
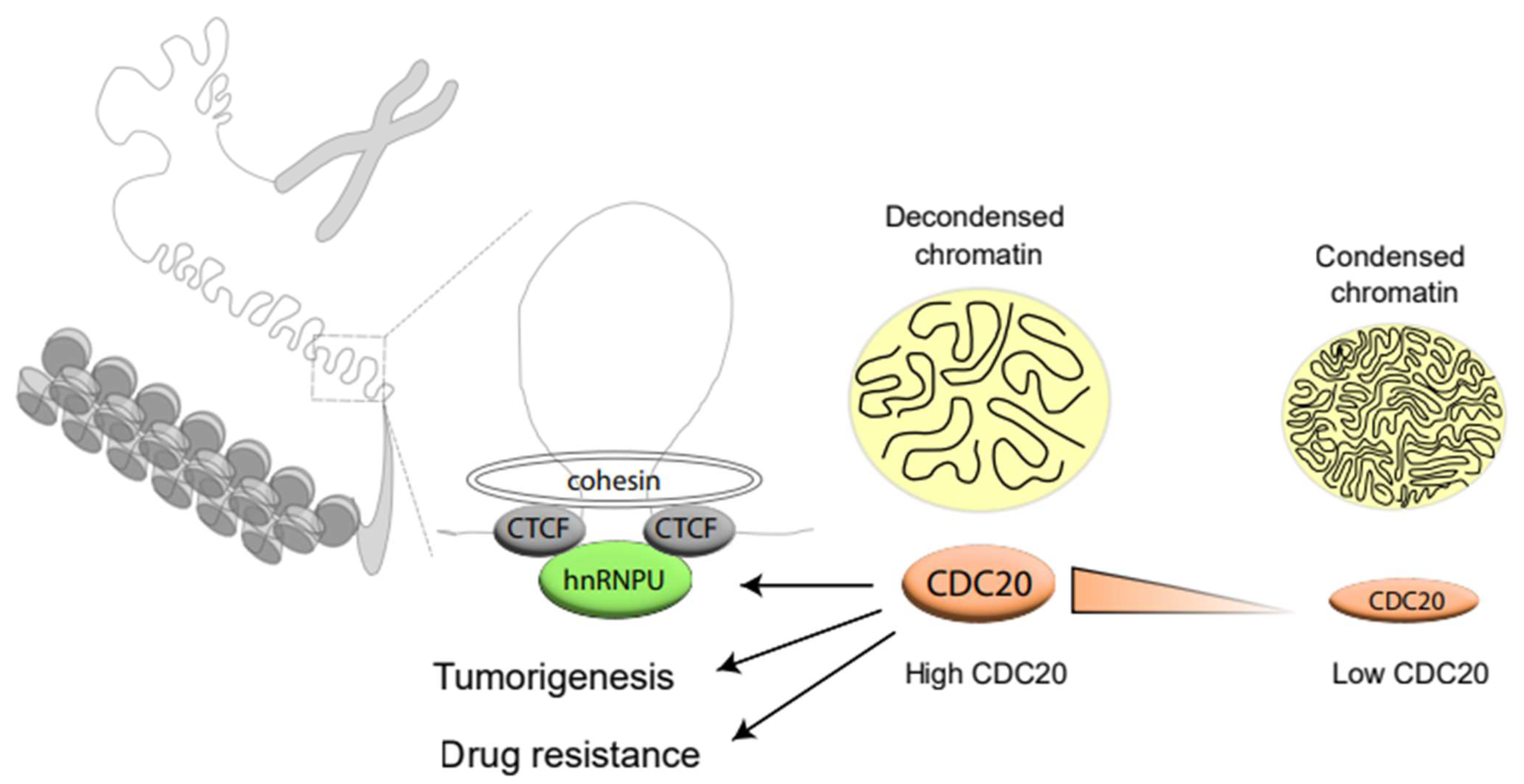
CDC20-Mediated hnRNPU Ubiquitination Regulates Chromatin Condensation and Anti-Cancer Drug Response

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Condition
2.2. Western Blot
2.3. Bioinformatics Analysis
2.4. Plasmid Constructions and Mutagenesis
2.5. Lentiviral Particles Production and Infection
2.6. Survival Assay
2.7. Microscopy, Immunofluorescence, and Images Analysis
2.8. Flow Cytometry
2.9. Crosslinking and Cell Fractionation
2.10. Purification and Mass Spectrometry
2.11. Immunoprecipitation
2.12. Clonogenic Assay
2.13. Statistical Analysis
3. Results
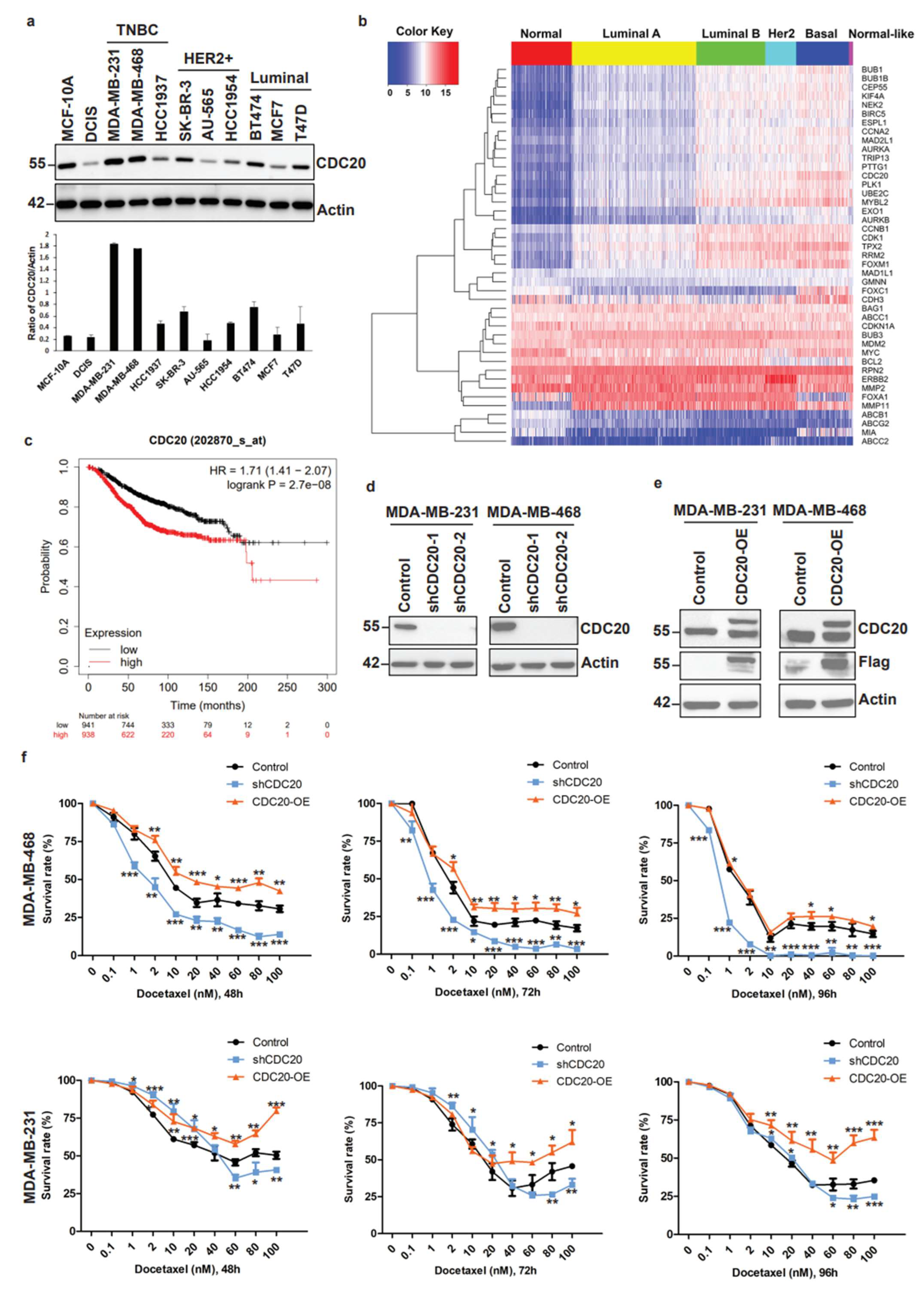
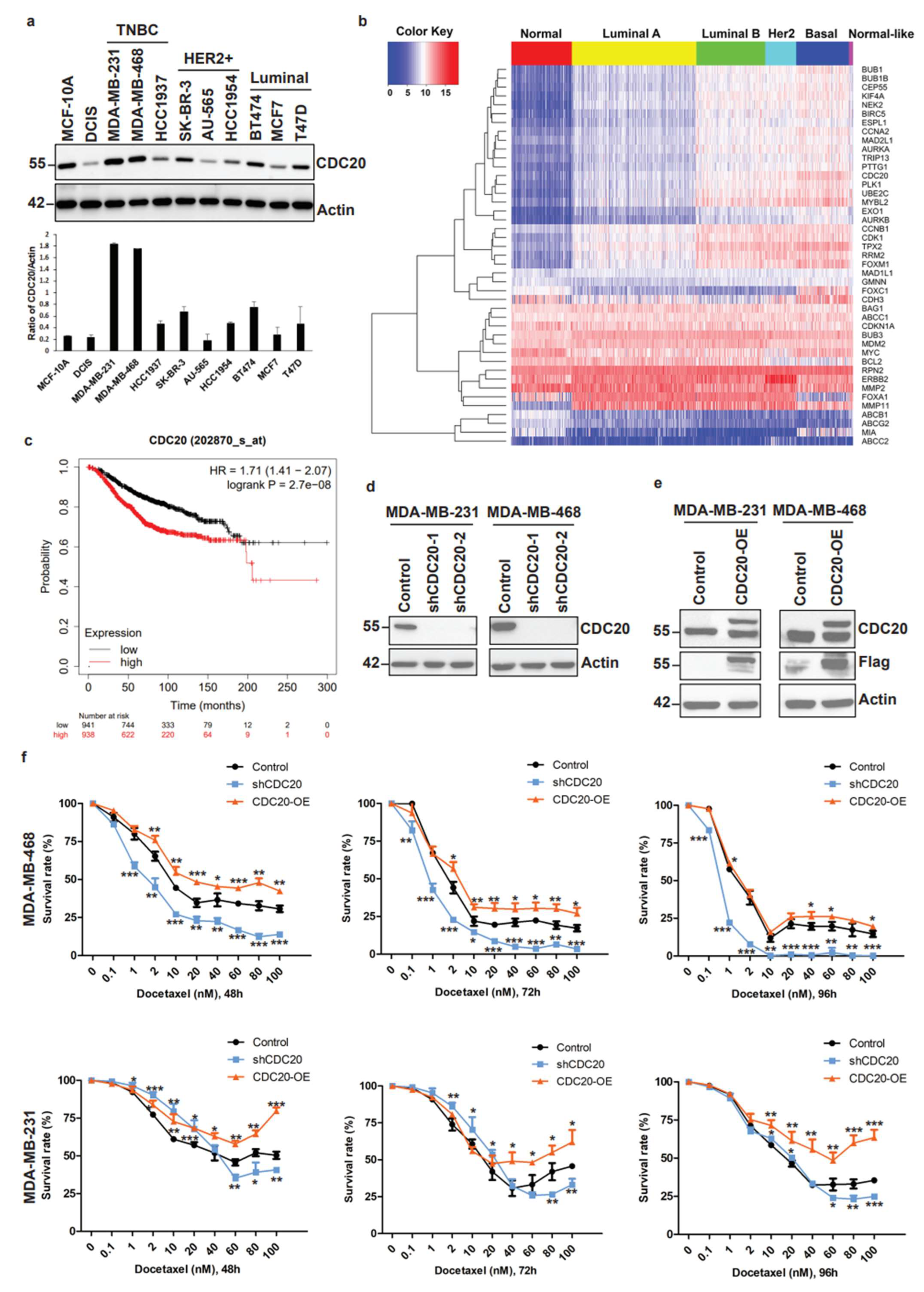
3.1. Elevated CDC20 Correlates with Poor Breast Cancer Prognosis and Drug Resistance
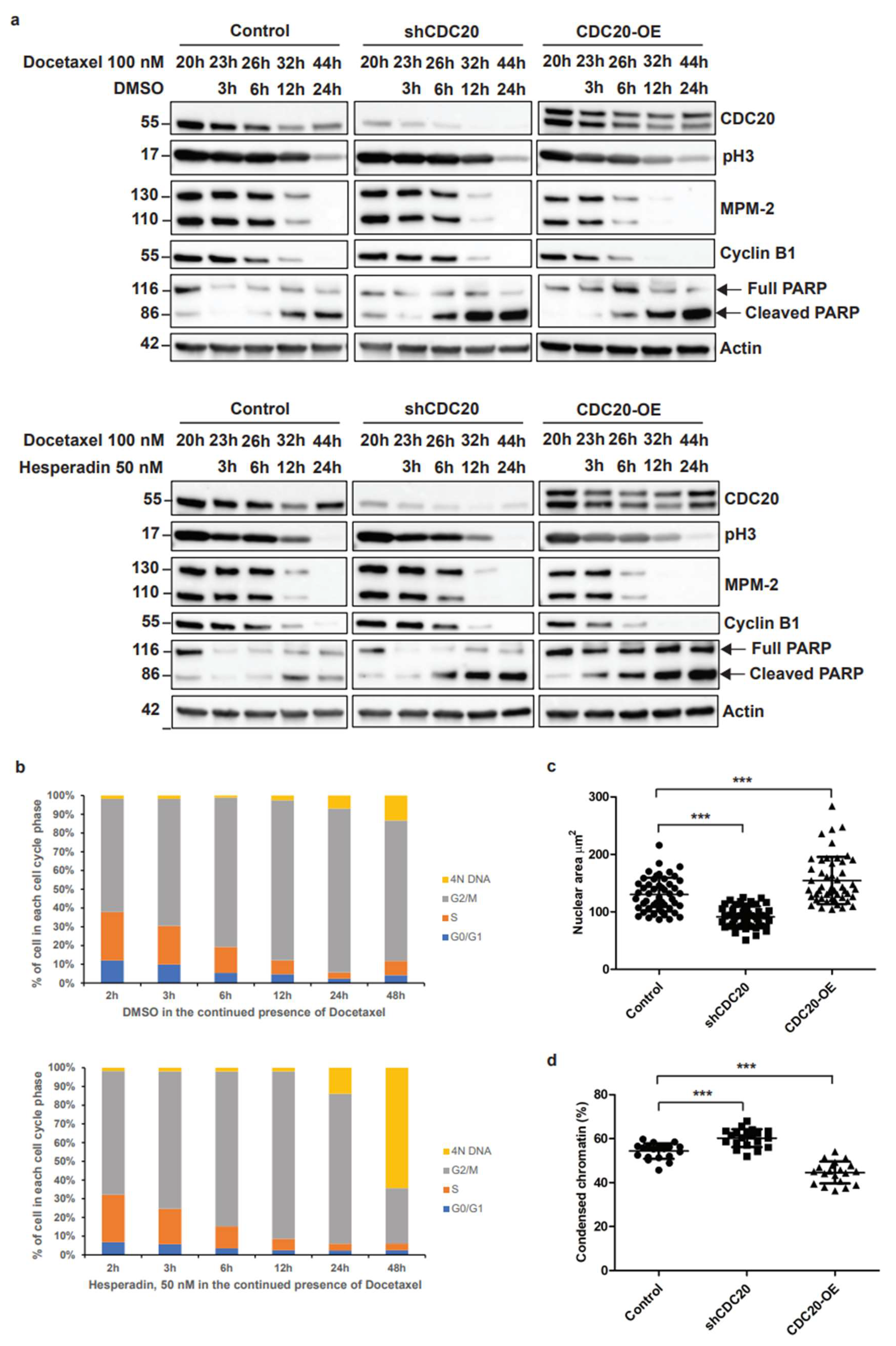
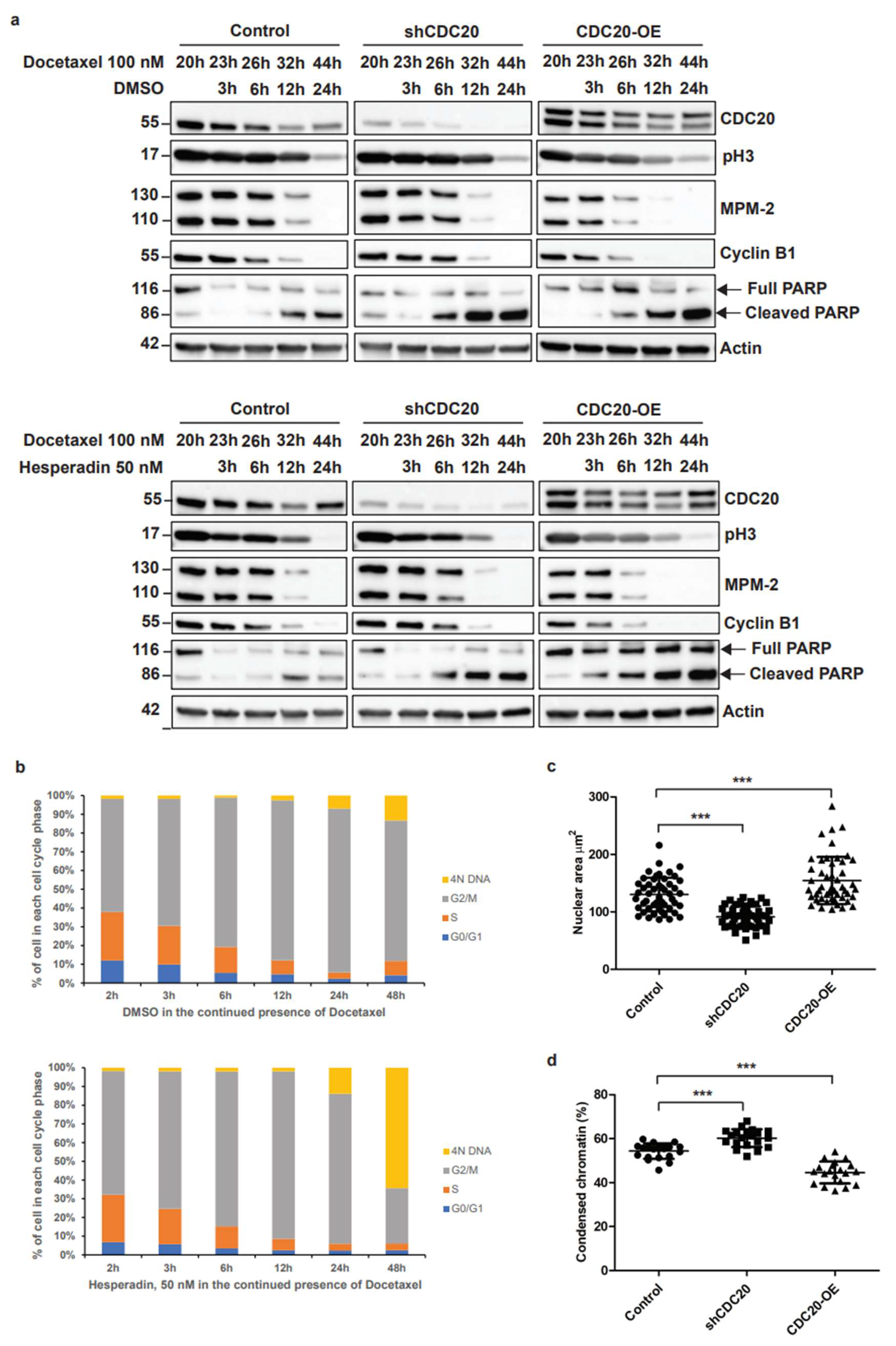
3.2. Elevated CDC20 Is Correlated with Mitotic Slippage, a Possible Reason Causing an Increase in Nuclear Size and Chromatin Decondensation in Cancer Cells
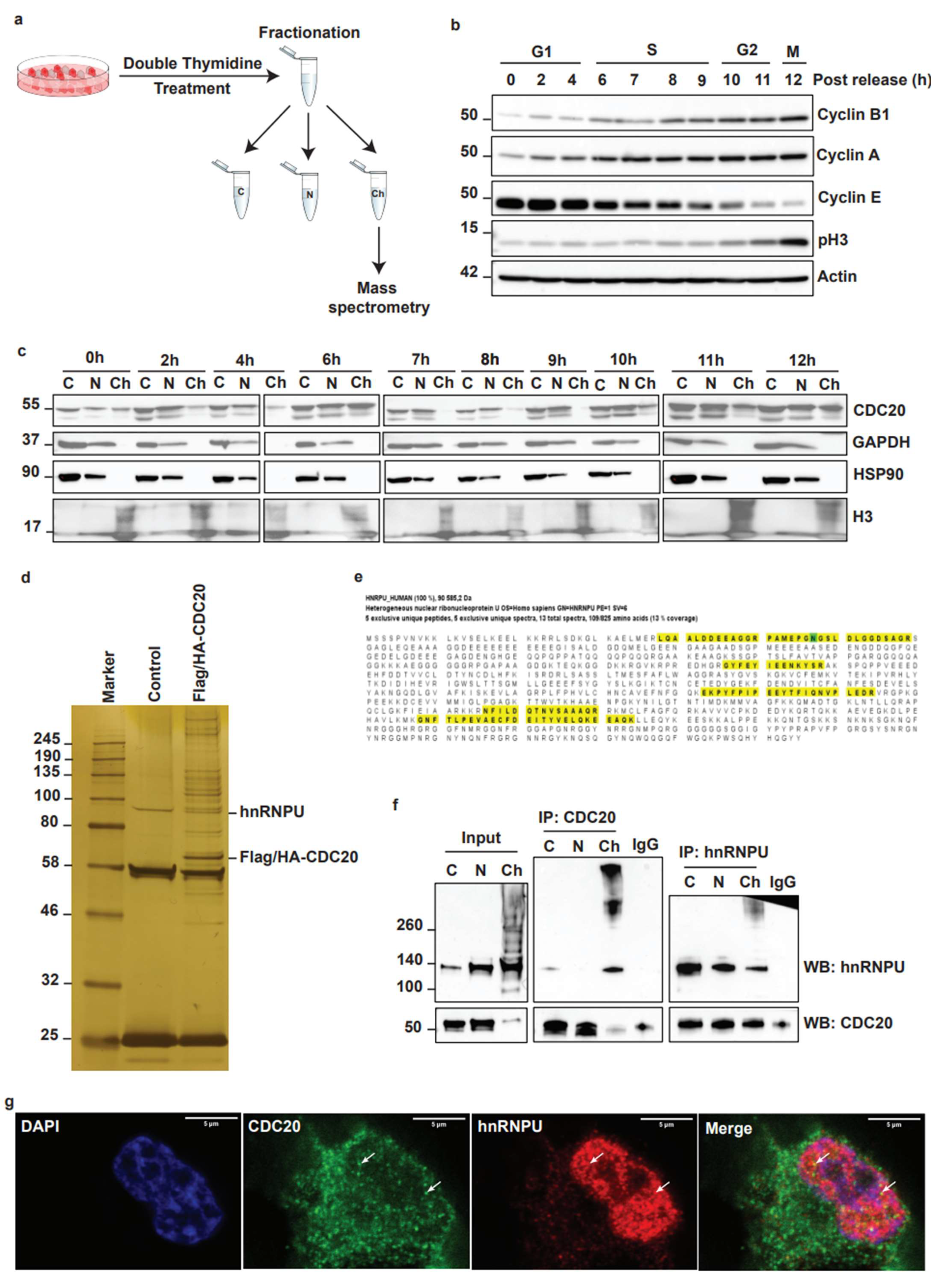
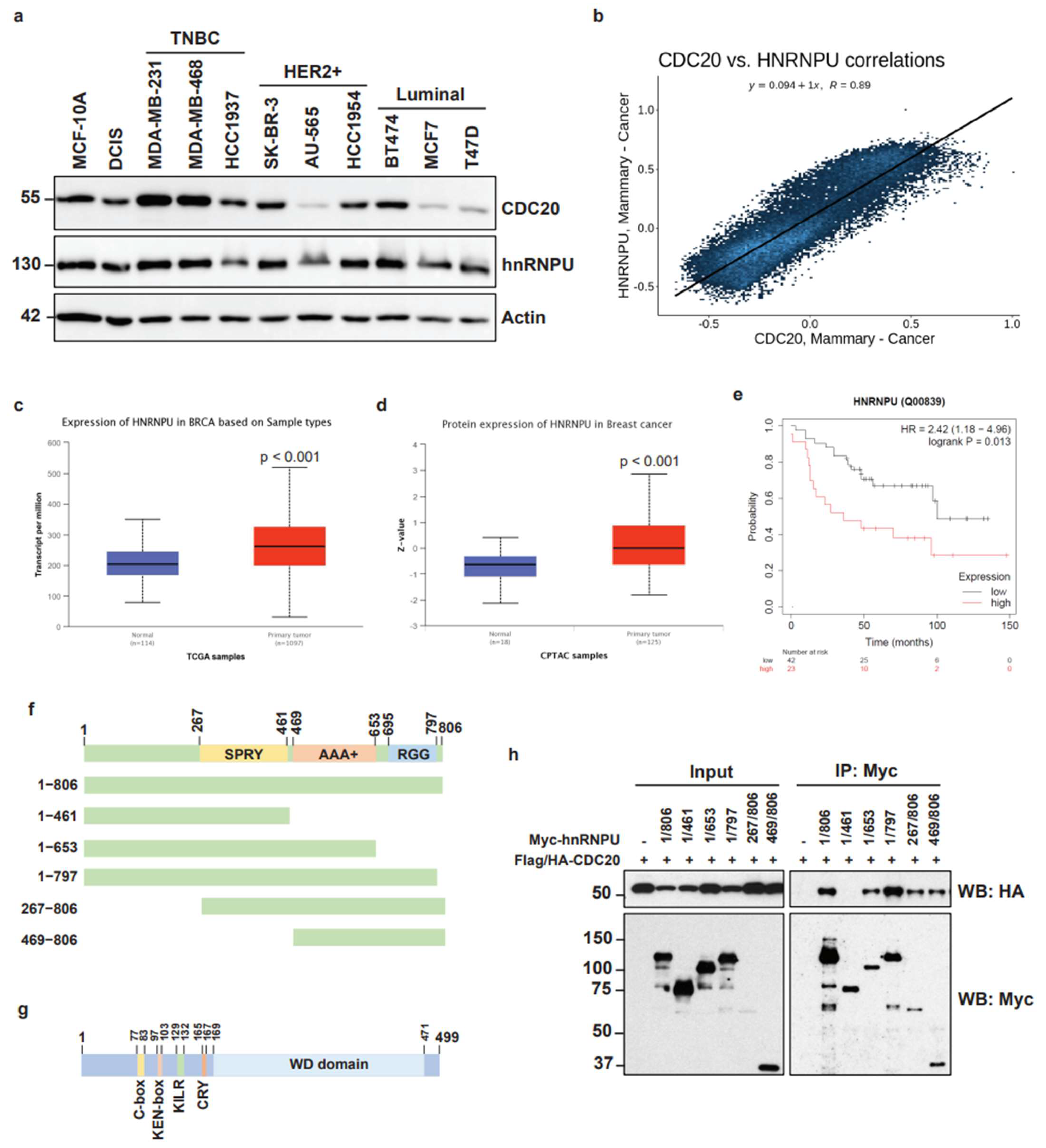
3.3. Identification of Nuclear Matrix Protein hnRNPU as a Target for CDC20 during Chromatin Dynamics
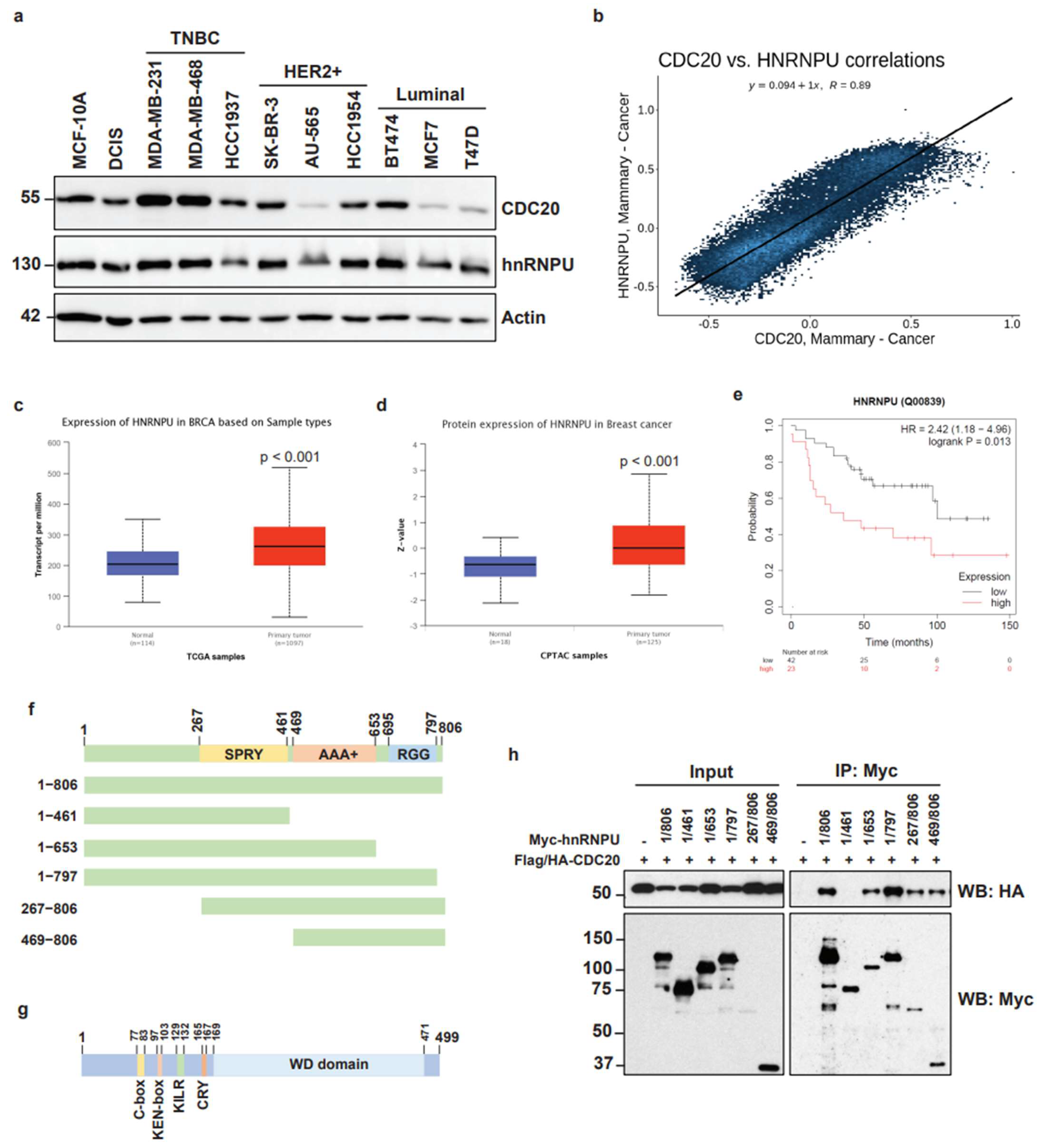
3.4. High Expression of hnRNPU in Breast Cancer and Mapping of Molecular Interaction between CDC20 and hnRNPU
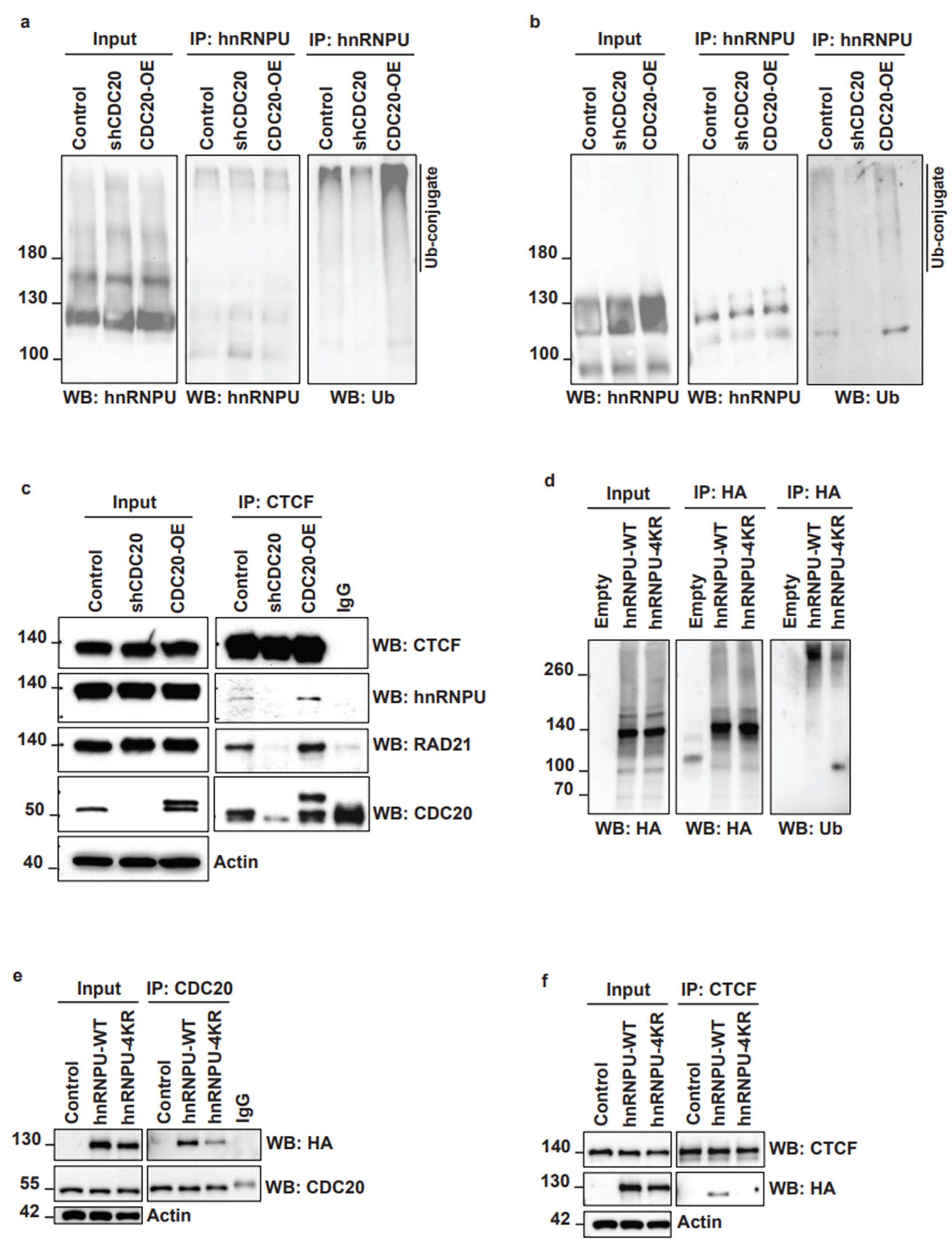
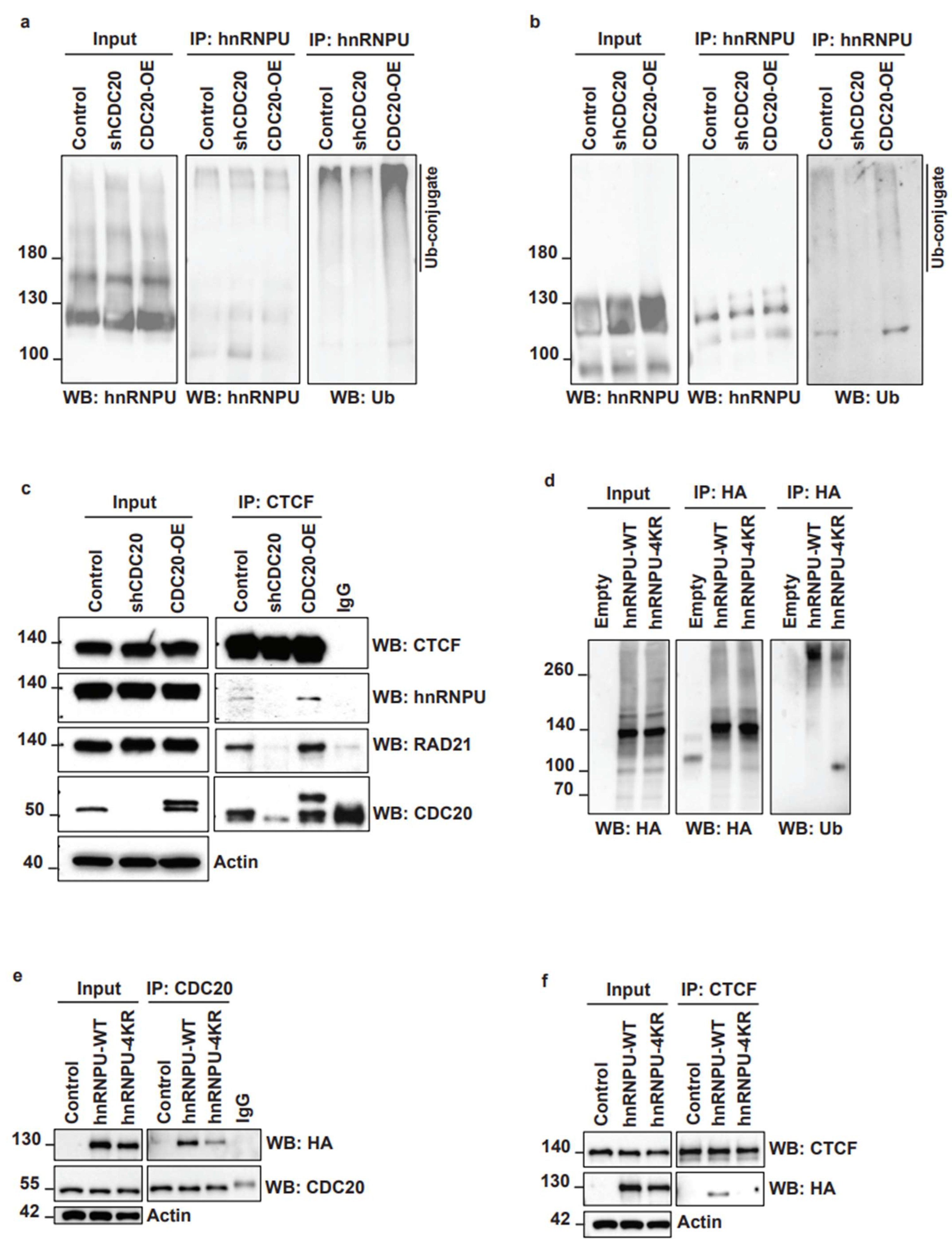
3.5. CDC20 Regulates hnRNPU Ubiquitination and Its Interaction with CTCF
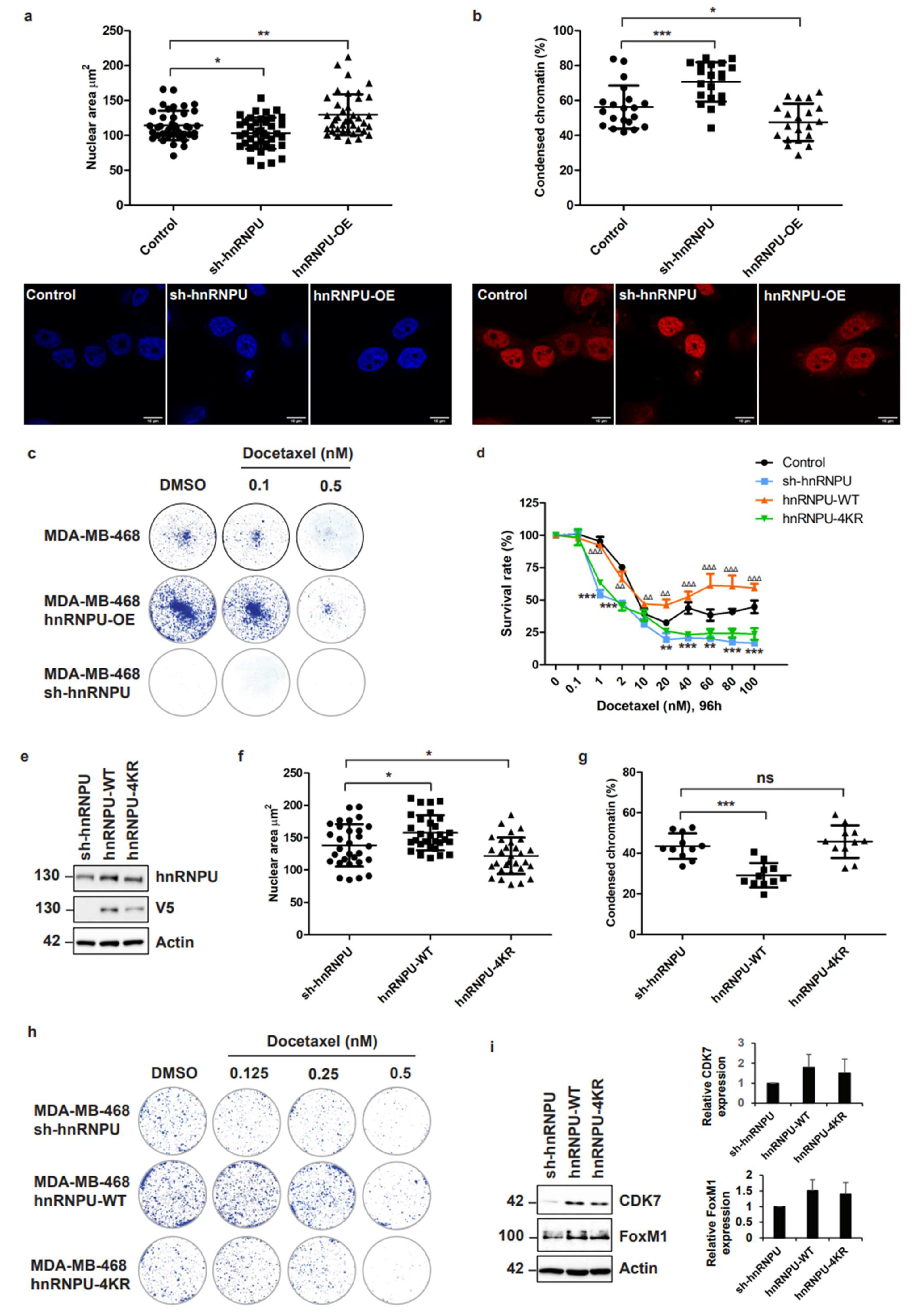
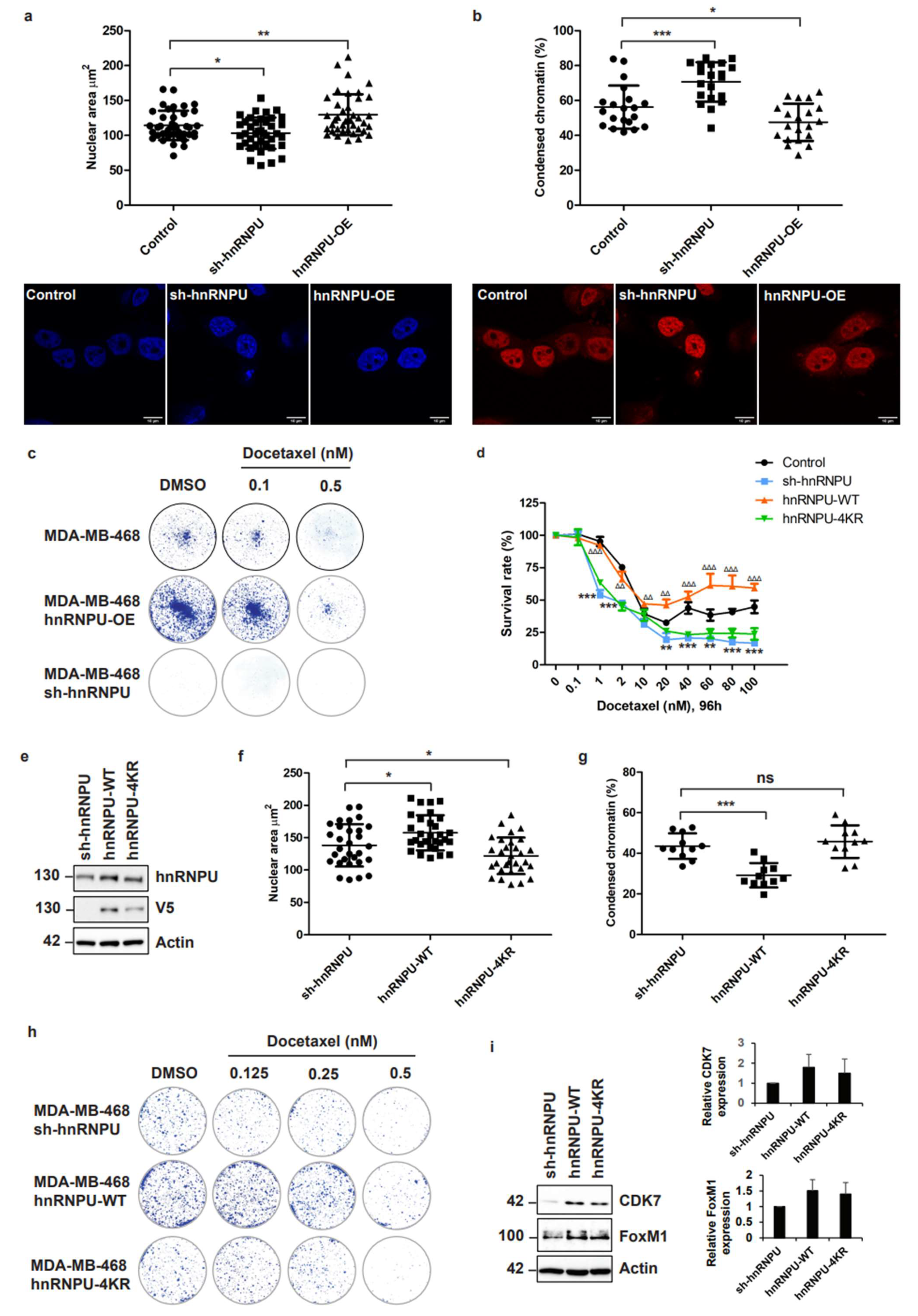
3.6. Inhibition of CDC20-Mediated hnRNPU Ubiquitination Results in the Decreased Nuclear Area, Enhanced Chromatin Condensation and Cellular Sensitization to Docetaxel Treatment
4. Discussion
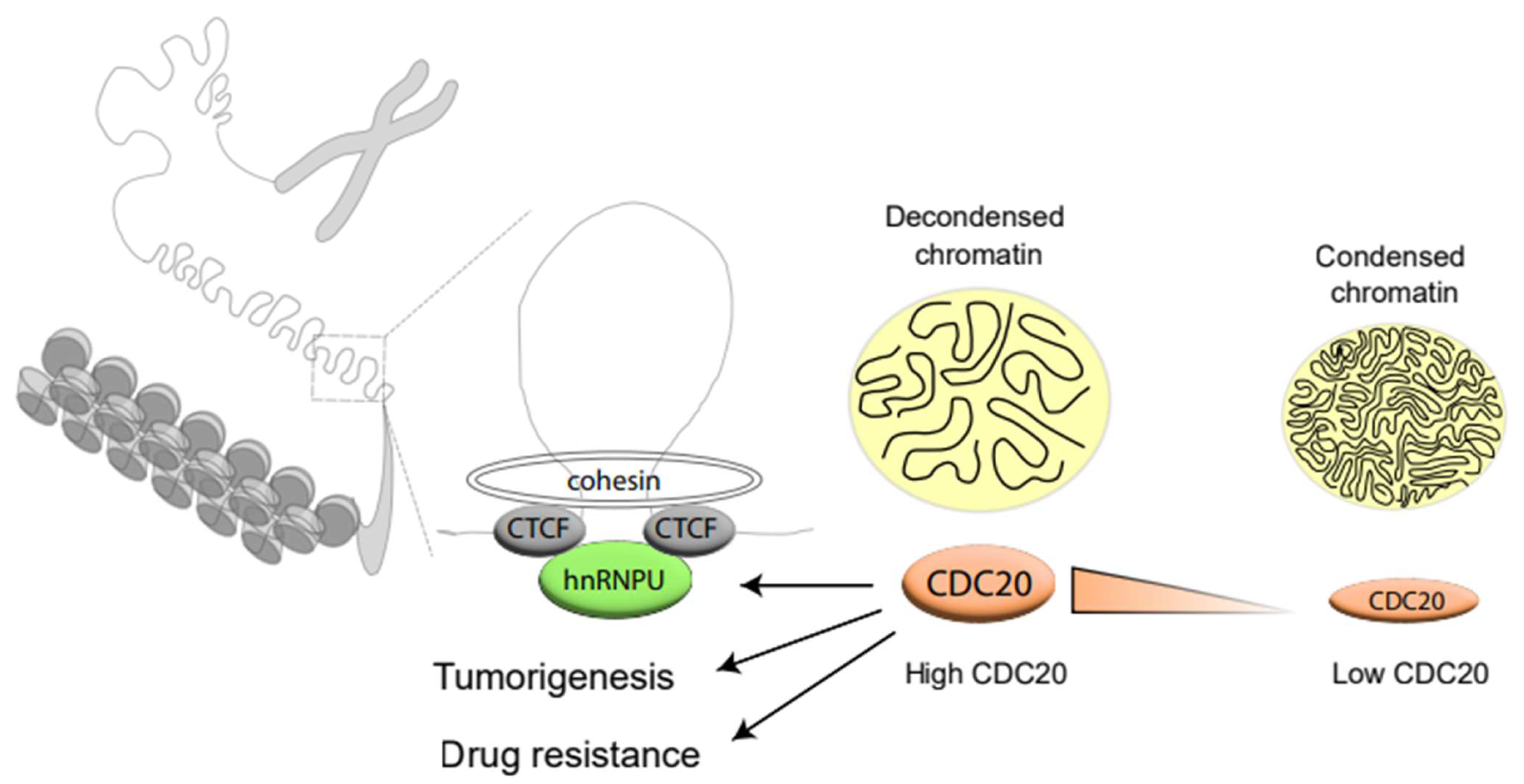
