
Receptor Tyrosine Kinases Amplified in Diffuse-Type Gastric Carcinoma: Potential Targeted Therapies and Novel Downstream Effectors



Simple Summary
Abstract
1. Introduction
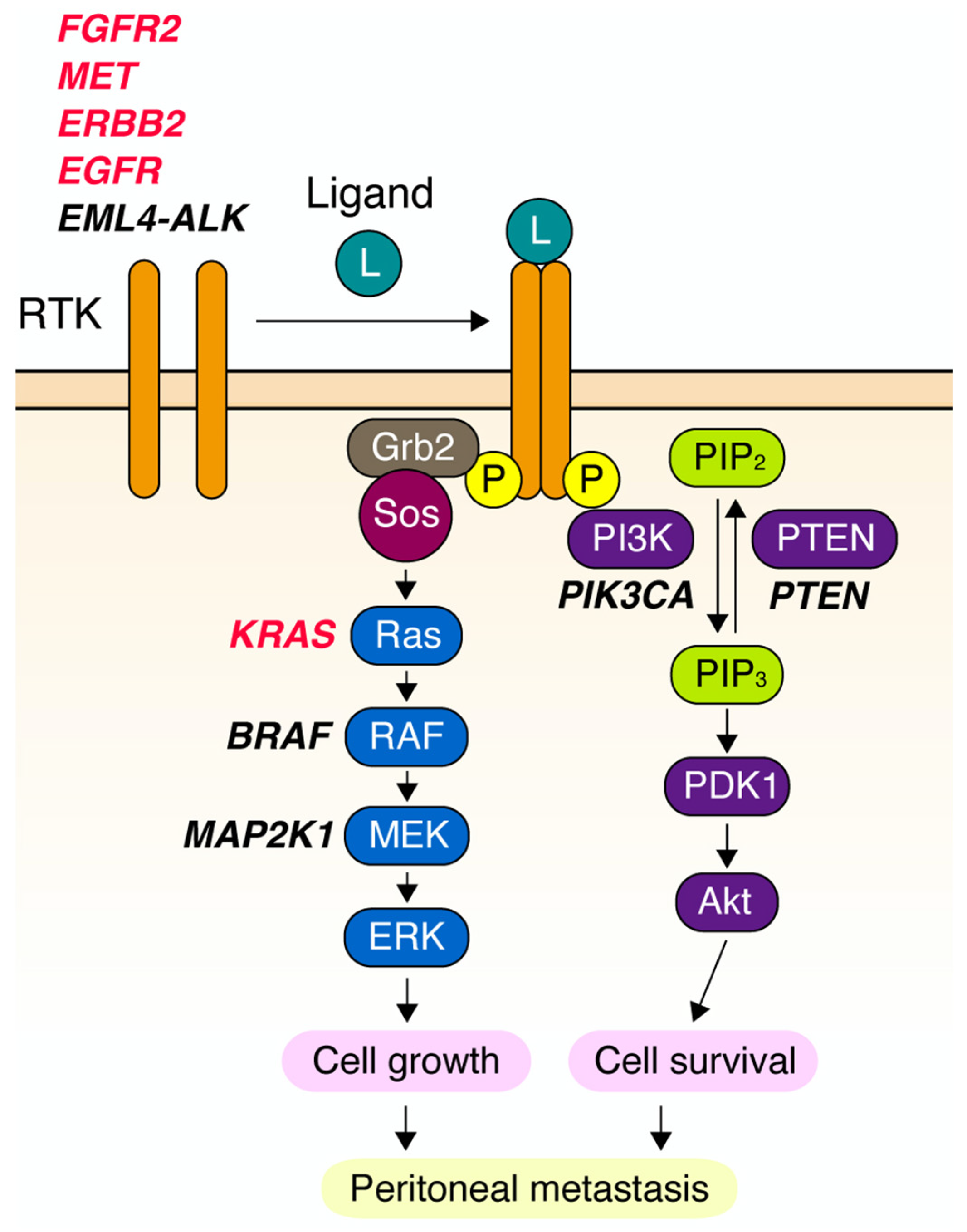
2. Gene Amplification of RTKs in DGC
3. Targeting RTKs for Peritoneal Dissemination of DGC
4. Novel Downstream Effectors of RTK in DGC
5. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Lauren, P. The two histological main types of gastric carcinoma: Diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol. Microbiol. Scand. 1965, 64, 31–49. [Google Scholar] [CrossRef] [PubMed]
- Ooki, A.; Yamaguchi, K. The dawn of precision medicine in diffuse-type gastric cancer. Ther. Adv. Med. Oncol. 2022, 14, 17588359221083049. [Google Scholar] [CrossRef]
- Yashiro, M.; Hirakawa, K. Cancer-stromal interactions in scirrhous gastric carcinoma. Cancer Microenviron. 2010, 3, 127–135. [Google Scholar] [CrossRef]
- Saraon, P.; Pathmanathan, S.; Snider, J.; Lyakisheva, A.; Wong, V.; Stagljar, I. Receptor tyrosine kinases and cancer: Oncogenic mechanisms and therapeutic approaches. Oncogene 2021, 40, 4079–4093. [Google Scholar] [CrossRef] [PubMed]
- Tsujino, T.; Yoshida, K.; Nakayama, H.; Ito, H.; Shimosato, T.; Tahara, E. Alterations of oncogenes in metastatic tumours of human gastric carcinomas. Br. J. Cancer 1990, 62, 226–230. [Google Scholar] [CrossRef]
- Tsugawa, K.; Yonemura, Y.; Hirono, Y.; Fushida, S.; Kaji, M.; Miwa, K.; Miyazaki, I.; Yamamoto, H. Amplification of the c-met, c-erbB-2 and epidermal growth factor receptor gene in human gastric cancers: Correlation to clinical features. Oncology 1998, 55, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Kwak, E.L.; Ackerman, A.; Michael, M.; Fox, S.B.; Bergethon, K.; Lauwers, G.Y.; Christensen, J.G.; Wilner, K.D.; Haber, D.A.; et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 2011, 29, 4803–4810. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuma, A.K.; Aizawa, M.; Kuwata, T.; Doi, T.; Ohtsu, A.; Fujii, H.; Ochiai, A. Expression profiles of HER2, EGFR, MET and FGFR2 in a large cohort of patients with gastric adenocarcinoma. Gastric Cancer 2015, 18, 227–238. [Google Scholar] [CrossRef]
- Tanaka, Y.; Chiwaki, F.; Kojima, S.; Kawazu, M.; Komatsu, M.; Ueno, T.; Inoue, S.; Sekine, S.; Matsusaki, K.; Matsushita, H.; et al. Multi-omic profiling of peritoneal metastases in gastric cancer identifies molecular subtypes and therapeutic vulnerabilities. Nat. Camcer 2021, 2, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Sawada, H.; Yamada, Y.; Watanabe, A.; Tatsumi, M.; Yamashita, J.; Matsuda, M.; Sakaguchi, T.; Hirao, T.; Nakano, H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer 1999, 85, 1894–1902. [Google Scholar] [CrossRef]
- Liu, Y.J.; Shen, D.; Yin, X.; Gavine, P.; Zhang, T.; Su, X.; Zhan, P.; Xu, Y.; Lv, J.; Qian, J.; et al. HER2, MET and FGFR2 oncogenic driver alterations define distinct molecular segments for targeted therapies in gastric carcinoma. Br. J. Cancer 2014, 110, 1169–1178. [Google Scholar] [CrossRef]
- Tajiri, R.; Ooi, A.; Fujimura, T.; Dobashi, Y.; Oyama, T.; Nakamura, R.; Ikeda, H. Intratumoral heterogeneous amplification of ERBB2 and subclonal genetic diversity in gastric cancers revealed by multiple ligation-dependent probe amplification and fluorescence in situ hybridization. Hum. Pathol. 2014, 45, 725–734. [Google Scholar] [CrossRef]
- Su, X.; Zhan, P.; Gavine, P.R.; Morgan, S.; Womack, C.; Ni, X.; Shen, D.; Bang, Y.J.; Im, S.A.; Ho Kim, W.; et al. FGFR2 amplification has prognostic significance in gastric cancer: Results from a large international multicentre study. Br. J. Cancer 2014, 110, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Bang, Y.J.; Feng-Yi, F.; Xu, J.M.; Lee, K.W.; Jiao, S.C.; Chong, J.L.; Lopez-Sanchez, R.I.; Price, T.; Gladkov, O.; et al. HER2 screening data from ToGA: Targeting HER2 in gastric and gastroesophageal junction cancer. Gastric Cancer 2015, 18, 476–484. [Google Scholar] [CrossRef]
- Wang, J.; Goetsch, L.; Tucker, L.; Zhang, Q.; Gonzalez, A.; Vaidya, K.S.; Oleksijew, A.; Boghaert, E.; Song, M.; Sokolova, I.; et al. Anti-c-Met monoclonal antibody ABT-700 breaks oncogene addiction in tumors with MET amplification. BMC Cancer 2016, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Phan, D.A.T.; Nguyen, V.T.; Hua, T.N.H.; Ngo, Q.D.; Doan, T.P.T.; Nguyen, S.T.; Thai, A.T.; Nguyen, V.T. HER2 status and its heterogeneity in gastric carcinoma of Vietnamese patient. J. Pathol. Transl. Med. 2017, 51, 396–402. [Google Scholar] [CrossRef]
