
Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies

 , , , , , , , , and
, , , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
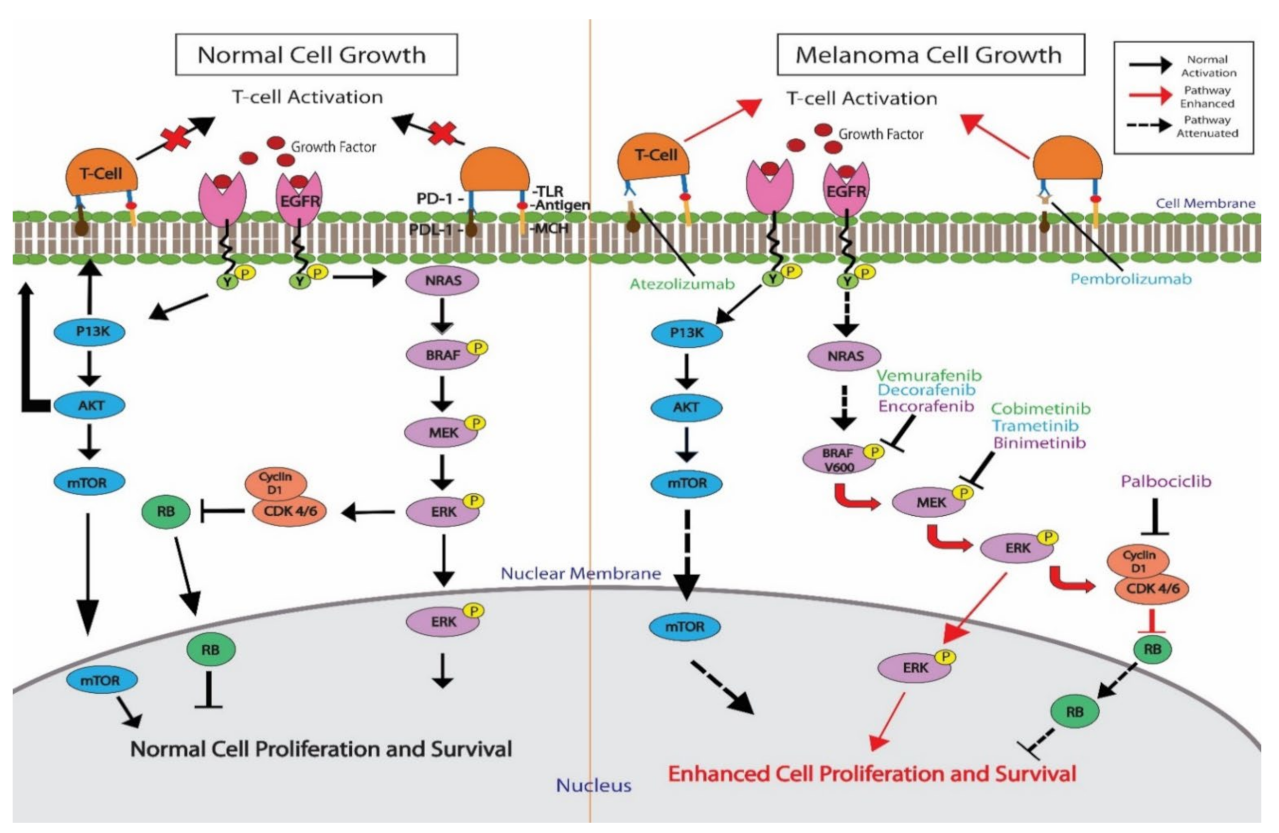
2. BRAF
3. MEK
4. NRAS
5. HRAS and KRAS
6. c-KIT
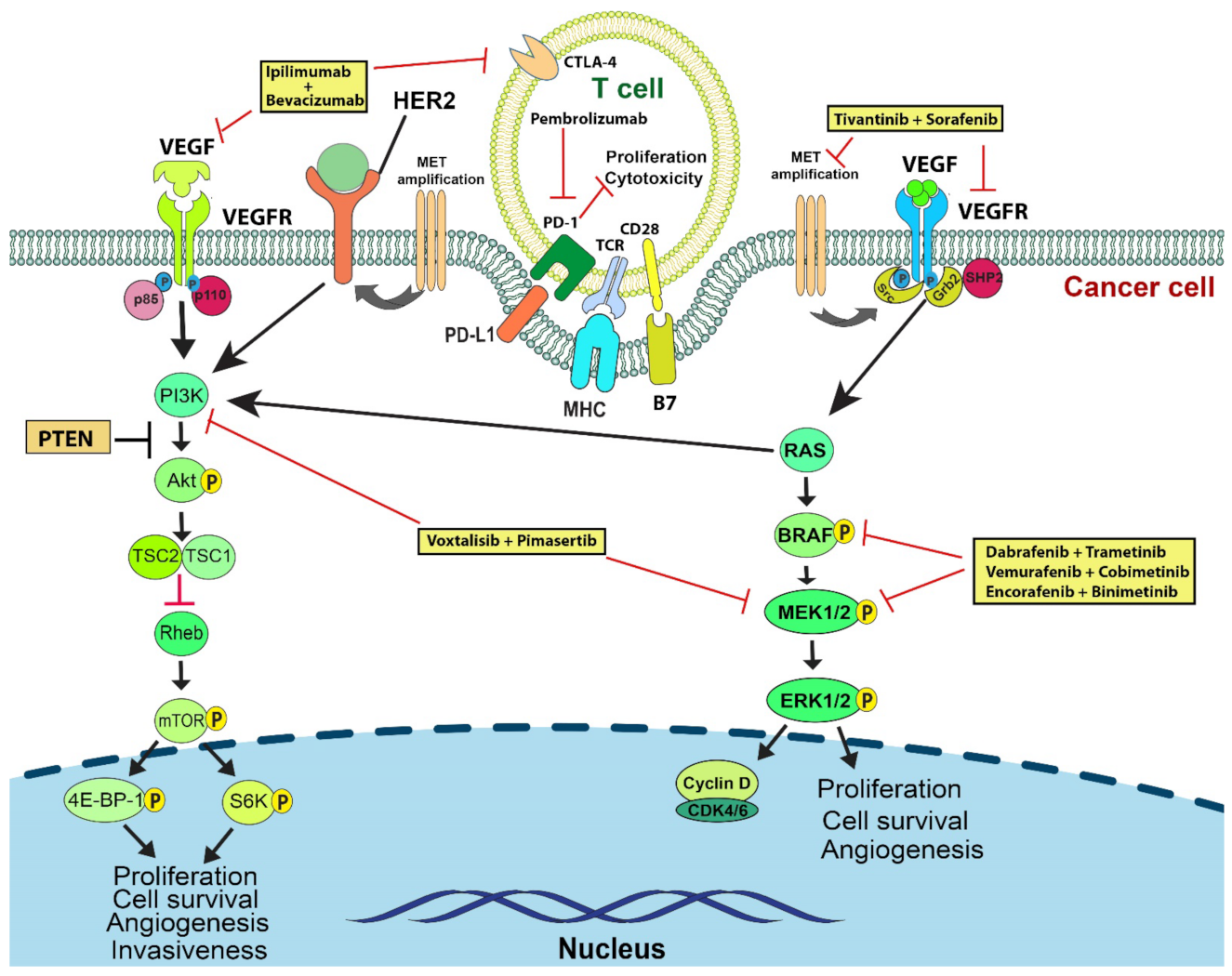
7. VEGFR
8. c-MET
9. PI3K/AKT
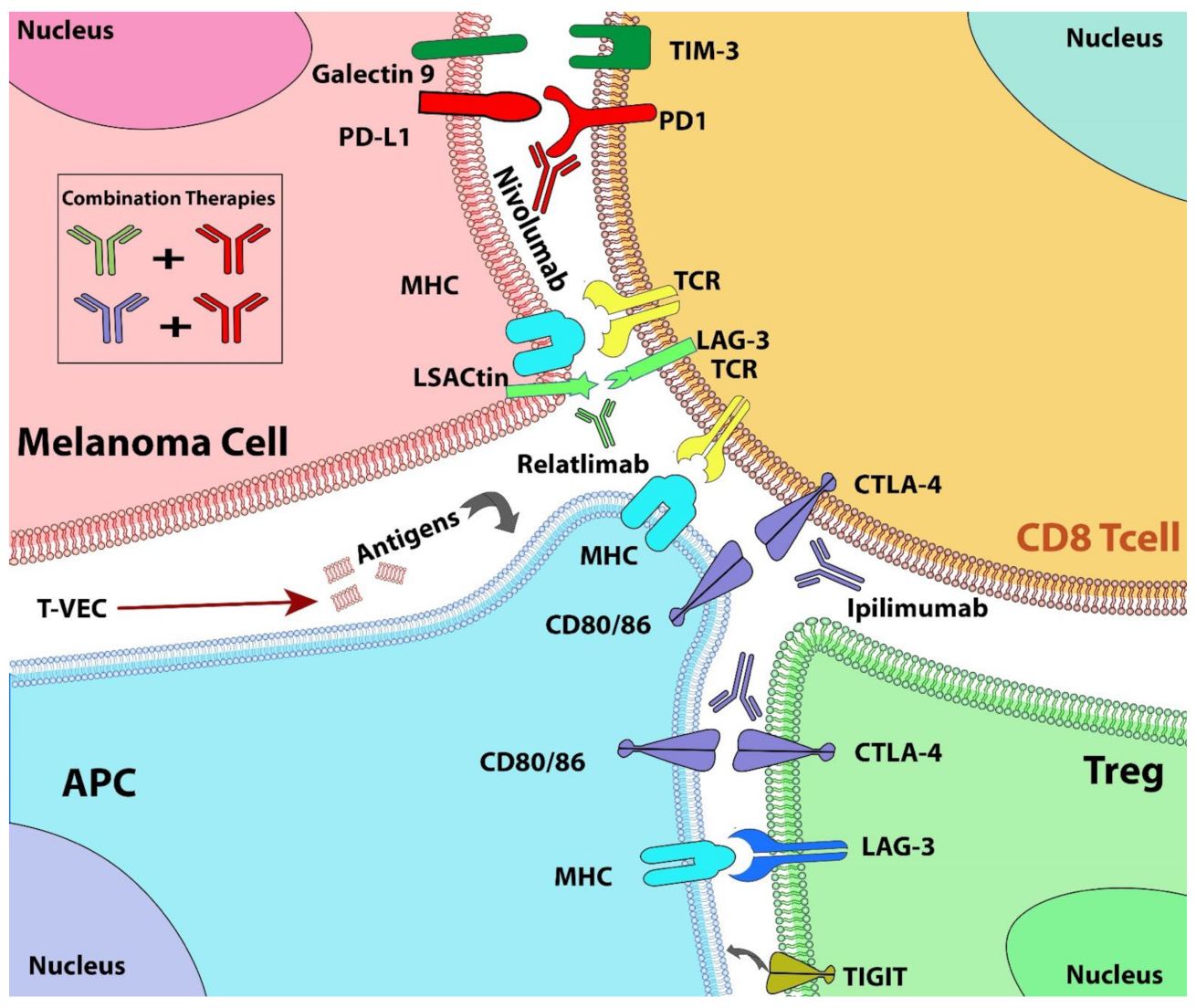
10. Combination Therapies with Immune Checkpoint Inhibitors
11. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Rajanna, S.; Rastogi, I.; Wojdyla, L.; Furo, H.; Kulesza, A.; Lin, L.; Sheu, B.; Frakes, M.; Ivanovich, M.; Puri, N. Current Molecularly Targeting Therapies in NSCLC and Melanoma. Anticancer Agents Med. Chem. 2015, 15, 856–868. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.Q.; Travers, J.B.; Kemp, M.G. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ. Mol. Mutagen. 2018, 59, 438–460. [Google Scholar] [CrossRef] [PubMed]
- Montor, W.R.; Salas, A.; Melo, F.H.M. Receptor tyrosine kinases and downstream pathways as druggable targets for cancer treatment: The current arsenal of inhibitors. Mol. Cancer 2018, 17, 55. [Google Scholar] [CrossRef]
- Schrank, Z.; Chhabra, G.; Lin, L.; Iderzorig, T.; Osude, C.; Khan, N.; Kuckovic, A.; Singh, S.; Miller, R.J.; Puri, N. Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance. Cancers 2018, 10, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabeta, P. Paradigms of vascularization in melanoma: Clinical significance and potential for therapeutic targeting. Biomed. Pharm. 2020, 127, 110135. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Del Vecchio, M.; Mandala, M.; Gogas, H.; Arance, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant nivolumab versus ipilimumab in resected stage IIIB-C and stage IV melanoma (CheckMate 238): 4-year results from a multicentre, double-blind, randomised, controlled, phase 3 trial. Lancet Oncol. 2020, 21, 1465–1477. [Google Scholar] [CrossRef]
- Weiss, S.A.; Wolchok, J.D.; Sznol, M. Immunotherapy of Melanoma: Facts and Hopes. Clin. Cancer Res. 2019, 25, 5191–5201. [Google Scholar] [CrossRef] [Green Version]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef] [PubMed]
- Iderzorig, T.; Kellen, J.; Osude, C.; Singh, S.; Woodman, J.A.; Garcia, C.; Puri, N. Comparison of EMT mediated tyrosine kinase inhibitor resistance in NSCLC. Biochem. Biophys. Res. Commun. 2018, 496, 770–777. [Google Scholar] [CrossRef]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namikawa, K.; Yamazaki, N. Targeted Therapy and Immunotherapy for Melanoma in Japan. Curr. Treat. Options Oncol. 2019, 20, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H., Jr.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Dummer, R.; Long, G.V.; Robert, C.; Tawbi, H.A.; Flaherty, K.T.; Ascierto, P.A.; Nathan, P.D.; Rutkowski, P.; Leonov, O.; Dutriaux, C.; et al. Randomized Phase III Trial Evaluating Spartalizumab Plus Dabrafenib and Trametinib for BRAF V600-Mutant Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2022, 40, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Gutzmer, R.; Stroyakovskiy, D.; Gogas, H.; Robert, C.; Lewis, K.; Protsenko, S.; Pereira, R.P.; Eigentler, T.; Rutkowski, P.; Demidov, L.; et al. Atezolizumab, vemurafenib, and cobimetinib as first-line treatment for unresectable advanced BRAF(V600) mutation-positive melanoma (IMspire150): Primary analysis of the randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 395, 1835–1844. [Google Scholar] [CrossRef]
- Atefi, M.; Titz, B.; Avramis, E.; Ng, C.; Wong, D.J.; Lassen, A.; Cerniglia, M.; Escuin-Ordinas, H.; Foulad, D.; Comin-Anduix, B.; et al. Combination of pan-RAF and MEK inhibitors in NRAS mutant melanoma. Mol. Cancer 2015, 14, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchberger, M.C.; Ugurel, S.; Mangana, J.; Heppt, M.V.; Eigentler, T.K.; Berking, C.; Schadendorf, D.; Schuler, G.; Dummer, R.; Heinzerling, L. MEK inhibition may increase survival of NRAS-mutated melanoma patients treated with checkpoint blockade: Results of a retrospective multicentre analysis of 364 patients. Eur. J. Cancer 2018, 98, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Sosman, J.; Santoro, A.; Saif, M.W.; Goff, L.; Dy, G.K.; Zucali, P.; Means-Powell, J.A.; Ma, W.W.; Simonelli, M.; et al. Phase 1 trial of tivantinib in combination with sorafenib in adult patients with advanced solid tumors. Investig. New Drugs 2015, 33, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Schuler, M.; Zimmer, L.; Kim, K.B.; Sosman, J.A.; Ascierto, P.A.; Postow, M.A.; De Vos, F.; van Herpen, C.M.L.; Carlino, M.S.; Johnson, D.B.; et al. Phase Ib/II Trial of Ribociclib in Combination with Binimetinib in Patients with NRAS-Mutant Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 3002–3010. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Fusi, L.; Weiss, N.; Goller, S.G.; Meder, K.; Frings, V.G.; Kneitz, H.; Goebeler, M.; Houben, R.; Schrama, D.; et al. Efficient Suppression of NRAS-Driven Melanoma by Co-Inhibition of ERK1/2 and ERK5 MAPK Pathways. J. Investig. Dermatol. 