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kapanidou, M.; Curtis, N.L.; Bolanos-Garcia, V.M. Cdc20: At the Crossroads between Chromosome Segregation and Mitotic Exit. Trends Biochem. Sci. 2017, 42, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Shirayama, M.; Toth, A.; Galova, M.; Nasmyth, K. APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 1999, 402, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Puram, S.V.; Bilimoria, P.M.; Ikeuchi, Y.; Keough, S.; Wong, M.; Rowitch, D.; Bonni, A. A centrosomal Cdc20-APC pathway controls dendrite morphogenesis in postmitotic neurons. Cell 2009, 136, 322–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.B.; Kim, J.J.; Nam, H.J.; Gao, B.; Yin, P.; Qin, B.; Yi, S.Y.; Ham, H.; Evans, D.; Kim, S.H.; et al. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol. Cell 2015, 60, 21–34. [Google Scholar] [CrossRef] [Green Version]
- Wan, L.; Tan, M.; Yang, J.; Inuzuka, H.; Dai, X.; Wu, T.; Liu, J.; Shaik, S.; Chen, G.; Deng, J.; et al. APC(Cdc20) suppresses apoptosis through targeting Bim for ubiquitination and destruction. Dev. Cell 2014, 29, 377–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Wu, Q.; Mack, S.C.; Yang, K.; Kim, L.; Hubert, C.G.; Flavahan, W.A.; Chu, C.; Bao, S.; Rich, J.N. CDC20 maintains tumor initiating cells. Oncotarget 2015, 6, 13241–13254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, G.; Sengupta, S.; Panda, C.K.; Gollin, S.M.; Saunders, W.S.; Roychoudhury, S. Overexpression of Cdc20 leads to impairment of the spindle assembly checkpoint and aneuploidization in oral cancer. Carcinogenesis 2007, 28, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, B.A.; Cleveland, D.W. The role of aneuploidy in promoting and suppressing tumors. J. Cell Biol. 2009, 185, 935–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, D.Z.; Ma, Y.; Ji, B.; Liu, Y.; Hwu, P.; Abbruzzese, J.L.; Logsdon, C.; Wang, H. Increased CDC20 expression is associated with pancreatic ductal adenocarcinoma differentiation and progression. J. Hematol. Oncol. 2012, 5, 15. [Google Scholar] [CrossRef] [Green Version]
