2011, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Modzelewska, K.; Kwong, L.; Keely, P.J. Focal adhesion regulation of cell behavior. Biochim. Biophys. Acta 2004, 1692, 103–119. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Tran, H.; Yamada, K.M. PTEN Gene and Integrin Signaling in Cancer. JNCI J. Natl. Cancer Inst. 1999, 91, 1820–1828. [Google Scholar] [CrossRef] [PubMed]
- Perumal, E.; Youn, K.S.; Sun, S.; Seung-Hyun, J.; Suji, M.; Jieying, L.; Yeun-Jun, C. PTEN inactivation induces epithelial-mesenchymal transition and metastasis by intranuclear translocation of β-catenin and snail/slug in non-small cell lung carcinoma cells. Lung Cancer 2019, 130, 25–34. [Google Scholar] [CrossRef]
- Tzenaki, N.; Aivaliotis, M.; Papakonstanti, E.A. Focal adhesion kinase phosphorylates the phosphatase and tensin homolog deleted on chromosome 10 under the control of p110δ phosphoinositide-3 kinase. FASEB J. 2015, 29, 4840–4852. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; del Río Hernández, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [PubMed]
- López-Colomé, A.M.; Lee-Rivera, I.; Benavides-Hidalgo, R.; López, E. Paxillin: A crossroad in pathological cell migration. J. Hematol. Oncol. 2017, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Haier, J.; Nicolson, G.L. PTEN regulates tumor cell adhesion of colon carcinoma cells under dynamic conditions of fluid flow. Oncogene 2002, 21, 1450–1460. [Google Scholar] [CrossRef]
- Kotelevets, L.; Van Hengel, J.; Bruyneel, E.; Mareel, M.; Van Roy, F.; Chastre, E. The lipid phosphatase activity of PTEN is critical for stabilizing intercellular junctions and reverting invasiveness. J. Cell Biol. 2001, 155, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Kotelevets, L.; Van Hengel, J.; Bruyneel, E.; Mareel, M.; Van Roy, F.; Chastre, E. Implication of the MAGI-1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J. 2004, 19, 115–117. [Google Scholar] [CrossRef]
- Gou, H.; Liang, J.Q.; Zhang, L.; Chen, H.; Zhang, Y.; Li, R.; Wang, X.; Ji, J.; Tong, J.H.; To, K.-F.; et al. TTPAL Promotes Colorectal Tumorigenesis by Stabilizing TRIP6 to Activate Wnt/β-Catenin Signaling. Cancer Res. 2019, 79, 3332–3346. [Google Scholar] [CrossRef] [PubMed]
- Chastre, E.; Abdessamad, M.; Kruglov, A.; Bruyneel, E.; Bracke, M.; Di Gioia, Y.; Beckerle, M.C.; Van Roy, F.; Kotelevets, L. TRIP6, a novel molecular partner of the MAGI-1 scaffolding molecule, promotes invasiveness. FASEB J. 2008, 23, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Javadi, A.; Deevi, R.K.; Evergren, E.; Blondel-Tepaz, E.; Baillie, G.S.; Scott, M.G.; Campbell, F.C. PTEN controls glandular morphogenesis through a juxtamembrane β-Arrestin1/ARHGAP21 scaffolding complex. eLife 2017, 6, e24578. [Google Scholar] [CrossRef] [PubMed]
- Lima-Fernandes, E.; Enslen, H.; Camand, E.; Kotelevets, L.; Boularan, C.; Achour, L.; Benmerah, A.; Gibson, L.C.D.; Baillie, G.S.; Pitcher, J.A.; et al. Distinct functional outputs of PTEN signalling are controlled by dynamic association with β-arrestins. EMBO J. 2011, 30, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Liu, J.; Chao, J.; Greer, P.A.; Li, S. PTEN dephosphorylates Abi1 to promote epithelial morphogenesis. J. Cell Biol. 2020, 219, e201910041. [Google Scholar] [CrossRef]
- Chen, B.; Brinkmann, K.; Chen, Z.; Pak, C.W.; Liao, Y.; Shi, S.; Henry, L.; Grishin, N.V.; Bogdan, S.; Rosen, M.K. The WAVE Regulatory Complex Links Diverse Receptors to the Actin Cytoskeleton. Cell 2014, 156, 195–207. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, J.; Chao, J.; Scheuerman, M.P.; Rahimi, S.A.; Lee, L.Y.; Li, S. PTEN suppresses epithelial–mesenchymal transition and cancer stem cell activity by downregulating Abi1. Sci. Rep. 2020, 10, 12685. [Google Scholar] [CrossRef]