- Rajadurai, P.; Fatt, H.K.; Ching, F.Y. Prevalence of HER2 positivity and its clinicopathological correlation in locally advanced/metastatic gastric cancer patients in Malaysia. J. Gastrointest. Cancer 2018, 49, 150–157. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Yasui, W.; Kitadai, Y.; Yokozaki, H.; Ito, H.; Tahara, E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem. Biophys. Res. Commun. 1992, 189, 227–232. [Google Scholar] [CrossRef]
- Hara, T.; Ooi, A.; Kobayashi, M.; Mai, M.; Yanagihara, K.; Nakanishi, I. Amplification of c-myc, K-sam, and c-met in gastric cancers: Detection by fluorescence in situ hybridization. Lab. Investig. 1998, 78, 1143–1153. [Google Scholar] [PubMed]
- Graziano, F.; Galluccio, N.; Lorenzini, P.; Ruzzo, A.; Canestrari, E.; D’Emidio, S.; Catalano, V.; Sisti, V.; Ligorio, C.; Andreoni, F.; et al. Genetic activation of the MET pathway and prognosis of patients with high-risk, radically resected gastric cancer. J. Clin. Oncol. 2011, 29, 4789–4795. [Google Scholar] [CrossRef]
- Janjigian, Y.Y.; Tang, L.H.; Coit, D.G.; Kelsen, D.P.; Francone, T.D.; Weiser, M.R.; Jhanwar, S.C.; Shah, M.A. MET expression and amplification in patients with localized gastric cancer. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Seo, J.W.; Jun, H.J.; Ki, C.S.; Park, S.H.; Park, Y.S.; Lim, H.Y.; Choi, M.G.; Bae, J.M.; Sohn, T.S.; et al. Impact of MET amplification on gastric cancer: Possible roles as a novel prognostic marker and a potential therapeutic target. Oncol. Rep. 2011, 25, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, H.; Okamoto, I.; Arao, T.; Okamoto, W.; Matsumoto, K.; Taniguchi, H.; Kuwata, K.; Yamaguchi, H.; Nishio, K.; Nakagawa, K.; et al. MET amplification as a potential therapeutic target in gastric cancer. Oncotarget 2012, 4, 9–17. [Google Scholar] [CrossRef]
- Shi, J.; Yao, D.; Liu, W.; Wang, N.; Lv, H.; He, N.; Shi, B.; Hou, P.; Ji, M. Frequent gene amplification predicts poor prognosis in gastric cancer. Int. J. Mol. Sci. 2012, 13, 4714–4726. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Song, X.; Zhu, M.; Ma, H. Overexpression of FGFR2 contributes to inherent resistance to MET inhibitors in MET-amplified patient-derived gastric cancer xenografts. Oncol. Lett. 2015, 10, 2003–2008. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Li, Z.; Gao, J.; Lu, M.; Gong, J.; Tang, E.T.; Oliner, K.S.; Hei, Y.J.; Zhou, H.; Shen, L. Tumor MET expression and gene amplification in Chinese patients with locally advanced or metastatic gastric or gastroesophageal junction cancer. Mol. Cancer Ther. 2015, 14, 2634–2641. [Google Scholar] [CrossRef]
- Liao, H.; Tian, T.; Sheng, Y.; Peng, Z.; Li, Z.; Wang, J.; Li, Y.; Zhang, C.; Gao, J. The significance of MET expression and strategies of targeting MET treatment in advanced gastric cancer. Front. Oncol. 2021, 11, 719217. [Google Scholar] [CrossRef]
- Nakatani, H.; Sakamoto, H.; Yoshida, T.; Yokota, J.; Tahara, E.; Sugimura, T.; Terada, M. Isolation of an amplified DNA sequence in stomach cancer. Jpn. J. Cancer Res. 1990, 81, 707–710. [Google Scholar] [CrossRef]
- Jung, E.J.; Jung, E.J.; Min, S.Y.; Kim, M.A.; Kim, W.H. Fibroblast growth factor receptor 2 gene amplification status and its clinicopathologic significance in gastric carcinoma. Hum. Pathol. 2012, 43, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Su, X.; Zhang, L.; Yin, X.; Tang, L.; Zhang, X.; Xu, Y.; Gao, Z.; Liu, K.; Zhou, M.; et al. FGFR2 gene amplification in gastric cancer predicts sensitivity to the selective FGFR inhibitor AZD4547. Clin. Cancer Res. 2013, 19, 2572–2583. [Google Scholar] [CrossRef] [PubMed]
- Betts, G.; Valentine, H.; Pritchard, S.; Swindell, R.; Williams, V.; Morgan, S.; Griffiths, E.A.; Welch, I.; West, C.; Womack, C. FGFR2, HER2 and cMet in gastric adenocarcinoma: Detection, prognostic significance and assessment of downstream pathway activation. Virchows Arch. 2014, 464, 145–156. [Google Scholar] [CrossRef]
- Kim, S.T.; Jang, H.L.; Lee, S.J.; Lee, J.; Choi, Y.L.; Kim, K.M.; Cho, J.; Park, S.H.; Park, Y.S.; Lim, H.Y.; et al. Pazopanib, a novel multitargeted kinase inhibitor, shows potent in vitro antitumor activity in gastric cancer cell lines with FGFR2 amplification. Mol. Cancer Ther. 2014, 13, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Kim, M.A.; Lee, H.S.; Kim, W.H. Evaluation of fibroblast growth factor receptor 2 expression, heterogeneity and clinical significance in gastric cancer. Pathobiology 2015, 82, 269–279. [Google Scholar] [CrossRef]