- Karra, H.; Repo, H.; Ahonen, I.; Loyttyniemi, E.; Pitkanen, R.; Lintunen, M.; Kuopio, T.; Soderstrom, M.; Kronqvist, P. Cdc20 and securin overexpression predict short-term breast cancer survival. Br. J. Cancer 2014, 110, 2905–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Sun, Y.; Chen, J.; Li, H.; Yao, K.; Liu, Y.; Liu, Q.; Lu, J. The Oncogenic Role of APC/C Activator Protein Cdc20 by an Integrated Pan-Cancer Analysis in Human Tumors. Front. Oncol. 2021, 11, 721797. [Google Scholar] [CrossRef] [PubMed]
- Bah, N.; Maillet, L.; Ryan, J.; Dubreil, S.; Gautier, F.; Letai, A.; Juin, P.; Barille-Nion, S. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death Dis. 2014, 5, e1291. [Google Scholar] [CrossRef] [Green Version]
- Brito, D.A.; Rieder, C.L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 2006, 16, 1194–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, Y.; Iimori, M.; Nakashima, Y.; Nakanishi, R.; Ando, K.; Ohgaki, K.; Kitao, H.; Saeki, H.; Oki, E.; Maehara, Y. Mitotic slippage and the subsequent cell fates after inhibition of Aurora B during tubulin-binding agent-induced mitotic arrest. Sci. Rep. 2017, 7, 16762. [Google Scholar] [CrossRef] [Green Version]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, J.A.; Margolis, R.L.; Fotedar, R. Substrate degradation by the anaphase promoting complex occurs during mitotic slippage. Cell Cycle 2010, 9, 1792–1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.C.; Shi, J.; Orth, J.D.; Mitchison, T.J. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell 2009, 16, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Lok, T.M.; Wang, Y.; Xu, W.K.; Xie, S.; Ma, H.T.; Poon, R.Y.C. Mitotic slippage is determined by p31(comet) and the weakening of the spindle-assembly checkpoint. Oncogene 2020, 39, 2819–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Mao, Y.; Lu, L.; Hu, C.; Wang, D.; Si-Tu, J.; Lu, M.; Peng, S.; Qiu, J.; Gao, X. Silencing of CDC20 suppresses metastatic castration-resistant prostate cancer growth and enhances chemosensitivity to docetaxel. Int. J. Oncol. 2016, 49, 1679–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, K.; Momiyama, N.; Ueda, M.; Matsuyama, R.; Mori, R.; Fujii, Y.; Ichikawa, Y.; Endo, I.; Togo, S.; Shimada, H. Targeting of CDC20 via small interfering RNA causes enhancement of the cytotoxicity of chemoradiation. Anticancer Res. 2008, 28, 1559–1563. [Google Scholar] [PubMed]