2020, 140, 2455–2465.e10. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Nguyen, M.Q.; Wilski, N.A.; Purwin, T.J.; Vernon, M.; Tiago, M.; Aplin, A.E. A Genome-wide screen identifies PDPK1 as a target to enhance the efficacy of MEK1/2 inhibitors in NRAS mutant melanoma. Cancer Res. 2022, 82, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Appleton, K.M.; Palsuledesai, C.C.; Misek, S.A.; Blake, M.; Zagorski, J.; Gallo, K.A.; Dexheimer, T.S.; Neubig, R.R. Inhibition of the Myocardin-Related Transcription Factor Pathway Increases Efficacy of Trametinib in NRAS-Mutant Melanoma Cell Lines. Cancers 2021, 13, 2012. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.Q.; Teh, J.L.F.; Purwin, T.J.; Chervoneva, I.; Davies, M.A.; Nathanson, K.L.; Cheng, P.F.; Levesque, M.P.; Dummer, R.; Aplin, A.E. Targeting PHGDH Upregulation Reduces Glutathione Levels and Resensitizes Resistant NRAS-Mutant Melanoma to MAPK Kinase Inhibition. J. Investig. Dermatol. 2020, 140, 2242–2252.e47. [Google Scholar] [CrossRef] [PubMed]
- Landras, A.; Reger de Moura, C.; Villoutreix, B.O.; Battistella, M.; Sadoux, A.; Dumaz, N.; Menashi, S.; Fernandez-Recio, J.; Lebbe, C.; Mourah, S. Novel treatment strategy for NRAS-mutated melanoma through a selective inhibitor of CD147/VEGFR-2 interaction. Oncogene 2022, 41, 2254–2264. [Google Scholar] [CrossRef] [PubMed]
- Portelinha, A.; Thompson, S.; Smith, R.A.; Da Silva Ferreira, M.; Asgari, Z.; Knezevic, A.; Seshan, V.; de Stanchina, E.; Gupta, S.; Denis, L.; et al. ASN007 is a selective ERK1/2 inhibitor with preferential activity against RAS-and RAF-mutant tumors. Cell Rep. Med. 2021, 2, 100350. [Google Scholar] [CrossRef] [PubMed]
- Abdou, Y.; Kapoor, A.; Hamad, L.; Ernstoff, M.S. Combination of pembrolizumab and imatinib in a patient with double KIT mutant melanoma: A case report. Medicine 2019, 98, e17769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algazi, A.P.; Weber, J.S.; Andrews, S.C.; Urbas, P.; Munster, P.N.; DeConti, R.C.; Hwang, J.; Sondak, V.K.; Messina, J.L.; McCalmont, T.; et al. Phase I clinical trial of the Src inhibitor dasatinib with dacarbazine in metastatic melanoma. Br. J. Cancer 2012, 106, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintas-Cardama, A.; Lazar, A.J.; Woodman, S.E.; Kim, K.; Ross, M.; Hwu, P. Complete response of stage IV anal mucosal melanoma expressing KIT Val560Asp to the multikinase inhibitor sorafenib. Nat. Clin. Pract. Oncol. 2008, 5, 737–740. [Google Scholar] [CrossRef]
- Jilaveanu, L.; Zito, C.; Lee, S.J.; Nathanson, K.L.; Camp, R.L.; Rimm, D.L.; Flaherty, K.T.; Kluger, H.M. Expression of sorafenib targets in melanoma patients treated with carboplatin, paclitaxel and sorafenib. Clin. Cancer Res. 2009, 15, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Liang, L.; Wen, Y.; Hu, R.; Wang, L.; Xia, Y.; Hu, C.; Qiao, Y.; Geng, X.; Chen, T.; Fei, J.; et al. Safety and efficacy of PD-1 blockade-activated multiple antigen-specific cellular therapy alone or in combination with apatinib in patients with advanced solid tumors: A pooled analysis of two prospective trials. Cancer Immunol. Immunother. 2019, 68, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Zhou, L.; Lian, B.; Si, L.; Sheng, X.; Chi, Z.; Kong, Y.; Wang, X.; Tang, B.; Mao, L.; et al. Safety and Efficacy of Apatinib Combined with Temozolomide in Advanced Melanoma Patients after Conventional Treatment Failure. Transl. Oncol. 2018, 11, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Yan, X.; Chi, Z.; Si, L.; Cui, C.; Tang, B.; Li, S.; Mao, L.; Lian, B.; Wang, X.; et al. Axitinib in Combination With Toripalimab, a Humanized Immunoglobulin G4 Monoclonal Antibody Against Programmed Cell Death-1, in Patients With Metastatic Mucosal Melanoma: An Open-Label Phase IB Trial. J. Clin. Oncol. 2019, 37, 2987–2999. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Lawrence, D.; Lezcano, C.; Wu, X.; Zhou, J.; Sasada, T.; Zeng, W.; Giobbie-Hurder, A.; Atkins, M.B.; Ibrahim, N.; et al. Bevacizumab plus ipilimumab in patients with metastatic melanoma. Cancer Immunol. Res. 2014, 2, 632–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Sheng, X.; Chi, Z.; Si, L.; Cui, C.; Kong, Y.; Tang, B.; Mao, L.; Wang, X.; Lian, B.; et al. Randomized Phase II Study of Bevacizumab in Combination With Carboplatin Plus Paclitaxel in Patients With Previously Untreated Advanced Mucosal Melanoma. J. Clin. Oncol. 2021, 39, 881–889. [Google Scholar] [CrossRef]
- Taylor, M.H.; Lee, C.H.; Makker, V.; Rasco, D.; Dutcus, C.E.; Wu, J.; Stepan, D.E.; Shumaker, R.C.; Motzer, R.J. Phase IB/II Trial of Lenvatinib Plus Pembrolizumab in Patients With Advanced Renal Cell Carcinoma, Endometrial Cancer, and Other Selected Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 1154–1163. [Google Scholar] [CrossRef] [PubMed]