- Ahn, S.; Lee, J.; Hong, M.; Kim, S.T.; Park, S.H.; Choi, M.G.; Lee, J.H.; Sohn, T.S.; Bae, J.M.; Kim, S.; et al. FGFR2 in gastric cancer: Protein overexpression predicts gene amplification and high H-index predicts poor survival. Mod. Pathol. 2016, 29, 1095–1103. [Google Scholar] [CrossRef]
- Teles, S.P.; Oliveira, P.; Ferreira, M.; Carvalho, J.; Ferreira, P.; Oliveira, C. Integrated analysis of structural variation and RNA expression of FGFR2 and its splicing modulator ESRP1 highlight the ESRP1(amp)-FGFR2(norm)-FGFR2-IIIc(high) axis in diffuse gastric cancer. Cancers 2019, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Schrumpf, T.; Behrens, H.M.; Haag, J.; Kruger, S.; Rocken, C. FGFR2 overexpression and compromised survival in diffuse-type gastric cancer in a large central European cohort. PLoS ONE 2022, 17, e0264011. [Google Scholar] [CrossRef]
- Fu, J.; Su, X.; Li, Z.; Deng, L.; Liu, X.; Feng, X.; Peng, J. HGF/c-MET pathway in cancer: From molecular characterization to clinical evidence. Oncogene 2021, 40, 4625–4651. [Google Scholar] [CrossRef]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- Recondo, G.; Che, J.; Janne, P.A.; Awad, M.M. Targeting MET dysregulation in cancer. Cancer Discov. 2020, 10, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Matsuoka, T. Fibroblast growth factor receptor signaling as therapeutic targets in gastric cancer. World J. Gastroenterol. 2016, 22, 2415–2423. [Google Scholar] [CrossRef]
- Hattori, Y.; Odagiri, H.; Nakatani, H.; Miyagawa, K.; Naito, K.; Sakamoto, H.; Katoh, O.; Yoshida, T.; Sugimura, T.; Terada, M. K-sam, an amplified gene in stomach cancer, is a member of the heparin-binding growth factor receptor genes. Proc. Natl. Acad. Sci. USA 1990, 87, 5983–5987. [Google Scholar] [CrossRef]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Graziano, F.; Humar, B.; Guilford, P. The role of the E-cadherin gene (CDH1) in diffuse gastric cancer susceptibility: From the laboratory to clinical practice. Ann. Oncol. 2003, 14, 1705–1713. [Google Scholar] [CrossRef]
- Wang, K.; Li, E.; Busuttil, R.A.; Kong, J.C.; Pattison, S.; Sung, J.J.Y.; Yu, J.; El-Omar, E.M.; Simpson, J.A.; Boussioutas, A. A cohort study and meta-analysis of the evidence for consideration of Lauren subtype when prescribing adjuvant or palliative chemotherapy for gastric cancer. Ther. Adv. Med. Oncol. 2020, 12, 1758835920930359. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Kim, J.; Park, K.E.; Jeong, Y.S.; Kim, Y.; Park, H.; Nam, J.H.; Jung, K.; Son, W.S.; Jung, H.S.; Lee, J.H.; et al. Therapeutic efficacy of ABN401, a highly potent and selective MET inhibitor, based on diagnostic biomarker test in MET-addicted cancer. Cancers 2020, 12, 1575. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.E.; Rex, K.; Caenepeel, S.; Yang, Y.; Zhang, Y.; Broome, M.A.; Kha, H.T.; Burgess, T.L.; Amore, B.; Kaplan-Lefko, P.J.; et al. In vitro and in vivo activity of AMG 337, a potent and selective MET kinase inhibitor, in MET-dependent cancer models. Mol. Cancer Ther. 2016, 15, 1568–1579. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Karaszewska, B.; Kang, Y.K.; Chung, H.C.; Shankaran, V.; Siena, S.; Go, N.F.; Yang, H.; Schupp, M.; Cunningham, D. A multicenter phase II study of AMG 337 in patients with MET-amplified gastric/gastroesophageal junction/esophageal adenocarcinoma and other MET-amplified solid tumors. Clin. Cancer Res. 2019, 25, 2414–2423. [Google Scholar] [CrossRef]
- Kasai, S.; Kuwayama, N.; Motoo, Y.; Kawashima, A.; Matsumoto, K.; Yano, S.; Matsushima, K.; Yasumoto, K. Dual blockade of MET and VEGFR2 signaling pathways as a potential therapeutic maneuver for peritoneal carcinomatosis in scirrhous gastric cancer. Biochem. Biophys. Res. Commun. 2022, 600, 80–86. [Google Scholar] [CrossRef]