- Wang, J.; Zhou, F.; Li, Y.; Li, Q.; Wu, Z.; Yu, L.; Yuan, F.; Liu, J.; Tian, Y.; Cao, Y.; et al. Cdc20 overexpression is involved in temozolomide-resistant glioma cells with epithelial-mesenchymal transition. Cell Cycle 2017, 16, 2355–2365. [Google Scholar] [CrossRef] [Green Version]
- Chi, J.J.; Li, H.; Zhou, Z.; Izquierdo-Ferrer, J.; Xue, Y.; Wavelet, C.M.; Schiltz, G.E.; Zhang, B.; Cristofanilli, M.; Lu, X.; et al. A novel strategy to block mitotic progression for targeted therapy. EBioMedicine 2019, 49, 40–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackton, K.L.; Dimova, N.; Zeng, X.; Tian, W.; Zhang, M.; Sackton, T.B.; Meaders, J.; Pfaff, K.L.; Sigoillot, F.; Yu, H.; et al. Synergistic blockade of mitotic exit by two chemical inhibitors of the APC/C. Nature 2014, 514, 646–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almassalha, L.M.; Bauer, G.M.; Wu, W.; Cherkezyan, L.; Zhang, D.; Kendra, A.; Gladstein, S.; Chandler, J.E.; VanDerway, D.; Seagle, B.L.; et al. Macrogenomic engineering via modulation of the scaling of chromatin packing density. Nat. Biomed. Eng. 2017, 1, 902–913. [Google Scholar] [CrossRef] [Green Version]
- Fu, D.; Collins, K. Purification of human telomerase complexes identifies factors involved in telomerase biogenesis and telomere length regulation. Mol. Cell 2007, 28, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Kukalev, A.; Nord, Y.; Palmberg, C.; Bergman, T.; Percipalle, P. Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat. Struct. Mol. Biol. 2005, 12, 238–244. [Google Scholar] [CrossRef]
- Xiao, R.; Tang, P.; Yang, B.; Huang, J.; Zhou, Y.; Shao, C.; Li, H.; Sun, H.; Zhang, Y.; Fu, X.D. Nuclear matrix factor hnRNP U/SAF-A exerts a global control of alternative splicing by regulating U2 snRNP maturation. Mol. Cell 2012, 45, 656–668. [Google Scholar] [CrossRef] [Green Version]