- Mo, D.C.; Luo, P.H.; Huang, S.X.; Wang, H.L.; Huang, J.F. Safety and efficacy of pembrolizumab plus lenvatinib versus pembrolizumab and lenvatinib monotherapies in cancers: A systematic review. Int. Immunopharmacol. 2021, 91, 107281. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; El-Masry, M.; Osann, K.; Parmakhtiar, B.; Yamamoto, M.; Jakowatz, J.G. Phase II study of pazopanib in combination with paclitaxel in patients with metastatic melanoma. Cancer Chemother. Pharmacol. 2018, 82, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etnyre, D.; Stone, A.L.; Fong, J.T.; Jacobs, R.J.; Uppada, S.B.; Botting, G.M.; Rajanna, S.; Moravec, D.N.; Shambannagari, M.R.; Crees, Z.; et al. Targeting c-Met in melanoma: Mechanism of resistance and efficacy of novel combinatorial inhibitor therapy. Cancer Biol. Ther. 2014, 15, 1129–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Chua, V.; Liao, C.; Purwin, T.J.; Terai, M.; Kageyama, K.; Davies, M.A.; Sato, T.; Aplin, A.E. Co-targeting HGF/cMET Signaling with MEK Inhibitors in Metastatic Uveal Melanoma. Mol. Cancer Ther. 2017, 16, 516–528. [Google Scholar] [CrossRef] [Green Version]
- Ohara, M.; Saito, K.; Kageyama, K.; Terai, M.; Cheng, H.; Aplin, A.E.; Sato, T. Dual Targeting of CDK4/6 and cMET in Metastatic Uveal Melanoma. Cancers 2021, 13, 1104. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.J.; Wu, Y.; Hou, W.H.; Wang, Y.X.; Yuan, Q.Y.; Wang, H.J.; Yu, M. A novel bispecific c-MET/PD-1 antibody with therapeutic potential in solid cancer. Oncotarget 2017, 8, 29067–29079. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Gandhi, L.; Mita, M.M.; Damstrup, L.; Campana, F.; Hidalgo, M.; Grande, E.; Hyman, D.M.; Heist, R.S. A phase Ib dose-escalation and expansion study of the oral MEK inhibitor pimasertib and PI3K/MTOR inhibitor voxtalisib in patients with advanced solid tumours. Br. J. Cancer 2018, 119, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Molife, L.R.; Yan, L.; Vitfell-Rasmussen, J.; Zernhelt, A.M.; Sullivan, D.M.; Cassier, P.A.; Chen, E.; Biondo, A.; Tetteh, E.; Siu, L.L.; et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J. Hematol. Oncol. 2014, 7, 1. [Google Scholar] [CrossRef] [Green Version]
- da Silveira Nogueira Lima, J.P.; Georgieva, M.; Haaland, B.; de Lima Lopes, G. A systematic review and network meta-analysis of immunotherapy and targeted therapy for advanced melanoma. Cancer Med. 2017, 6, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S. Understanding melanoma signaling networks as the basis for molecular targeted therapy. J. Investig. Dermatol. 2010, 130, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to Molecularly Targeted Therapies in Melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef]
- Yu, C.; Liu, X.; Yang, J.; Zhang, M.; Jin, H.; Ma, X.; Shi, H. Combination of Immunotherapy With Targeted Therapy: Theory and Practice in Metastatic Melanoma. Front. Immunol. 2019, 10, 990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; McArthur, G.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Di Giacomo, A.M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): Updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016, 17, 1248–1260. [Google Scholar] [CrossRef]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbiah, V.; Baik, C.; Kirkwood, J.M. Clinical Development of BRAF plus MEK Inhibitor Combinations. Trends Cancer 2020, 6, 797–810. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef]
- Louveau, B.; Resche-Rigon, M.; Lesimple, T.; Da Meda, L.; Pracht, M.; Baroudjian, B.; Delyon, J.; Amini-Adle, M.; Dutriaux, C.; Reger de Moura, C.; et al. Phase I-II Open-Label Multicenter Study of Palbociclib + Vemurafenib in BRAF (V600MUT) Metastatic Melanoma Patients: Uncovering CHEK2 as a Major Response Mechanism. Clin. Cancer Res. 2021, 27, 3876–3883. [Google Scholar] [CrossRef] [PubMed]
- Pelster, M.S.; Amaria, R.N. Combined targeted therapy and immunotherapy in melanoma: A review of the impact on the tumor microenvironment and outcomes of early clinical trials. Ther. Adv. Med. Oncol. 2019, 11, 1758835919830826. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Atkinson, V.; Sandhu, S.; Hospers, G.; Long, G.V.; Aglietta, M.; Ferrucci, P.F.; Tulyte, S.; Cappellini, G.C.A.; Soriano, V.; Ali, S.; et al. Dabrafenib plus trametinib is effective in the treatment of BRAF V600-mutated metastatic melanoma patients: Analysis of patients from the dabrafenib plus trametinib Named Patient Program (DESCRIBE II). Melanoma Res. 2020, 30, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bergholz, J.S.; Wang, Q.; Kabraji, S.; Zhao, J.J. Integrating Immunotherapy and Targeted Therapy in Cancer Treatment: Mechanistic Insights and Clinical Implications. Clin. Cancer Res. 2020, 26, 5557–5566. [Google Scholar] [CrossRef] [PubMed]
- Koelblinger, P.; Thuerigen, O.; Dummer, R. Development of encorafenib for BRAF-mutated advanced melanoma. Curr. Opin. Oncol. 2018, 30, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zager, J.S.; Eroglu, Z. Encorafenib/binimetinib for the treatment of BRAF-mutant advanced, unresectable, or metastatic melanoma: Design, development, and potential place in therapy. OncoTargets Ther. 2018, 11, 9081–9089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogas, H.J.; Flaherty, K.T.; Dummer, R.; Ascierto, P.A.; Arance, A.; Mandala, M.; Liszkay, G.; Garbe, C.; Schadendorf, D.; Krajsova, I.; et al. Adverse events associated with encorafenib plus binimetinib in the COLUMBUS study: Incidence, course and management. Eur. J. Cancer 2019, 119, 97–106. [Google Scholar] [CrossRef]