- Sohn, S.H.; Kim, B.; Sul, H.J.; Kim, Y.J.; Kim, H.S.; Kim, H.; Seo, J.B.; Koh, Y.; Zang, D.Y. INC280 inhibits Wnt/beta-catenin and EMT signaling pathways and its induce apoptosis in diffuse gastric cancer positive for c-MET amplification. BMC Res. Notes 2019, 12, 125. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, W.; Okamoto, I.; Arao, T.; Kuwata, K.; Hatashita, E.; Yamaguchi, H.; Sakai, K.; Yanagihara, K.; Nishio, K.; Nakagawa, K. Antitumor action of the MET tyrosine kinase inhibitor crizotinib (PF-02341066) in gastric cancer positive for MET amplification. Mol. Cancer Ther. 2012, 11, 1557–1564. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Takanashi, M.; Yoshida, N.; Ito, Y.; Kamata, R.; Fukami, K.; Yanagihara, K.; Sakai, R. Saracatinib impairs the peritoneal dissemination of diffuse-type gastric carcinoma cells resistant to Met and fibroblast growth factor receptor inhibitors. Cancer Sci. 2014, 105, 528–536. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tohyama, O.; Yamaguchi, A.; Matsushima, T.; Takahashi, K.; Funasaka, S.; Shirotori, S.; Asada, M.; Obaishi, H. E7050: A dual c-Met and VEGFR-2 tyrosine kinase inhibitor promotes tumor regression and prolongs survival in mouse xenograft models. Cancer Sci. 2010, 101, 210–215. [Google Scholar] [CrossRef]
- Kataoka, Y.; Mukohara, T.; Tomioka, H.; Funakoshi, Y.; Kiyota, N.; Fujiwara, Y.; Yashiro, M.; Hirakawa, K.; Hirai, M.; Minami, H. Foretinib (GSK1363089), a multi-kinase inhibitor of MET and VEGFRs, inhibits growth of gastric cancer cell lines by blocking inter-receptor tyrosine kinase networks. Investig. New Drugs 2012, 30, 1352–1360. [Google Scholar] [CrossRef]
- Kim, H.J.; Kang, S.K.; Kwon, W.S.; Kim, T.S.; Jeong, I.; Jeung, H.C.; Kragh, M.; Horak, I.D.; Chung, H.C.; Rha, S.Y. Forty-nine gastric cancer cell lines with integrative genomic profiling for development of c-MET inhibitor. Int. J. Cancer 2018, 143, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Fujita, R.; Blot, V.; Wong, E.; Stewart, C.; Lieuw, V.; Richardson, R.; Banah, A.; Villicana, J.; Timmer, A.; Coronella, J.; et al. A novel non-agonist c-Met antibody drug conjugate with superior potency over a c-Met tyrosine kinase inhibitor in c-Met amplified and non-amplified cancers. Cancer Biol. Ther. 2020, 21, 549–559. [Google Scholar] [CrossRef]
- Smolen, G.A.; Sordella, R.; Muir, B.; Mohapatra, G.; Barmettler, A.; Archibald, H.; Kim, W.J.; Okimoto, R.A.; Bell, D.W.; Sgroi, D.C.; et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. USA 2006, 103, 2316–2321. [Google Scholar] [CrossRef] [PubMed]
- Burbridge, M.F.; Bossard, C.J.; Saunier, C.; Fejes, I.; Bruno, A.; Leonce, S.; Ferry, G.; Da Violante, G.; Bouzom, F.; Cattan, V.; et al. S49076 is a novel kinase inhibitor of MET, AXL, and FGFR with strong preclinical activity alone and in association with bevacizumab. Mol. Cancer Ther. 2013, 12, 1749–1762. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.D.O.; Grondine, M.; Borodovsky, A.; San Martin, M.; DuPont, M.; D’Cruz, C.; Schuller, A.; Henry, R.; Barry, E.; Castriotta, L.; et al. A pharmacokinetic-pharmacodynamic model for the MET tyrosine kinase inhibitor, savolitinib, to explore target inhibition requirements for anti-tumour activity. Br. J. Pharmacol. 2021, 178, 600–613. [Google Scholar] [CrossRef]
- Gavine, P.R.; Ren, Y.; Han, L.; Lv, J.; Fan, S.; Zhang, W.; Xu, W.; Liu, Y.J.; Zhang, T.; Fu, H.; et al. Volitinib, a potent and highly selective c-Met inhibitor, effectively blocks c-Met signaling and growth in c-MET amplified gastric cancer patient-derived tumor xenograft models. Mol. Oncol. 2015, 9, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, S.T.; Kim, K.; Lee, H.; Kozarewa, I.; Mortimer, P.G.S.; Odegaard, J.I.; Harrington, E.A.; Lee, J.; Lee, T.; et al. Tumor genomic profiling guides patients with metastatic gastric cancer to targeted treatment: The VIKTORY umbrella trial. Cancer Discov. 2019, 9, 1388–1405. [Google Scholar] [CrossRef] [PubMed]
- Toiyama, Y.; Yasuda, H.; Saigusa, S.; Matushita, K.; Fujikawa, H.; Tanaka, K.; Mohri, Y.; Inoue, Y.; Goel, A.; Kusunoki, M. Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. Int. J. Cancer 2012, 130, 2912–2921. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Kim, Y.J.; Sohn, S.H.; Kim, B.; Sul, H.J.; Kim, H.S.; Zang, D.Y. Tivantinib inhibits the VEGF signaling pathway and induces apoptosis in gastric cancer cells with c-MET or VEGFA amplification. Investig. New Drugs 2020, 38, 1633–1640. [Google Scholar] [CrossRef]
- Okuno, T.; Yashiro, M.; Masuda, G.; Togano, S.; Kuroda, K.; Miki, Y.; Hirakawa, K.; Ohsawa, M.; Wanibuchi, H.; Ohira, M. Establishment of a new scirrhous gastric cancer cell line with FGFR2 overexpression, OCUM-14. Ann. Surg. Oncol. 2019, 26, 1093–1102. [Google Scholar] [CrossRef]