- Annese, T.; Tamma, R.; De Giorgis, M.; Ribatti, D. microRNAs Biogenesis, Functions and Role in Tumor Angiogenesis. Front. Oncol. 2020, 10, 581007. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Abak, A.; Shoorei, H.; Mohaqiq, M.; Majidpoor, J.; Sayad, A.; Taheri, M. Regulatory role of microRNAs on PTEN signaling. Biomed. Pharmacother. 2020, 133, 110986. [Google Scholar] [CrossRef]
- Yuan, Y.; Liao, H.; Pu, Q.; Ke, X.; Hu, X.; Ma, Y.; Luo, X.; Jiang, Q.; Gong, Y.; Wu, M.; et al. miR-410 induces both epithelial–mesenchymal transition and radioresistance through activation of the PI3K/mTOR pathway in non-small cell lung cancer. Signal Transduct. Target. Ther. 2020, 5, 85. [Google Scholar] [CrossRef]
- Zhang, J.-G.; Wang, J.-J.; Zhao, F.; Liu, Q.; Jiang, K.; Yang, G.-H. MicroRNA-21 (miR-21) represses tumor suppressor PTEN and promotes growth and invasion in non-small cell lung cancer (NSCLC). Clin. Chim. Acta 2010, 411, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dong, Z.; Chen, Y.; Yang, L.; Lai, D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013, 46, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharmacother. 2016, 79, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Kia, V.; Beigli, M.S.; Hosseini, V.; Koochaki, A.; Paryan, M.; Mohammadi-Yeganeh, S. Is miR-144 an effective inhibitor of PTEN mRNA: A controversy in breast cancer. In Vitro Cell Dev. Biol. Anim. 2018, 54, 621–628. [Google Scholar] [CrossRef]
- Li, N.; Miao, Y.; Shan, Y.; Liu, B.; Li, Y.; Zhao, L.; Jia, L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017, 8, e2796. [Google Scholar] [CrossRef]
- Liu, T.; Guo, J.; Zhang, X. MiR-202-5p/PTEN mediates doxorubicin-resistance of breast cancer cells via PI3K/Akt signaling pathway. Cancer Biol. Ther. 2019, 20, 989–998. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Sasi, A.K.; Abak, A.; Shoorei, H.; Khoshkar, A.; Taheri, M. Contribution of miRNAs in the Pathogenesis of Breast Cancer. Front. Oncol. 2021, 11, 768949. [Google Scholar] [CrossRef]
- Liu, Z.; Liang, X.; Li, X.; Liu, X.; Zhu, M.; Gu, Y.; Zhou, P. MiRNA-21 functions in ionizing radiation-induced epithelium-to-mesenchymal transition (EMT) by downregulating PTEN. Toxicol. Res. 2019, 8, 328–340. [Google Scholar] [CrossRef]
- Guo, Y.; Yuan, X.; Hong, L.; Wang, Q.; Liu, S.; Li, Z.; Huang, L.; Jiang, S.; Shi, J. Promotor Hypomethylation Mediated Upregulation of miR-23b-3p Targets PTEN to Promote Bronchial Epithelial-Mesenchymal Transition in Chronic Asthma. Front. Immunol. 2022, 12, 771216. [Google Scholar] [CrossRef]
- Ouyang, L.; Liu, R.-D.; Lei, D.-Q.; Shang, Q.-C.; Li, H.-F.; Hu, X.-G.; Zheng, H.; Jin, G. MiR-499a-5p promotes 5-FU resistance and the cell proliferation and migration through activating PI3K/Akt signaling by targeting PTEN in pancreatic cancer. Ann. Transl. Med. 2021, 9, 1798. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Q.; Liu, Y.; Wang, X.; Ma, C.; Zhu, W. LncRNA HOTAIR Promotes Chemoresistance by Facilitating Epithelial to Mesenchymal Transition through miR-29b/PTEN/PI3K Signaling in Cervical Cancer. Cells Tissues Organs 2021, 211, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Gurbuz, V.; Sozen, S.; Bilen, C.Y.; Konac, E. miR-148a, miR-152 and miR-200b promote prostate cancer metastasis by targeting DNMT1 and PTEN expression. Oncol. Lett. 2021, 22, 805. [Google Scholar] [CrossRef]
- Qian, X.-L.; Zhou, F.; Xu, S.; Jiang, J.; Chen, Z.-P.; Wang, S.-K.; Zuo, Y.; Ni, C. MiR-454-3p Promotes Oxaliplatin Resistance by Targeting PTEN in Colorectal Cancer. Front. Oncol. 2021, 11, 638537. [Google Scholar] [CrossRef]
- Wu, Z.; Luo, J.; Huang, T.; Yi, R.; Ding, S.; Xie, C.; Xu, A.; Zeng, Y.; Wang, X.; Song, Y.; et al. MiR-4310 induced by SP1 targets PTEN to promote glioma progression. Cancer Cell Int. 2020, 20, 567. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, L.; Ding, X.; Sui, X. LINC00861 inhibits the progression of cervical cancer cells by functioning as a ceRNA for miR-513b-5p and regulating the PTEN/AKT/mTOR signaling pathway. Mol. Med. Rep. 2020, 23, 11662. [Google Scholar] [CrossRef]