- McHugh, C.A.; Chen, C.K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Ye, R.; Morrice, N.; Britton, S.; Trinkle-Mulcahy, L.; Lees-Miller, S.P. Phosphorylation of SAF-A/hnRNP-U Serine 59 by Polo-Like Kinase 1 Is Required for Mitosis. Mol. Cell Biol. 2015, 35, 2699–2713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Fan, H.; Lv, P.; Huo, X.; Wu, J.; Wang, Q.; Cheng, L.; Liu, Y.; Tang, Q.Q.; Zhang, L.; Zhang, F.; et al. The nuclear matrix protein HNRNPU maintains 3D genome architecture globally in mouse hepatocytes. Genome Res. 2018, 28, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, R.S.; Boteva, L.; Soares, D.C.; Naughton, C.; Dun, A.R.; Buckle, A.; Ramsahoye, B.; Bruton, P.C.; Saleeb, R.S.; Arnedo, M.; et al. SAF-A Regulates Interphase Chromosome Structure through Oligomerization with Chromatin-Associated RNAs. Cell 2017, 169, 1214–1227.e1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Li, D.; Wang, X.; Fang, E.; Yang, F.; Hu, A.; Wang, J.; Guo, Y.; Liu, Y.; Li, H.; et al. HNF4A-AS1/hnRNPU/CTCF axis as a therapeutic target for aerobic glycolysis and neuroblastoma progression. J. Hematol. Oncol. 2020, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Guo, Y.; Huo, Z.; Xing, Y.; Fang, J.; Ma, G.; Han, Q.; Wang, M.; Xu, Q. Identification of therapeutic targets and prognostic biomarkers from the hnRNP family in invasive breast carcinoma. Aging 2021, 13, 4503–4521. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.; Varambally, S. UALCAN: A Portal for Facilitating Tumor Subgroup Gene Expression and Survival Analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.J.; Rickels, R.A.; Collings, C.K.; He, X.; Cao, K.; Herz, H.M.; Cozzolino, K.A.; Abshiru, N.A.; Marshall, S.A.; Rendleman, E.J.; et al. A cryptic Tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev. 2017, 31, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- Hernandez-Vargas, H.; Palacios, J.; Moreno-Bueno, G. Molecular profiling of docetaxel cytotoxicity in breast cancer cells: Uncoupling of aberrant mitosis and apoptosis. Oncogene 2007, 26, 2902–2913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braadland, P.R.; Urbanucci, A. Chromatin reprogramming as an adaptation mechanism in advanced prostate cancer. Endocr. Relat. Cancer 2019, 26, R211–R235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunham, W.H.; Mullin, M.; Gingras, A.C. Affinity-purification coupled to mass spectrometry: Basic principles and strategies. Proteomics 2012, 12, 1576–1590. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.D.; Hao, L.; Han, X.X.; Wu, Z.X.; Pang, K.; Dong, Y.; Qin, J.X.; Wang, G.Y.; Zhang, X.M.; Xia, T.; et al. Targeting HNRNPU to overcome cisplatin resistance in bladder cancer. Mol. Cancer 2022, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Miller, H.E.; Bishop, A.J.R. Correlation AnalyzeR: Functional predictions from gene co-expression correlations. BMC Bioinform. 2021, 22, 206. [Google Scholar] [CrossRef] [PubMed]
- Attia, Y.M.; Shouman, S.A.; Salama, S.A.; Ivan, C.; Elsayed, A.M.; Amero, P.; Rodriguez-Aguayo, C.; Lopez-Berestein, G. Blockade of CDK7 Reverses Endocrine Therapy Resistance in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, T.; Kwiatkowski, N.; Abraham, B.J.; Lee, T.I.; Xie, S.; Yuzugullu, H.; Von, T.; Li, H.; Lin, Z.; et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 2015, 163, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, G.B.; Li, X.Z.; Zeng, S.; Liu, C.; Yang, S.M.; Yang, L.; Hu, C.J.; Bai, J.Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef] [Green Version]
- Okuda, S.; Sato, M.; Kato, S.; Nagashima, S.; Inatome, R.; Yanagi, S.; Fukuda, T. Oscillation of Cdc20-APC/C-mediated CAMDI stability is critical for cortical neuron migration. J. Biol. Chem. 2021, 297, 100986. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, A.H.; Yamada, T.; Wu, B.; Bilimoria, P.M.; Ikeuchi, Y.; de la Iglesia, N.; Shen, J.; Bonni, A. A Cdc20-APC ubiquitin signaling pathway regulates presynaptic differentiation. Science 2009, 326, 575–578. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Guo, C.; Fu, S.; Cheng, Y.; Song, C. Downregulation of CDC20 suppressed cell proliferation, induced apoptosis, triggered cell cycle arrest in osteosarcoma cells, and enhanced chemosensitivity to cisplatin. Neoplasma 2021, 68, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; Ghelli Luserna di Rora, A.; Napolitano, R.; Soverini, S.; Martinelli, G.; Simonetti, G. CDC20 in and out of mitosis: A prognostic factor and therapeutic target in hematological malignancies. J. Exp. Clin. Cancer Res. 2022, 41, 159. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Li, F.; Ge, S.; Zhang, F.; Jia, R.; Fan, X. CDC20 Knockdown and Acidic Microenvironment Collaboratively Promote Tumorigenesis through Inhibiting Autophagy and Apoptosis. Mol. Ther. Oncolytics 2020, 17, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, J.; Wan, L.; Zhou, X.; Wang, Z.; Wei, W. Targeting Cdc20 as a novel cancer therapeutic strategy. Pharmacol. Ther. 2015, 151, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubert, F.; Srivas, R.; Spacek, D.V.; Kasowski, M.; Ruiz-Velasco, M.; Sinnott-Armstrong, N.; Greenside, P.; Narasimha, A.; Liu, Q.; Geller, B.; et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature 2020, 583, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Kubo, N.; Loukinov, D.; Tajmul, M.; Kang, S.; Kovalchuk, A.L.; Strunnikov, A.V.; Zentner, G.E.; Ren, B.; Lobanenkov, V.V. CTCF mediates chromatin looping via N-terminal domain-dependent cohesin retention. Proc. Natl. Acad. Sci. USA 2020, 117, 2020–2031. [Google Scholar] [CrossRef] [Green Version]
- Tark-Dame, M.; Jerabek, H.; Manders, E.M.; van der Wateren, I.M.; Heermann, D.W.; van Driel, R. Depletion of the chromatin looping proteins CTCF and cohesin causes chromatin compaction: Insight into chromatin folding by polymer modelling. PLoS Comput. Biol. 2014, 10, e1003877. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, G.; Misteli, T. Functional implications of genome topology. Nat. Struct. Mol. Biol. 2013, 20, 290–299. [Google Scholar] [CrossRef]
- Gibcus, J.H.; Dekker, J. The hierarchy of the 3D genome. Mol Cell 2013, 49, 773–782. [Google Scholar] [CrossRef] [Green Version]
- Pombo, A.; Branco, M.R. Functional organisation of the genome during interphase. Curr. Opin. Genet. Dev. 2007, 17, 451–455. [Google Scholar] [CrossRef]