- Ran, K.; Zeng, J.; Wan, G.; He, X.; Feng, Z.; Xiang, W.; Wei, W.; Hu, X.; Wang, N.; Liu, Z.; et al. Design, synthesis and biological evaluations of a series of Pyrido[1,2-a]pyrimidinone derivatives as novel selective FGFR inhibitors. Eur. J. Med. Chem. 2021, 220, 113499. [Google Scholar] [CrossRef]
- Perera, T.P.S.; Jovcheva, E.; Mevellec, L.; Vialard, J.; De Lange, D.; Verhulst, T.; Paulussen, C.; Van De Ven, K.; King, P.; Freyne, E.; et al. Discovery and pharmacological characterization of JNJ-42756493 (erdafitinib), a functionally selective small-molecule FGFR family inhibitor. Mol. Cancer Ther. 2017, 16, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib is a novel irreversible FGFR 1-4 inhibitor that shows selective antitumor activity against FGFR-deregulated tumors. Cancer Res. 2020, 80, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Yashiro, M.; Matsuoka, T.; Tendo, M.; Shimizu, T.; Miwa, A.; Hirakawa, K. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology 2006, 131, 1530–1541. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, M.; Shinto, O.; Nakamura, K.; Tendo, M.; Matsuoka, T.; Matsuzaki, T.; Kaizaki, R.; Miwa, A.; Hirakawa, K. Synergistic antitumor effects of FGFR2 inhibitor with 5-fluorouracil on scirrhous gastric carcinoma. Int. J. Cancer 2010, 126, 1004–1016. [Google Scholar] [CrossRef]
- Zhao, G.; Li, W.Y.; Chen, D.; Henry, J.R.; Li, H.Y.; Chen, Z.; Zia-Ebrahimi, M.; Bloem, L.; Zhai, Y.; Huss, K.; et al. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Mol. Cancer Ther. 2011, 10, 2200–2210. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Tontsch-Grunt, U.; Baum, A.; Le, A.T.; Doebele, R.C.; Lieb, S.; Gianni, D.; Voss, T.; Garin-Chesa, P.; Haslinger, C.; et al. Triple angiokinase inhibitor nintedanib directly inhibits tumor cell growth and induces tumor shrinkage via blocking oncogenic receptor tyrosine kinases. J. Pharmacol. Exp. Ther. 2018, 364, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Kunii, K.; Davis, L.; Gorenstein, J.; Hatch, H.; Yashiro, M.; Di Bacco, A.; Elbi, C.; Lutterbach, B. FGFR2-amplified gastric cancer cell lines require FGFR2 and Erbb3 signaling for growth and survival. Cancer Res. 2008, 68, 2340–2348. [Google Scholar] [CrossRef] [PubMed]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.F.; Dai, Y.; Peng, X.; Shen, Y.Y.; Su, Y.; Wei, M.M.; Liu, W.R.; Ding, Z.B.; Zhang, A.; Shi, Y.H.; et al. SOMCL-085, a novel multi-targeted FGFR inhibitor, displays potent anticancer activity in FGFR-addicted human cancer models. Acta Pharmacol. Sin. 2018, 39, 243–250. [Google Scholar] [CrossRef]
- Du, G.; Jiang, J.; Wu, Q.; Henning, N.J.; Donovan, K.A.; Yue, H.; Che, J.; Lu, W.; Fischer, E.S.; Bardeesy, N.; et al. Discovery of a potent degrader for fibroblast growth factor receptor 1/2. Angew. Chem. Int. Ed. Engl. 2021, 60, 15905–15911. [Google Scholar] [CrossRef]
- Xiang, H.; Chan, A.G.; Ahene, A.; Bellovin, D.I.; Deng, R.; Hsu, A.W.; Jeffry, U.; Palencia, S.; Powers, J.; Zanghi, J.; et al. Preclinical characterization of bemarituzumab, an anti-FGFR2b antibody for the treatment of cancer. MAbs 2021, 13, 1981202. [Google Scholar] [CrossRef]
- Kim, S.T.; Lee, I.K.; Rom, E.; Sirkis, R.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K.; Kim, K.M.; et al. Neutralizing antibody to FGFR2 can act as a selective biomarker and potential therapeutic agent for gastric cancer with FGFR2 amplification. Am. J. Transl. Res. 2019, 11, 4508–4515. [Google Scholar]
- Sommer, A.; Kopitz, C.; Schatz, C.A.; Nising, C.F.; Mahlert, C.; Lerchen, H.G.; Stelte-Ludwig, B.; Hammer, S.; Greven, S.; Schuhmacher, J.; et al. Preclinical efficacy of the auristatin-based antibody-drug conjugate BAY 1187982 for the treatment of FGFR2-positive solid tumors. Cancer Res. 2016, 76, 6331–6339. [Google Scholar] [CrossRef]
- Nagamura, Y.; Miyazaki, M.; Nagano, Y.; Tomiyama, A.; Ohki, R.; Yanagihara, K.; Sakai, R.; Yamaguchi, H. SHP2 as a potential therapeutic target in diffuse-type gastric carcinoma addicted to receptor tyrosine kinase signaling. Cancers 2021, 13, 4309. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Liu, X.; Wu, Y.; Fang, X.; Huang, G. Overexpression of PI3K p110alpha contributes to acquired resistance to MET inhibitor, in MET-amplified SNU-5 gastric xenografts. Drug Des. Dev. Ther. 2015, 9, 5697–5704. [Google Scholar] [CrossRef]