- Cao, H.-L.; Gu, M.-Q.; Sun, Z.; Chen, Z.-J. miR-144-3p Contributes to the Development of Thyroid Tumors Through the PTEN/PI3K/AKT Pathway. Cancer Manag. Res. 2020, 12, 9845–9855. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, G.-X.; Che, L.-S.; Shi, S.-H.; Lin, W.-Y. miR-19 promotes development of renal fibrosis by targeting PTEN-mediated epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 2020, 13, 642–654. [Google Scholar] [PubMed]
- Zhang, J.; Chen, D.; Liang, S.; Wang, J.; Liu, C.; Nie, C.; Shan, Z.; Wang, L.; Fan, Q.; Wang, F. miR-106b promotes cell invasion and metastasis via PTEN mediated EMT in ESCC. Oncol. Lett. 2018, 15, 4619–4626. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Yan, M.-D.; Wu, A.T.; Yuan, K.S.-P.; Liu, S.H. Brown Seaweed Fucoidan Inhibits Cancer Progression by Dual Regulation of mir-29c/ADAM12 and miR-17-5p/PTEN Axes in Human Breast Cancer Cells. J. Cancer 2016, 7, 2408–2419. [Google Scholar] [CrossRef]
- Zhu, L.; Zhang, S.; Chen, S.; Wu, H.; Jiang, M.; Liu, A. Exosomal miR-552-5p promotes tumorigenesis and disease progression via the PTEN/TOB1 axis in gastric cancer. J. Cancer 2022, 13, 890–905. [Google Scholar] [CrossRef]
- Yang, Y.; Ban, D.; Zhang, C.; Shen, L. Downregulation of circ_0000673 Promotes Cell Proliferation and Migration in Endometriosis via the Mir-616-3p/PTEN Axis. Int. J. Med Sci. 2021, 18, 3506–3515. [Google Scholar] [CrossRef]
- Chang, S.; Chen, B.; Wang, X.; Wu, K.; Sun, Y. Long non-coding RNA XIST regulates PTEN expression by sponging miR-181a and promotes hepatocellular carcinoma progression. BMC Cancer 2017, 17, 248. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, L.; Li, G.; Ma, D.; Sun, C.; Gao, S.; Zhang, P.; Gao, F. miR-221 Promotes Epithelial-Mesenchymal Transition through Targeting PTEN and Forms a Positive Feedback Loop with β-catenin/c-Jun Signaling Pathway in Extra-Hepatic Cholangiocarcinoma. PLoS ONE 2015, 10, e0141168. [Google Scholar] [CrossRef] [PubMed]
- Yao, R.-W.; Wang, Y.; Chen, L.-L. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019, 21, 542–551. [Google Scholar] [CrossRef]
- Hu, Q.; Li, C.; Wang, S.; Li, Y.; Wen, B.; Zhang, Y.; Liang, K.; Yao, J.; Ye, Y.; Hsiao, H.; et al. LncRNAs-directed PTEN enzymatic switch governs epithelial–mesenchymal transition. Cell Res. 2019, 29, 286–304. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Wang, X.-Y.; Jin, Z.-L. Linc00702 inhibits cell growth and metastasis through regulating PTEN in colorectal cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3624–3632. [Google Scholar]
- Yu, W.; Li, D.; Ding, X.; Sun, Y.; Liu, Y.; Cong, J.; Yang, J.; Sun, J.; Ning, X.; Wang, H. LINC00702 suppresses proliferation and invasion in non-small cell lung cancer through regulating miR-510/PTEN axis. Aging 2019, 11, 1471. [Google Scholar] [CrossRef]
- Yan, J.; Huang, X.; Zhang, X.; Chen, Z.; Ye, C.; Xiang, W.; Huang, Z. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem. Biophys. Res. Commun. 2019, 521, 887–893. [Google Scholar] [CrossRef]
- Luongo, F.; Colonna, F.; Calapà, F.; Vitale, S.; Fiori, M.E.; De Maria, R. PTEN Tumor-Suppressor: The Dam of Stemness in Cancer. Cancers 2019, 11, 1076. [Google Scholar] [CrossRef]
- Fischer, T.; Hartmann, O.; Reissland, M.; Prieto-Garcia, C.; Klann, K.; Pahor, N.; Schülein-Völk, C.; Baluapuri, A.; Polat, B.; Abazari, A.; et al. PTEN mutant non-small cell lung cancer require ATM to suppress pro-apoptotic signalling and evade radiotherapy. Cell Biosci. 2022, 12, 50. [Google Scholar] [CrossRef]
- Rivas, J.D.L.; Brozovic, A.; Izraely, S.; Casas-Pais, A.; Witz, I.P.; Figueroa, A. Cancer drug resistance induced by EMT: Novel therapeutic strategies. Arch. Toxicol. 2021, 95, 2279–2297. [Google Scholar] [CrossRef] [PubMed]