- Garutti, M.; Targato, G.; Buriolla, S.; Palmero, L.; Minisini, A.M.; Puglisi, F. CDK4/6 Inhibitors in Melanoma: A Comprehensive Review. Cells 2021, 10, 1334. [Google Scholar] [CrossRef] [PubMed]
- Hartsough, E.J.; Kugel, C.H., 3rd; Vido, M.J.; Berger, A.C.; Purwin, T.J.; Goldberg, A.; Davies, M.A.; Schiewer, M.J.; Knudsen, K.E.; Bollag, G.; et al. Response and Resistance to Paradox-Breaking BRAF Inhibitor in Melanomas In Vivo and Ex Vivo. Mol. Cancer Ther. 2018, 17, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tutuka, C.S.A.; Andrews, M.C.; Mariadason, J.M.; Ioannidis, P.; Hudson, C.; Cebon, J.; Behren, A. PLX8394, a new generation BRAF inhibitor, selectively inhibits BRAF in colonic adenocarcinoma cells and prevents paradoxical MAPK pathway activation. Mol. Cancer 2017, 16, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aida, S.; Sonobe, Y.; Tanimura, H.; Oikawa, N.; Yuhki, M.; Sakamoto, H.; Mizuno, T. MITF suppression improves the sensitivity of melanoma cells to a BRAF inhibitor. Cancer Lett. 2017, 409, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Nepote, A.; Avallone, G.; Ribero, S.; Cavallo, F.; Roccuzzo, G.; Mastorino, L.; Conforti, C.; Paruzzo, L.; Poletto, S.; Carnevale Schianca, F.; et al. Current Controversies and Challenges on BRAF V600K-Mutant Cutaneous Melanoma. J. Clin. Med. 2022, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; LoRusso, P.; Boerner, S.; Dummer, R. Achievements and challenges of molecular targeted therapy in melanoma. Am. Soc. Clin. Oncol. Educ. Book 2015, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.; Fedorenko, I.V.; Cantini, L.P.; Munko, A.C.; Hall, M.; Sondak, V.K.; Messina, J.L.; Flaherty, K.T.; Smalley, K.S. Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 2010, 102, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Lee, K.M. Current Insights into Combination Therapies with MAPK Inhibitors and Immune Checkpoint Blockade. Int. J. Mol. Sci. 2020, 21, 2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Hauschild, A.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; Haydon, A.; et al. Adjuvant Dabrafenib plus Trametinib in Stage III BRAF-Mutated Melanoma. N. Engl. J. Med. 2017, 377, 1813–1823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauschild, A.; Dummer, R.; Schadendorf, D.; Santinami, M.; Atkinson, V.; Mandala, M.; Chiarion-Sileni, V.; Larkin, J.; Nyakas, M.; Dutriaux, C.; et al. Longer Follow-Up Confirms Relapse-Free Survival Benefit With Adjuvant Dabrafenib Plus Trametinib in Patients with Resected BRAF V600-Mutant Stage III Melanoma. J. Clin. Oncol. 2018, 36, 3441–3449. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.W.; Hintzsche, J.D.; Bagby, S.M.; Espinoza, V.; Langouet-Astrie, C.; Amato, C.M.; Chimed, T.S.; Fujita, M.; Robinson, W.; Tan, A.C.; et al. Targeting CDK4/6 Represents a Therapeutic Vulnerability in Acquired BRAF/MEK Inhibitor-Resistant Melanoma. Mol Cancer Ther 2021, 20, 2049–2060. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devitt, B.; Liu, W.; Salemi, R.; Wolfe, R.; Kelly, J.; Tzen, C.Y.; Dobrovic, A.; McArthur, G. Clinical outcome and pathological features associated with NRAS mutation in cutaneous melanoma. Pigment. Cell Melanoma Res. 2011, 24, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.E.; Edmiston, S.N.; Alexander, A.; Groben, P.A.; Parrish, E.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; From, L.; et al. Association Between NRAS and BRAF Mutational Status and Melanoma-Specific Survival Among Patients With Higher-Risk Primary Melanoma. JAMA Oncol. 2015, 1, 359–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, F.C.; McArthur, G.A. Targeting NRAS in melanoma. Cancer J. 2012, 18, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.B.; Puzanov, I. Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol. 2015, 16, 15. [Google Scholar] [CrossRef]
- Randic, T.; Kozar, I.; Margue, C.; Utikal, J.; Kreis, S. NRAS mutant melanoma: Towards better therapies. Cancer Treat. Rev. 2021, 99, 102238. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Kittaneh, M.; Lolkema, M.P.J.K.; Postow, M.A.; Schwartz, G.; Matano, C.F.; Bhansali, S.; Parasuraman, S.; Kim, K. A phase 1b/2 study of LEE011 in combination with binimetinib (MEK162) in patients with NRAS-mutant melanoma: Early encouraging clinical activity. J. Clin. Oncol. 2014, 32, 9009. [Google Scholar] [CrossRef]
- Schuler, M.H.; Ascierto, P.A.; de Vos, F.Y.F.L.; Postow, M.A.; van Herpen, C.M.L.; Carlino, M.S.; Sosman, J.A.; Berking, C.; Long, G.V.; Weise, A.; et al. Phase 1b/2 trial of ribociclib+binimetinib in metastatic NRAS-mutant melanoma: Safety, efficacy, and recommended phase 2 dose (RP2D). J. Clin. Oncol. 2017, 35, 9519. [Google Scholar] [CrossRef]