- Lengyel, C.G.; Hussain, S.; Seeber, A.; Jamil Nidhamalddin, S.; Trapani, D.; Habeeb, B.S.; Elfaham, E.; Mazher, S.A.; Seid, F.; Khan, S.Z.; et al. FGFR pathway inhibition in gastric cancer: The golden era of an old target? Life 2022, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Tsimafeyeu, I.; Ludes-Meyers, J.; Stepanova, E.; Daeyaert, F.; Khochenkov, D.; Joose, J.B.; Solomko, E.; Van Akene, K.; Peretolchina, N.; Yin, W.; et al. Targeting FGFR2 with alofanib (RPT835) shows potent activity in tumour models. Eur. J. Cancer 2016, 61, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kwak, E.L.; Ahronian, L.G.; Siravegna, G.; Mussolin, B.; Borger, D.R.; Godfrey, J.T.; Jessop, N.A.; Clark, J.W.; Blaszkowsky, L.S.; Ryan, D.P.; et al. Molecular heterogeneity and receptor coamplification drive resistance to targeted therapy in MET-amplified esophagogastric cancer. Cancer Discov. 2015, 5, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Petti, C.; Picco, G.; Martelli, M.L.; Trisolini, E.; Bucci, E.; Perera, T.; Isella, C.; Medico, E. Truncated RAF kinases drive resistance to MET inhibition in MET-addicted cancer cells. Oncotarget 2015, 6, 221–233. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ahn, T.; Bang, H.; Ham, J.S.; Kim, J.; Kim, S.T.; Jang, J.; Shim, M.; Kang, S.Y.; Park, S.H.; et al. Acquired resistance to LY2874455 in FGFR2-amplified gastric cancer through an emergence of novel FGFR2-ACSL5 fusion. Oncotarget 2017, 8, 15014–15022. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lau, W.M.; Teng, E.; Huang, K.K.; Tan, J.W.; Das, K.; Zang, Z.; Chia, T.; Teh, M.; Kono, K.; Yong, W.P.; et al. Acquired resistance to FGFR inhibitor in diffuse-type gastric cancer through an AKT-independent PKC-mediated phosphorylation of GSK3beta. Mol. Cancer Ther. 2018, 17, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an irreversible FGFR1-4 inhibitor, in patients with advanced solid tumors harboring FGF/FGFR aberrations: A phase I dose-expansion study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Nagamura, Y.; Miyazaki, M.; Nagano, Y.; Yuki, M.; Fukami, K.; Yanagihara, K.; Sasaki, K.; Sakai, R.; Yamaguchi, H. PLEKHA5 regulates the survival and peritoneal dissemination of diffuse-type gastric carcinoma cells with Met gene amplification. Oncogenesis 2021, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793. [Google Scholar] [CrossRef]
- Chen, Y.N.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.; Chen, Z.; Cooke, V.G.; et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Guo, W.; Wu, Y.; Yang, C.; Zhong, L.; Deng, G.; Zhu, Y.; Liu, W.; Gu, Y.; Lu, Y.; et al. SHP2 inhibition triggers anti-tumor immunity and synergizes with PD-1 blockade. Acta Pharm. Sin. B 2019, 9, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Li, X.; Dong, D.; Zhang, B.; Xue, Y.; Shang, P. Transferrin receptor 1 in cancer: A new sight for cancer therapy. Am. J. Cancer Res. 2018, 8, 916–931. [Google Scholar] [PubMed]
- Shirakihara, T.; Yamaguchi, H.; Kondo, T.; Yashiro, M.; Sakai, R. Transferrin receptor 1 promotes the fibroblast growth factor receptor-mediated oncogenic potential of diffused-type gastric cancer. Oncogene 2022, 41, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Candelaria, P.V.; Leoh, L.S.; Penichet, M.L.; Daniels-Wells, T.R. Antibodies targeting the transferrin receptor 1 (TfR1) as direct anti-cancer agents. Front. Immunol. 2021, 12, 607692. [Google Scholar] [CrossRef] [PubMed]
- Reddavid, R.; Dagatti, S.; Franco, C.; Puca, L.; Tomatis, M.; Corso, S.; Giordano, S.; Degiuli, M. Molecularly targeted therapies for gastric cancer. State of the art. Cancers 2021, 13, 4094. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene | Sample (1) | Frequency (%) (2) | Technique (3) | Classification (4) | Associated Phenotypes (5) | Ref. |
|---|---|---|---|---|---|---|
| EGFR | Early GC Advanced GC Metastatic GC | 0/20 (0%) 1/69 (1.4%) 3/32 (9.3%) | Southern blot | Metastatic tumor | [6] | |
| EGFR | GC | 6/70 (8.5%) | Slot blot | >2-fold | Large tumor, advanced stage, poor survival | [7] |
| EGFR | GEC | 23/489 (4.7%) | FISH | EGFR/CEP7 > 2.2 | Squamous cell carcinoma, poor survival | [8] |
| EGFR | GC | 15/193 (7.7%) | SNP array | CNA | [9] | |
| EGFR | GC | 17/293 (5.8%) | SNP array | CNA | [10] | |
| EGFR | GC | 23/950 (2.4%) | FISH | EGFR/CEP7 ≥ 2 | [11] | |
| EGFR | GC ascites | 4/98 (4.0%) | WGS | CNA > 5 × ploidy | [12] | |
| HER2 | Early GC Advanced GC Metastatic GC | 0/20 (0%) 4/69 (5.7%) 8/32 (25%) | Southern blot | Metastatic tumor | [6] | |