- Song, K.-A.; Faber, A.C. Epithelial-to-mesenchymal transition and drug resistance: Transitioning away from death. J. Thorac. Dis. 2019, 11, E82–E85. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRs | Interaction with PTEN | Role in EMT Process |
|---|---|---|
| miR-21 | downregulation of PTEN | promotes EMT progression in lung epithelial cells [158] |
| miR-410 | downregulation of PTEN | activation of EMT in non-small cell lung cancer [150] |
| miR-106b and miR-93 | inhibits PTEN | promotes cell migration, invasion, and proliferation in vitro (in breast cancer cells) and tumor growth in vivo (breast cancer) [155] |
| miR-202-5p | downregulation of PTEN | increases DOX resistance and cell proliferation as well as inhibiting apoptosis (in breast cancer cells) [156] |
| miR-23b-3p | silence of PTEN | promotes EMT and migration in bronchial epithelium (lung) [159] |
| miR-499a-5p | decreases the expression levels of PTEN mRNA and protein | promotes 5-FU resistance and cell proliferation and migration in pancreatic cancer [160] |
| miR-29b | targeting the 3′-UTR of PTEN mRNA upregulation of PTEN and downregulation of PI3K in cervical cancer cells | decreases cell proliferation, migration, and invasion abilities of NSCLC cells [161] |
| miR-148a, miR-152 and miR-200b | downregulation of PTEN | presence of these miRs correlates with metastasis in patients with prostate cancer and metastatic prostate cancer [162] |
| miR-454-3p | suppresses PTEN | promotes oxaliplatin resistance and inhibits apoptosis in Colorectal cancer cell line [163] |
| miR-4310 | suppresses PTEN SP1 activates miR-4310 gene expression | promotes proliferation, migration, and invasion in glioma tissues [164] |
| miR-513b-5p | decreases the level of PTEN | migration and invasion in cervical cancer tissues and cell lines [165] |
| miR-144-3p | inhibition of miR-144-3p expression can up-regulate PTEN | induces cell proliferation and invasion, and reduces apoptosis, in thyroid cancer [166] |
| miR-19 | inhibits PTEN mRNA expression | decreases the expression of E-cadherin and increases the expression of α-SMA and fibronectin, while inhibition of miR-19 reverses TGF-β1-induced EMT in renal tubular epithelial cells, thereby promoting renal fibrosis [167] |
| miR-106b | suppresses the expression of PTEN | induces EMT in esophageal squamous cell carcinoma [168] |
| miR-17-5p | inhibits PTEN | enchases survival of breast cancer cells [169] |
| miR-552-5p | inhibits PTEN | promotes proliferation and migration and inhibits apoptosis in gastric cancer cells and stimulates metastasis in vivo; upregulation of miR-552-5p led to an increase in N-cadherin and vimentin and a reduction in E-cadherin [170] |
| miR-616-3p | reduced the mRNA and protein levels of PTEN | promotes proliferation and migration of endometrial stromal cells [171] |
| miR-181a | inhibits PTEN | promotes the proliferation and metastasis of Hepatocellular carcinoma cells in vitro and in vivo [172] |
| miR-221 | regulates PTEN | promotes invasion and metastasis in Extrahepatic cholangiocarcinoma [173] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fedorova, O.; Parfenyev, S.; Daks, A.; Shuvalov, O.; Barlev, N.A. The Role of PTEN in Epithelial–Mesenchymal Transition. Cancers 2022, 14, 3786. https://doi.org/10.3390/cancers14153786
Fedorova O, Parfenyev S, Daks A, Shuvalov O, Barlev NA. The Role of PTEN in Epithelial–Mesenchymal Transition. Cancers. 2022; 14(15):3786. https://doi.org/10.3390/cancers14153786
Chicago/Turabian StyleFedorova, Olga, Sergey Parfenyev, Alexandra Daks, Oleg Shuvalov, and Nickolai A. Barlev. 2022. "The Role of PTEN in Epithelial–Mesenchymal Transition" Cancers 14, no. 15: 3786. https://doi.org/10.3390/cancers14153786
APA StyleFedorova, O., Parfenyev, S., Daks, A., Shuvalov, O., & Barlev, N. A. (2022). The Role of PTEN in Epithelial–Mesenchymal Transition. Cancers, 14(15), 3786. https://doi.org/10.3390/cancers14153786