- Means-Powell, J.A.; Adjei, A.A.; Puzanov, I.; Dy, G.K.; Goff, L.W.; Fetterly, W.W.M.J.; Michael, S.A.; Chai, F.; Lamar, M.; Schwartz, B.E.; et al. Safety and efficacy of MET inhibitor tivantinib (ARQ 197) combined with sorafenib in patients (pts) with NRAS wild-type or mutant melanoma from a phase I study. J. Clin. Oncol. 2012, 30, 8519. [Google Scholar] [CrossRef]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF-->MEK-->ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Vanni, I.; Tanda, E.T.; Dalmasso, B.; Pastorino, L.; Andreotti, V.; Bruno, W.; Boutros, A.; Spagnolo, F.; Ghiorzo, P. Non-BRAF Mutant Melanoma: Molecular Features and Therapeutical Implications. Front. Mol. Biosci. 2020, 7, 172. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, R.; Li, Z. The Prognostic Value of HRAS mRNA Expression in Cutaneous Melanoma. Biomed. Res. Int. 2017, 2017, 5356737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, D.D.M.; Guhan, S.; Tsao, H. KIT and Melanoma: Biological Insights and Clinical Implications. Yonsei Med. J. 2020, 61, 562–571. [Google Scholar] [CrossRef]
- Delyon, J.; Chevret, S.; Jouary, T.; Dalac, S.; Dalle, S.; Guillot, B.; Arnault, J.P.; Avril, M.F.; Bedane, C.; Bens, G.; et al. STAT3 Mediates Nilotinib Response in KIT-Altered Melanoma: A Phase II Multicenter Trial of the French Skin Cancer Network. J. Investig. Dermatol. 2018, 138, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Kunisada, T.; Grimm, T.; Nishimura, E.K.; Nishioka, E.; Nishikawa, S.I. Review: Melanocyte migration and survival controlled by SCF/c-kit expression. J. Investig. Dermatol. Symp. Proc. 2001, 6, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosier, P.S.; Ricciardi, S.T.; Hall, L.R.; Vitas, M.R.; Clark, S.C.; Crosier, K.E. Expression of isoforms of the human receptor tyrosine kinase c-kit in leukemic cell lines and acute myeloid leukemia. Blood 1993, 82, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarlomo-Rikala, M.; Kovatich, A.J.; Barusevicius, A.; Miettinen, M. CD117: A sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod. Pathol. 1998, 11, 728–734. [Google Scholar] [PubMed]
- Maulik, G.; Bharti, A.; Khan, E.; Broderick, R.J.; Kijima, T.; Salgia, R. Modulation of c-Kit/SCF pathway leads to alterations in topoisomerase-I activity in small cell lung cancer. J. Environ. Pathol. Toxicol. Oncol. 2004, 23, 237–251. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Kim, T.M.; Kim, Y.J.; Jang, K.T.; Lee, H.J.; Lee, S.N.; Ahn, M.S.; Hwang, I.G.; Lee, S.; Lee, M.H.; et al. Phase II Trial of Nilotinib in Patients With Metastatic Malignant Melanoma Harboring KIT Gene Aberration: A Multicenter Trial of Korean Cancer Study Group (UN10-06). Oncologist 2015, 20, 1312–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.Z.; Zheng, H.Y.; Li, J. The clinical significance of KIT mutations in melanoma: A meta-analysis. Melanoma Res. 2018, 28, 259–270. [Google Scholar] [CrossRef]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F., 3rd; Azar, S.; et al. KIT gene mutations and copy number in melanoma subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic activation of KIT in distinct subtypes of melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Lawrence, D.P.; Weber, J.S.; Gajewski, T.F.; Gonzalez, R.; Lutzky, J.; O’Day, S.J.; Hamid, O.; Wolchok, J.D.; Chapman, P.B.; et al. Phase II Study of Nilotinib in Melanoma Harboring KIT Alterations Following Progression to Prior KIT Inhibition. Clin. Cancer Res. 2015, 21, 2289–2296. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Sosman, J.A. Therapeutic Advances and Treatment Options in Metastatic Melanoma. JAMA Oncol. 2015, 1, 380–386. [Google Scholar] [CrossRef] [Green Version]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: Biological and clinical implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Mao, L.; Chi, Z.; Sheng, X.; Cui, C.; Kong, Y.; Dai, J.; Wang, X.; Li, S.; Tang, B.; et al. Efficacy Evaluation of Imatinib for the Treatment of Melanoma: Evidence From a Retrospective Study. Oncol. Res. 2019, 27, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, C.; Natoli, A.; Hui, Y.; Conus, N.; Jackson, S.; Bruggen, J.; Manley, P.W.; McArthur, G.A. Preclinical evaluation of nilotinib efficacy in an imatinib-resistant KIT-driven tumor model. Mol. Cancer Ther. 2010, 9, 1461–1468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blay, J.Y.; von Mehren, M. Nilotinib: A novel, selective tyrosine kinase inhibitor. Semin. Oncol. 2011, 38 (Suppl. S1), S3–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, D.; Carvajal, R.D. KIT as an Oncogenic Driver in Melanoma: An Update on Clinical Development. Am. J. Clin. Dermatol. 2019, 20, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Todd, J.R.; Rizos, H. Resistance to c-Kit inhibitors in melanoma: Insights for future therapies. Oncoscience 2014, 1, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Kim, K.H.; Baek, E.J.; Park, B.; So, M.K.; Ko, B.J.; Ko, H.J.; Park, S.G. A novel anti-c-Kit antibody-drug conjugate to treat wild-type and activating-mutant c-Kit-positive tumors. Mol. Oncol. 2022, 16, 1290–1308. [Google Scholar] [CrossRef] [PubMed]