- Schoenfelder, S.; Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 2019, 20, 437–455. [Google Scholar] [CrossRef]
- Wang, Z.; Wan, L.; Zhong, J.; Inuzuka, H.; Liu, P.; Sarkar, F.H.; Wei, W. Cdc20: A potential novel therapeutic target for cancer treatment. Curr. Pharm. Des. 2013, 19, 3210–3214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Li, M.; Feng, Y.; Sun, F.; Zhang, L.; Xu, Y.; Lu, S.; Zhu, J.; Huang, J.; Wang, J.; et al. MDM2-P53 Signaling Pathway-Mediated Upregulation of CDC20 Promotes Progression of Human Diffuse Large B-Cell Lymphoma. Onco. Targets Ther. 2020, 13, 10475–10487. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Sigoillot, F.; Gaur, S.; Choi, S.; Pfaff, K.L.; Oh, D.C.; Hathaway, N.; Dimova, N.; Cuny, G.D.; King, R.W. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 2010, 18, 382–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.; Oh, C.; Yoo, K.H. Functional roles of CTCF in breast cancer. BMB Rep. 2017, 50, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wavelet-Vermuse, C.; Odnokoz, O.; Xue, Y.; Lu, X.; Cristofanilli, M.; Wan, Y. CDC20-Mediated hnRNPU Ubiquitination Regulates Chromatin Condensation and Anti-Cancer Drug Response. Cancers 2022, 14, 3732. https://doi.org/10.3390/cancers14153732
Wavelet-Vermuse C, Odnokoz O, Xue Y, Lu X, Cristofanilli M, Wan Y. CDC20-Mediated hnRNPU Ubiquitination Regulates Chromatin Condensation and Anti-Cancer Drug Response. Cancers. 2022; 14(15):3732. https://doi.org/10.3390/cancers14153732
Chicago/Turabian StyleWavelet-Vermuse, Cindy, Olena Odnokoz, Yifan Xue, Xinghua Lu, Massimo Cristofanilli, and Yong Wan. 2022. "CDC20-Mediated hnRNPU Ubiquitination Regulates Chromatin Condensation and Anti-Cancer Drug Response" Cancers 14, no. 15: 3732. https://doi.org/10.3390/cancers14153732
APA StyleWavelet-Vermuse, C., Odnokoz, O., Xue, Y., Lu, X., Cristofanilli, M., & Wan, Y. (2022). CDC20-Mediated hnRNPU Ubiquitination Regulates Chromatin Condensation and Anti-Cancer Drug Response. Cancers, 14(15), 3732. https://doi.org/10.3390/cancers14153732