| HER2 | GC | 9/70 (12.8%) | Slot blot | >2-fold | Lymph node metastasis, poor survival | [7] |
| HER2 | GC | 15/128 (11.7%) | Southern blot | >2-fold | IGC, poor survival | [13] |
| HER2 | GEC | 45/489 (9.2%) | FISH | HER2/CEP17 > 2.2 | [8] | |
| HER2 | GC | 14/193 (7.2%) | SNP array | CNA | Poor survival | [9] |
| HER2 | GC | 38/293 (12.9%) | SNP array | CNA | [10] | |
| HER2 | Chinese GC | 33/219 (15.0%) | FISH | HER2/CEP17 > 2 | [14] | |
| HER2 | GC | 51/475 (10.7%) | FISH | HER2/CEP17 > 2.2 | Differentiated | [15] |
| HER2 | Chinese GC Korean GC | 30/204 (14.7%) 27/338 (7.9%) | FISH | HER2/CEP17 ≥ 2 | [16] | |
| HER2 | GC | 90/950 (9.4%) | FISH | HER2/CEP17 ≥ 2 | [11] | |
| HER2 | GC/GEJC | 756/3280 (23.0%) | FISH | HER2/CEP17 ≥ 2 | IGC | [17] |
| HER2 | Asian GC | 32/134 (23.8%) | FISH | HER2/CEP17 ≥ 2 | 9/32 have Met coamplification | [18] |
| HER2 | GC | 33/208 (15.8%) | FISH/SISH | HER2/CEP17 ≥ 2 | IGC, differentiated, heterogeneity is associated with DGC | [19] |
| HER2 | GC/GEC | 40/228 (17.5%) | FISH | [20] | ||
| HER2 | GC ascites | 5/98 (5.1%) | WGS | CNA > 5 × ploidy | [12] | |
| MET | GC cell line Early GC Advanced GC SGC | 6/11 (54.5%) 0/11 (0%) 15/64 (23.4%) 5/13 (38.4%) | Southern blot | ≥3-fold | [21] | |
| MET | GC | 6/154 (3.8%) | FISH | [22] | ||
| MET | GC | 7/70 (10%) | Slot blot | >2-fold | Infiltrative invasion, peritoneal dissemination, poor survival | [7] |
| MET | GC | 13/128 (10.1%) | Southern blot | >2-fold | Lymph node metastasis, poor survival | [13] |
| MET | Stage II/III GC | 21/216 (9.7%) | qPCR | ≥5 copies | Poor survival | [23] |
| MET | Western GC | 0/38 (0%) | FISH | MET/CEP7 > 2 | [24] | |
| MET | GC | 100/472 (21.1%) | qPCR | >4 copies | Poor survival | [25] |
| MET | GEC | 10/489 (2.0%) | FISH | MET/CEP7 > 2.2 | High-grade, advanced stages, poor survival | [8] |
| MET | GC | 8/193 (4.1%) | SNP array | CNA | Poor survival | [9] |
| MET | GC GC cell line | 4/266 (1.5%) 3/11 (27.2%) | qPCR/FISH | ≥4 copies | [26] | |
| MET | GC | 39/128 (30.4%) | qPCR | ≥4 copies | Invasion, poor survival | [27] |
| MET | GC | 12/293 (4.1%) | SNP array | CNA | [10] | |
| MET | Chinese GC | 12/196 (6.1%) | FISH | MET/CEP7 > 2 | Lymph node and distant metastasis, Poor survival | [14] |
| MET | GC xenograft | 5/30 (16.6%) | SNP array | CNA | [28] | |
| MET | GC | 12/950 (1.2%) | FISH | MET/CEP7 ≥ 2 | [11] | |
| MET | Chinese advanced or metastatic GC or GEJC | 8/113 (7.0%) | FISH | MET/CEP7 > 2 | DGC | [29] |
| MET | Asian GC | 13/134 (9.7%) | FISH | MET/CEP7 ≥ 2 | 9/13 have HER2 coamplification | [18] |
| MET | GC | 7/49 (14.2%) | CISH | MET/CEP7 ≥ 2 | [30] | |
| MET | GC ascites | 7/98 (7.1%) | WGS | CNA > 5 × ploidy | [12] | |
| FGFR2 | GC GC xenograft | 3/24 (12.5%) 2/13 (15.3%) | Southern blot | [31] | ||
| FGFR2 | GC | 3/154 (1.9%) | FISH | [22] | ||
| FGFR2 | GC | 18/193 (9.3%) | SNP array | CNA | [9] | |
| FGFR2 | GC | 14/313 (4.4%) | FISH | FGFR2/CEP10 ≥ 2 | Invasion, metastasis, poor survival | [32] |
| FGFR2 | Chinese GC Chinese GC Caucasian GC | 3/131 (2.2%) 9/197 (4.5%) 7/97 (7.2%) | aCGH FISH | log ratio > 0.8 FGFR2/CEP10 ≥ 2 | [33] | |
| FGFR2 | GC | 3/171 (1.7%) | FISH | FGFR2/CEP10 ≥ 2 | Poor survival | [34] |
| FGFR2 | GC | 15/293 (5.1%) | SNP array | CNA | [10] | |
| FGFR2 | GC cell line GC | 4/38 (10.5%) 24/482 (4.9%) | FISH qRT-PCR | FGFR2/CEP10 ≥ 2 > 4 copies | [35] | |
| FGFR2 | Chinese GC | 10/198 (5.0%) | FISH | FGFR2/CEP10 > 2 | [14] | |
| FGFR2 | UK GC Chinese GC Korean GC | 30/408 (7.3%) 9/197 (4.4%) 15/356 (4.2%) | FISH | FGFR2/CEP10 ≥ 2 | Lymph node metastasis and poor survival | [16] |
| FGFR2 | GC | 5/188 (2.6%) | FISH | FGFR2/CEP10 ≥ 2 | [36] | |
| FGFR2 | GC | 67/1974 (3.3%) | FISH | FGFR2/CEP10 > 2 | [37] | |
| FGFR2 | GC (TCGA) | 63/338 (18.6%) | WGS | CNA | [38] | |
| FGFR2 | GC ascites | 11/98 (11.2%) | WGS | CNA > 5 × ploidy | [12] | |
| FGFR2 | Non-Asian GC | 20/493 (4.0%) | CISH | FGFR2/CEP10 > 2 | [39] |
| Gene | Dataset | Amplified/Total Tumors | Frequency |
|---|---|---|---|
| EGFR | ICGC_TCGA2020 | 10/68 | 14.7% |
| MSKCC2017 | 6/100 | 6% | |
| OrigiMed2020 | 23/850 | 2.7% | |
| TCGA_PanCancerAtlas_STAD | 23/438 | 5.2% | |
| MSK2021 | 16/320 | 5% | |
| TCGA2014 | 17/293 | 5.8% | |
| ERBB2 | ICGC_TCGA2020 | 15/68 | 22.0% |
| MSKCC2017 | 18/100 | 18% | |
| OrigiMed2020 | 68/850 | 8% | |
| TCGA_PanCancerAtlas_STAD | 58/438 | 13.2% | |
| MSK2021 | 37/320 | 11.5% | |