- Melincovici, C.S.; Bosca, A.B.; Susman, S.; Marginean, M.; Mihu, C.; Istrate, M.; Moldovan, I.M.; Roman, A.L.; Mihu, C.M. Vascular endothelial growth factor (VEGF—Key factor in normal and pathological angiogenesis. Rom. J. Morphol. Embryol. 2018, 59, 455–467. [Google Scholar]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, C.; Scapulatempo Neto, C.; Viana, C.R.; Vazquez, V.L. PD-1/PD-L1 and VEGF-A/VEGF-C expression in lymph node microenvironment and association with melanoma metastasis and survival. Melanoma Res. 2017, 27, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulieres, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin(R)) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef] [PubMed]
- Dithmer, M.; Kirsch, A.M.; Grafenstein, L.; Wang, F.; Schmidt, H.; Coupland, S.E.; Fuchs, S.; Roider, J.; Klettner, A.K. Uveal Melanoma Cell Under Oxidative Stress—Influence of VEGF and VEGF-Inhibitors. Klin Monbl Augenheilkd 2019, 236, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jiao, D.; Qin, S.; Chu, Q.; Wu, K.; Li, A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol. Cancer 2019, 18, 60. [Google Scholar] [CrossRef]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef]
- Kudo, M. Scientific Rationale for Combined Immunotherapy with PD-1/PD-L1 Antibodies and VEGF Inhibitors in Advanced Hepatocellular Carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacal, P.M.; Atzori, M.G.; Ruffini, F.; Scimeca, M.; Bonanno, E.; Cicconi, R.; Mattei, M.; Bernardini, R.; D’Atri, S.; Tentori, L.; et al. Targeting the vascular endothelial growth factor receptor-1 by the monoclonal antibody D16F7 to increase the activity of immune checkpoint inhibitors against cutaneous melanoma. Pharmacol. Res. 2020, 159, 104957. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Algazi, A.P.; Cha, E.; Ortiz-Urda, S.M.; McCalmont, T.; Bastian, B.C.; Hwang, J.; Pampaloni, M.H.; Behr, S.; Chong, K.; Cortez, B.; et al. The combination of axitinib followed by paclitaxel/carboplatin yields extended survival in advanced BRAF wild-type melanoma: Results of a clinical/correlative prospective phase II clinical trial. Br. J. Cancer 2015, 112, 1326–1331. [Google Scholar] [CrossRef]
- Tarhini, A.A.; Frankel, P.; Ruel, C.; Ernstoff, M.S.; Kuzel, T.M.; Logan, T.F.; Khushalani, N.I.; Tawbi, H.A.; Margolin, K.A.; Awasthi, S.; et al. NCI 8628: A randomized phase 2 study of ziv-aflibercept and high-dose interleukin 2 or high-dose interleukin 2 alone for inoperable stage III or IV melanoma. Cancer 2018, 124, 4332–4341. [Google Scholar] [CrossRef] [Green Version]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef] [PubMed]
- Atzori, M.G.; Ceci, C.; Ruffini, F.; Trapani, M.; Barbaccia, M.L.; Tentori, L.; D’Atri, S.; Lacal, P.M.; Graziani, G. Role of VEGFR-1 in melanoma acquired resistance to the BRAF inhibitor vemurafenib. J. Cell Mol. Med. 2020, 24, 465–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, J.H.; Kim, J.; Lin, A.; Folberg, R.; Iyer, S.; Abramson, D.H. Growth of Uveal Melanoma following Intravitreal Bevacizumab. Ocul. Oncol. Pathol. 2017, 3, 117–121. [Google Scholar] [CrossRef]
- Lima, B.R.; Schoenfield, L.R.; Singh, A.D. The impact of intravitreal bevacizumab therapy on choroidal melanoma. Am. J. Ophthalmol. 2011, 151, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Becherirat, S.; Valamanesh, F.; Karimi, M.; Faussat, A.M.; Launay, J.M.; Pimpie, C.; Therwath, A.; Pocard, M. Discontinuous Schedule of Bevacizumab in Colorectal Cancer Induces Accelerated Tumor Growth and Phenotypic Changes. Transl. Oncol. 2018, 11, 406–415. [Google Scholar] [CrossRef]
- Champiat, S.; Ferrara, R.; Massard, C.; Besse, B.; Marabelle, A.; Soria, J.C.; Ferte, C. Hyperprogressive disease: Recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018, 15, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Schuiveling, M.; Tonk, E.H.J.; Verheijden, R.J.; Suijkerbuijk, K.P.M. Hyperprogressive disease rarely occurs during checkpoint inhibitor treatment for advanced melanoma. Cancer Immunol. Immunother. 2021, 70, 1491–1496. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Tsao, M.S. c-MET as a potential therapeutic target and biomarker in cancer. Ther. Adv. Med. Oncol. 2011, 3, S21–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef]
- Peschard, P.; Park, M. From Tpr-Met to Met, tumorigenesis and tubes. Oncogene 2007, 26, 1276–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benvenuti, S.; Comoglio, P.M. The MET receptor tyrosine kinase in invasion and metastasis. J. Cell Physiol. 2007, 213, 316–325. [Google Scholar] [CrossRef]
- Knudsen, B.S.; Vande Woude, G. Showering c-MET-dependent cancers with drugs. Curr. Opin. Genet. Dev. 2008, 18, 87–96. [Google Scholar] [CrossRef]
- Puri, N.; Ahmed, S.; Janamanchi, V.; Tretiakova, M.; Zumba, O.; Krausz, T.; Jagadeeswaran, R.; Salgia, R. c-Met is a potentially new therapeutic target for treatment of human melanoma. Clin. Cancer Res. 2007, 13, 2246–2253. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef]
- Cheng, H.; Terai, M.; Kageyama, K.; Ozaki, S.; McCue, P.A.; Sato, T.; Aplin, A.E. Paracrine Effect of NRG1 and HGF Drives Resistance to MEK Inhibitors in Metastatic Uveal Melanoma. Cancer Res. 2015, 75, 2737–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, M.A. The role of the PI3K-AKT pathway in melanoma. Cancer J. 2012, 18, 142–147. [Google Scholar] [CrossRef]
- Karachaliou, N.; Gonzalez-Cao, M.; Sosa, A.; Berenguer, J.; Bracht, J.W.P.; Ito, M.; Rosell, R. The combination of checkpoint immunotherapy and targeted therapy in cancer. Ann. Transl. Med. 2017, 5, 388. [Google Scholar] [CrossRef] [Green Version]
- Frampton, A.E.; Sivakumar, S. A New Combination Immunotherapy in Advanced Melanoma. N. Engl. J. Med. 2022, 386, 91–92. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2018, 18, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M. Scientific Rationale for Combination Immunotherapy of Hepatocellular Carcinoma with Anti-PD-1/PD-L1 and Anti-CTLA-4 Antibodies. Liver Cancer 2019, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Weber, J.S.; Gibney, G.; Sullivan, R.J.; Sosman, J.A.; Slingluff, C.L., Jr.; Lawrence, D.P.; Logan, T.F.; Schuchter, L.M.; Nair, S.; Fecher, L.; et al. Sequential administration of nivolumab and ipilimumab with a planned switch in patients with advanced melanoma (CheckMate 064): An open-label, randomised, phase 2 trial. Lancet Oncol. 2016, 17, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Neoadjuvant PD-1 Blockade in Resectable Lung Cancer; Nivolumab and Ipilimumab in Advanced Melanoma; Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma; Prolonged Survival in Stage III Melanoma with Ipilimumab Adjuvant Therapy; Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma; Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma; Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma; Rapid Eradication of a Bulky Melanoma Mass with One Dose of Immunotherapy; Genetic Basis for Clinical Response to CTLA-4 Blockade; Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma; Nivolumab plus Ipilimumab in Advanced Melanoma; Safety and Tumor Responses with Lambrolizumab (Anti-PD-1) in Melanoma; Hepatotoxicity with Combination of Vemurafenib and Ipilimumab. N. Engl. J. Med. 2018, 379, 2185. [CrossRef]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignali, D.A.A. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Huang, X.; Chen, X.; Liu, J.; Wu, C.; Pu, Q.; Wang, Y.; Kang, X.; Zhou, L. Characterization of a novel anti-human lymphocyte activation gene 3 (LAG-3) antibody for cancer immunotherapy. mAbs 2019, 11, 1139–1148. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Arkhypov, I.; Weber, R.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Modern Aspects of Immunotherapy with Checkpoint Inhibitors in Melanoma. Int. J. Mol. Sci. 2020, 21, 2367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Drug 1 (Target) | Drug 2 (Target) | Drug 3 (Target) | Type of Study | Reference |
|---|---|---|---|---|
| BRAF/MEK | ||||
| Dabrafenib (BRAF) | Trametinib (MEK) | Pembrolizumab (PD-1) | Phase IIPhase I/II | [15,16] |
| Dabrafenib (BRAF) | Trametinib (MEK) | Spartalizumab (PD-1) | Phase III | [17] |
| Vemurafenib (BRAF) | Cobimetinib (MEK) | Atezolizumab (PD-L1) | Phase III | [18] |
| NRAS | ||||
| Compd A (RAF) | Trametinib (MEK) | Pre-clinical | [19] | |
| Ipilimumab (CTLA-4) or anti-PD-1 (PD-1) | Binimetinib, Pimasertinib, or Trametinib (MEK) | Case control studies | [20] | |
| Tivantinib (cMET) | Sorafenib (VEGFR/PDGFR/RAF/other) | Phase I | [21] | |
| Binimetinib (MEK) | Ribociclib (CDK4/6) | Phase Ib/II | [22] | |
| Trametinib (MEK) | XMD9-92 (ERK5) | Pre-clinical | [23] | |
| Trametinib (MEK) | GSK2334470 (PDPK1) | Pre-clinical | [24] | |
| Trametinib (MEK) | CCG-222740 (MRTF) | Pre-clinical | [25] | |
| Trametinib or PD901 (MEK) | PHGDH siRNA | Pre-clinical | [26] | |
| Cobimetinib (MEK) | CD147 inhibitor | Pre-clinical | [27] | |
| HRAS | ||||
| ASN007 (ERK1/2) | Copanlisib (PI3K) | Pre-clinical | [28] | |
| KIT | ||||
| Imatinib (KIT/other) | Pembrolizumab (PD-1) | Case Report | [29] | |
| Dasatinib (KIT) | Dacarbazine | Phase 1 | [30] | |
| Sorafenib (KIT/other) | Temozolomide | Case Report | [31] | |
| Sorafenib (KIT) | Carboplatin | Paclitaxel | Phase I/II | [32] |
| VEGFR | ||||
| Apatinib (VEGFR) | Camrelizumab/SHR-1210 (PD-1) | Phase II/III | [33] | |
| Apatinib (VEGFR) | Temozolomide (Antineoplastic) | Clinical trial (escalation study) | [34] | |
| Axitinib (VEGFR) | Toripalimab (PD-1) | Phase Ib | [35] | |
| Bevacizumab (VEGFR) | Ipilimumab (CTLA-4) | Phase I | [36] | |
| Bevacizumab (VEGFR) | Paclitaxel (Antineoplastic) | Carboplatin (Antineoplastic) | Phase II | [37] |
| Lenvatinib (VEGFR) | Pembrolizumab (PD-1) | Phase Ib/II | [38,39] | |
| Pazopanib (VEGFR/PDGFR/c-KIT) | Paclitaxel (Antineoplastic) | Phase II | [40] | |