| TCGA2014 | 38/293 | 12.9% | |
| MET | ICGC_TCGA2020 | 6/68 | 8.8% |
| MSKCC2017 | 4/100 | 4% | |
| OrigiMed2020 | 19/850 | 2.2% | |
| TCGA_PanCancerAtlas_STAD | 12/438 | 2.7% | |
| MSK2021 | 11/320 | 3.4% | |
| TCGA2014 | 12/293 | 4.0% | |
| FGFR2 | ICGC_TCGA2020 | 2/68 | 2.9% |
| MSKCC2017 | 2/100 | 2% | |
| OrigiMed2020 | 46/850 | 5.4% | |
| TCGA_PanCancerAtlas_STAD | 19/438 | 4.3% | |
| MSK2021 | 12/320 | 3.7% | |
| TCGA2014 | 15/293 | 5.1% |
| Drug | Type (1) | Target | Inhibited Functions and Phenotypes | Refs. |
|---|---|---|---|---|
| ABN401 | SMI | Met | Cell growth, survival, tumor growth | [49] |
| AMG 337 | SMI | Met | Cell growth, survival, tumor growth | [50,51] |
| Cabozantinib | SMI | Met/VEGFR2 | Cell growth | [52] |
| Capmatinib/ INC280 | SMI | Met | Cell growth, peritoneal metastasis | [12,53] |
| Crizotinib/PF-02341066 | SMI | Met/ALK | Cell growth, survival, tumor growth | [54,55] |
| E7050 | SMI | Met/VEGFR2 | Cell growth, tumor growth, angiogenesis, peritoneal metastasis | [56] |
| Foretinib/GSK1363089 | SMI | Met/VEGFR/PDGFRβ/Tie-2/RON/AXL | Cell growth | [57,58] |
| JNJ38877605 | SMI | Met | Cell growth, survival | [26,55] |
| PHA-665752 | SMI | Met | Cell growth, survival, tumor growth, peritoneal metastasis, ascites formation | [25,55,59,60] |
| S49076 | SMI | Met/FGFR1-3/AXL | Cell growth, tumor growth | [61] |
| Savolitinib/Volitinib | SMI | Met | Cell growth, tumor growth | [30,62,63,64] |
| SGX523 | SMI | Met | Cell growth, survival | [26] |
| SU11274 | SMI | Met | Cell growth, survival, migration, peritoneal metastasis | [65] |
| Tivantinib/ARQ197 | SMI | Met | Cell growth, survival | [58,66] |
| ABT-700 | mAb | Met | Cell growth, survival, tumor growth | [18] |
| SAIT301 | mAb | Met | Cell growth | [58] |
| Sym015 | mAb | Met | Cell growth | [58] |
| P3D12-vc-MMAF | ADC | Met | Cell survival, tumor growth | [59] |
| AZD4547 | SMI | FGFR1-3 | Cell growth, tumor growth | [33,67] |
| Compound 23d | SMI | FGFR1-4 | Cell growth, survival, tumor growth | [68] |
| Dovitinib | SMI | FGFR/VEGFR | Cell growth, survival, tumor growth | [9] |
| Erdafitinib/JNJ-42756493 | SMI | FGFR1-4 | Cell growth, tumor growth | [68,69] |
| Futibatinib | SMI | FGFR1-4 | Cell growth, tumor growth | [70] |
| Infigratinib/BGJ398 | SMI | FGFR1-3 | Cell growth, peritoneal metastasis | [12,67] |
| Ki23057 | SMI | FGFR1, 2/VEGFR/PDGFR/c-Kit | Cell growth, survival, tumor growth, peritoneal metastasis, lymph node metastasis, ascites formation | [71,72] |
| LY2874455 | SMI | FGFR1-4 | Tumor growth | [73] |
| Nintedanib | SMI | FGFR1-3/VEGFR1-3/PDGFRα, β | Cell growth | [74] |
| Pazopanib | SMI | FGFR/VEGFR/PDGFR/c-Kit | Cell growth, cell survival | [35] |
| PD173074 | SMI | FGFR1-3 | Cell growth, survival | [35,55,75] |
| Ponatinib/AP24534 | SMI | FGFR/Bcr-Abl/VEGFR/PDGFR/Src | Cell growth, tumor growth | [76] |
| SOMCL-085 | SMI | FGFR/VEGFR/PDGFR | Cell growth, tumor growth | [77] |
| DGY-09-192 | PROTAC | FGFR1, 2 | Cell growth | [78] |
| Bemarituzumab | mAb | FGFR2b | Cell growth, tumor growth | [79] |
| PRO-007 | mAb | FGFR2 | Cell growth, invasion | [80] |
| BAY 1187982 | ADC | FGFR2 | Tumor growth | [81] |
| Osimertinib | SMI | EGFR | Cell growth | [12] |
| SHP099 | SMI | SHP2 | Cell growth, migration, invasion, peritoneal metastasis, ascites formation | [82] |
| PI-103 | SMI | PI3K | Tumor growth | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, H.; Nagamura, Y.; Miyazaki, M. Receptor Tyrosine Kinases Amplified in Diffuse-Type Gastric Carcinoma: Potential Targeted Therapies and Novel Downstream Effectors. Cancers 2022, 14, 3750. https://doi.org/10.3390/cancers14153750
Yamaguchi H, Nagamura Y, Miyazaki M. Receptor Tyrosine Kinases Amplified in Diffuse-Type Gastric Carcinoma: Potential Targeted Therapies and Novel Downstream Effectors. Cancers. 2022; 14(15):3750. https://doi.org/10.3390/cancers14153750
Chicago/Turabian StyleYamaguchi, Hideki, Yuko Nagamura, and Makoto Miyazaki. 2022. "Receptor Tyrosine Kinases Amplified in Diffuse-Type Gastric Carcinoma: Potential Targeted Therapies and Novel Downstream Effectors" Cancers 14, no. 15: 3750. https://doi.org/10.3390/cancers14153750
APA StyleYamaguchi, H., Nagamura, Y., & Miyazaki, M. (2022). Receptor Tyrosine Kinases Amplified in Diffuse-Type Gastric Carcinoma: Potential Targeted Therapies and Novel Downstream Effectors. Cancers, 14(15), 3750. https://doi.org/10.3390/cancers14153750