| C-MET | ||||
| Everolimus (mTOR) | XAV939 (Wnt) | SU11274 (cMET) | Pre-clinical | [41] |
| LY2801653 (cMET) | Trametinib (MEK1/2) | Pre-clinical | [42] | |
| Abemaciclib (CDK4/6) | Merestinib (cMET) | Pre-clinical | [43] | |
| Bi-Specific antibody (cMET & PD1) | Pre-clinical | [44] | ||
| PI3K/AKT | ||||
| Pimasertib (MEK1/2) | Voxtalisib (pan-PI3K) | Phase Ib | [45] | |
| MK-2206 (AKT) | Carboplatin/Paclitaxel, Docetaxel, or Erlotinib | Phase I | [46] | |
| Clinical Trials | Phase/Status | Participants | Conditions | Drug Intervention (Drug Target) | Primary Outcome Measures | Estimated Completion Date |
|---|---|---|---|---|---|---|
| NCT04720768 (CELEBRATE) | Ib, Recruiting | 78 | Metastatic BRAF Mutant Melanoma | Encorafenib (BRAF) + Binimetinib (MEK) + Palbociclib (CDK4/6) | Dose-Limiting Toxicity | 12/04/2023 |
| NCT04835805 | Ib, Recruiting | 98 | Advanced NRAS Mutant Melanoma, Had received anti-PD-1/PD-L1 therapy | Belvarafenib (RAF) + Cobimetinib (MEK) with/without Atezolizumab (PD-L1) | Dose-Limiting Toxicity, Adverse Events | 11/11/2024 |
| NCT04109456 | Ib, Recruiting | 52 | Metastatic Uveal Melanoma, Metastatic NRAS Mutant Melanoma | IN10018 (FAK) + Cobimetinib (MEK) | Safety, Tolerability | 06/30/2023 |
| NCT02974725 | Ib, Active, not recruiting | 241 | Metastatic/Advanced KRAS or BRAF Mutant Non-Small Cell Lung Cancer or NRAS Mutant Melanoma | Naporafenib (RAF) + LTT462 (ERK1/2)/Trametinib (MEK)/Ribociclib (CDK4/6) | Adverse Events, Dose-Limiting Toxicities, Tolerability | 11/25/2022 |
| NCT04417621 | II, Recruiting | 320 | Previously Treated Unresectable or Metastatic BRAFV600 or NRAS Mutant Melanoma | Naporafenib (RAF) + LTT462 (ERK1/2)/Trametinib (MEK)/Ribociclib (CDK4/6) | Overall Response Rate | 09/08/2023 |
| NCT03979651 (CHLOROTRAMMEL) | I, Recruiting | 29 | Metastatic/Advanced NRAS Melanoma | Trametinib (MEK) + Hydroxychloroquine (autophagy) | Dose-Limiting Toxicities, Partial or Complete response | 03/31/2022 |
| NCT04903119 | I, Recruiting | 15 | Metastatic or Unresectable melanoma with BRAF V600 | Nilotinib (cKIT) + Dabrafenib (BRAF) + Trametinib (MEK) | Dose-Limiting Toxicities | 03/31/2027 |
| NCT02298959 | I, Recruiting | 78 | Advanced Solid Tumors | Aflibercept (VEGFR) + Pembrolizumab (PD-1) | Safety, Recommended Phase II Dosing | 11/31/2022 |
| NCT02159066 (LOGIC-2) | II, Active, not recruiting | 160 | Locally Advanced or Metastatic BRAF V600 Melanoma | Encorafenib (BRAF) + Binimetinib (MEK) + Ribociclib (CDK4/6)/Infigratinib (FGFR kinase)/Buparlisib (PI3K)/ Capmatinib (MET) | Overall Response Rate | 01/17/2023 |
| NCT03957551 | Ib/II, Recruiting | 39 | Advanced Melanoma | Cabozantinib (VEGFR/cMET/AXL) + Pembrolizumab (PD-1) | Dose-Limiting Toxicities, Overall Response Rate | 07/01/2024 |
| NCT03131908 | I/II, Active, not recruiting | 36 | Refractory Metastatic Melanoma with loss of PTEN | GSK2636771 (PI3Kβ) + Pembrolizumab (PD-1) | Maximum Tolerated Dose, Objective Response Rate | 12/31/2022 |
| NCT02637531 | I, Active, not recruiting | 219 | Advanced Melanoma | IPI-549 (PI3K-gamma) + Nivolumab (PD-1) | Dose-limiting Toxicity, Adverse Events | 12/2022 |
| NCT03673787 (IceCAP) | I/II, Recruiting | 87 | Solid Tumors with Hyperactive PI3K | Ipatasertib (AKT) + Atezolizumab (PD-L1) | Maximum Tolerated Dose, Adverse Events | 11/2023 |
| NCT01480154 | I, Active, not recruiting | 62 | Advanced Solid Tumors | MK-2206 (AKT) + Hydroxychloroquine | Maximum Tolerated Dose, Dose-Limiting Rate | 02/14/2020, not published |
| NCT03470922 (RELATIVITY-047) | II/III, Active, not recruiting | 714 | Previously Untreated Metastatic or Unresectable Melanoma | Relatlimab (LAG-3) + Nivolumab (PD-1) | Progression Free Survival | 11/30/2023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rager, T.; Eckburg, A.; Patel, M.; Qiu, R.; Gantiwala, S.; Dovalovsky, K.; Fan, K.; Lam, K.; Roesler, C.; Rastogi, A.; et al. Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies. Cancers 2022, 14, 3779. https://doi.org/10.3390/cancers14153779
Rager T, Eckburg A, Patel M, Qiu R, Gantiwala S, Dovalovsky K, Fan K, Lam K, Roesler C, Rastogi A, et al. Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies. Cancers. 2022; 14(15):3779. https://doi.org/10.3390/cancers14153779
Chicago/Turabian StyleRager, Taylor, Adam Eckburg, Meet Patel, Rong Qiu, Shahina Gantiwala, Katrina Dovalovsky, Kelly Fan, Katie Lam, Claire Roesler, Aayush Rastogi, and et al. 2022. "Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies" Cancers 14, no. 15: 3779. https://doi.org/10.3390/cancers14153779
APA StyleRager, T., Eckburg, A., Patel, M., Qiu, R., Gantiwala, S., Dovalovsky, K., Fan, K., Lam, K., Roesler, C., Rastogi, A., Gautam, S., Dube, N., Morgan, B., Nasifuzzaman, S. M., Ramaswami, D., Gnanasekar, V., Smith, J., Merchant, A., & Puri, N. (2022). Treatment of Metastatic Melanoma with a Combination of Immunotherapies and Molecularly Targeted Therapies. Cancers, 14(15), 3779. https://doi.org/10.3390/cancers14153779