
Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology


Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bidimensional Cultures of Cell Lines: Traditional Cancer Models
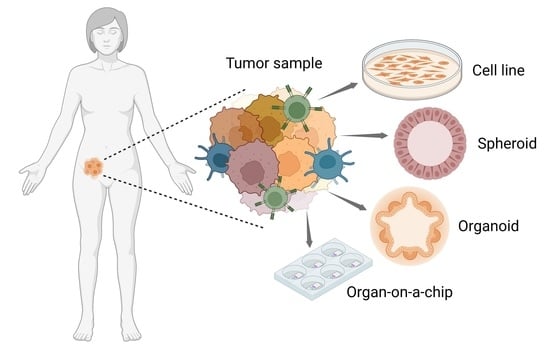
3. Tridimensional Cell Cultures: New Models in Cancer Research
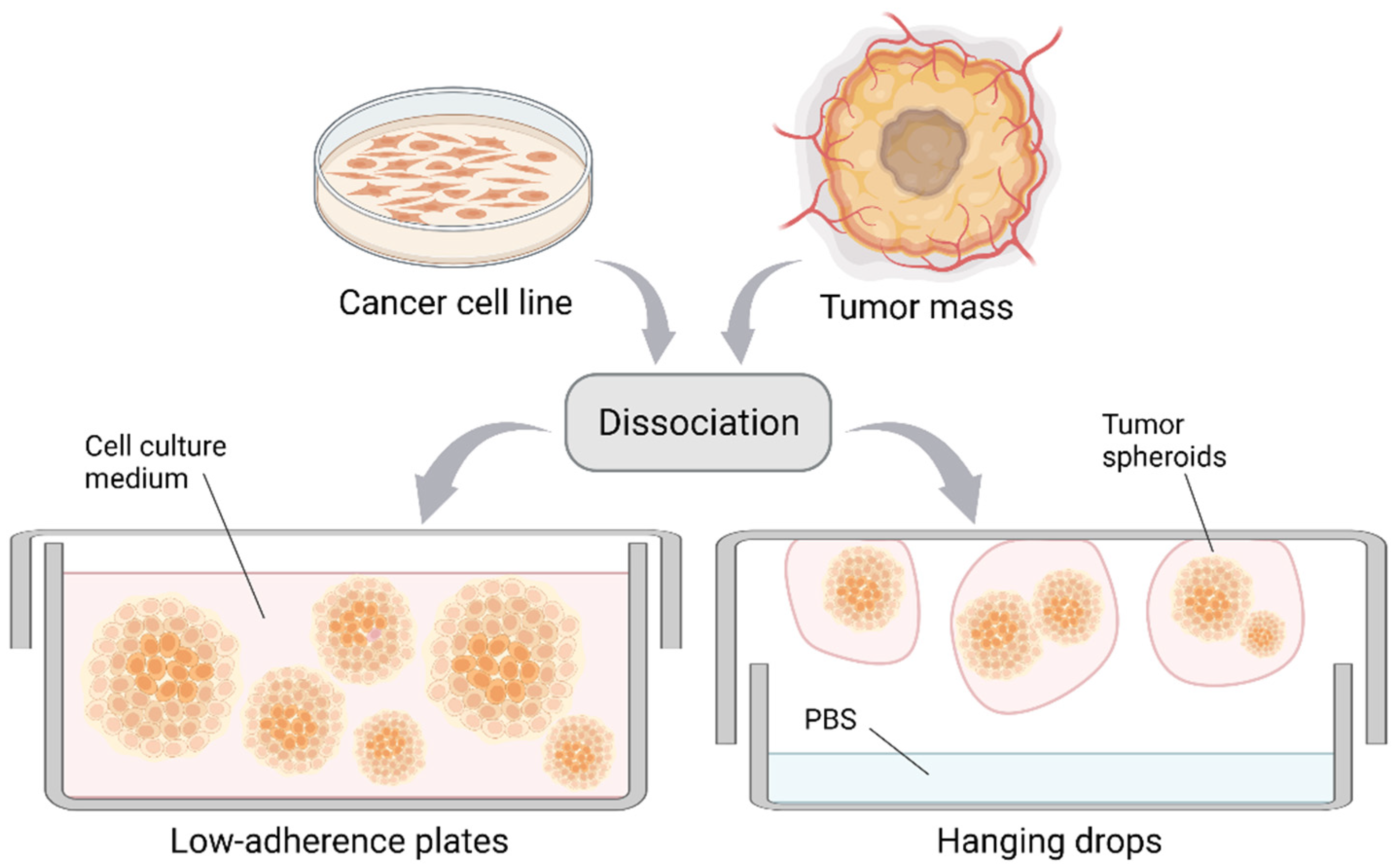
3.1. Multicellular Tumor Spheroids and Tumorshperes
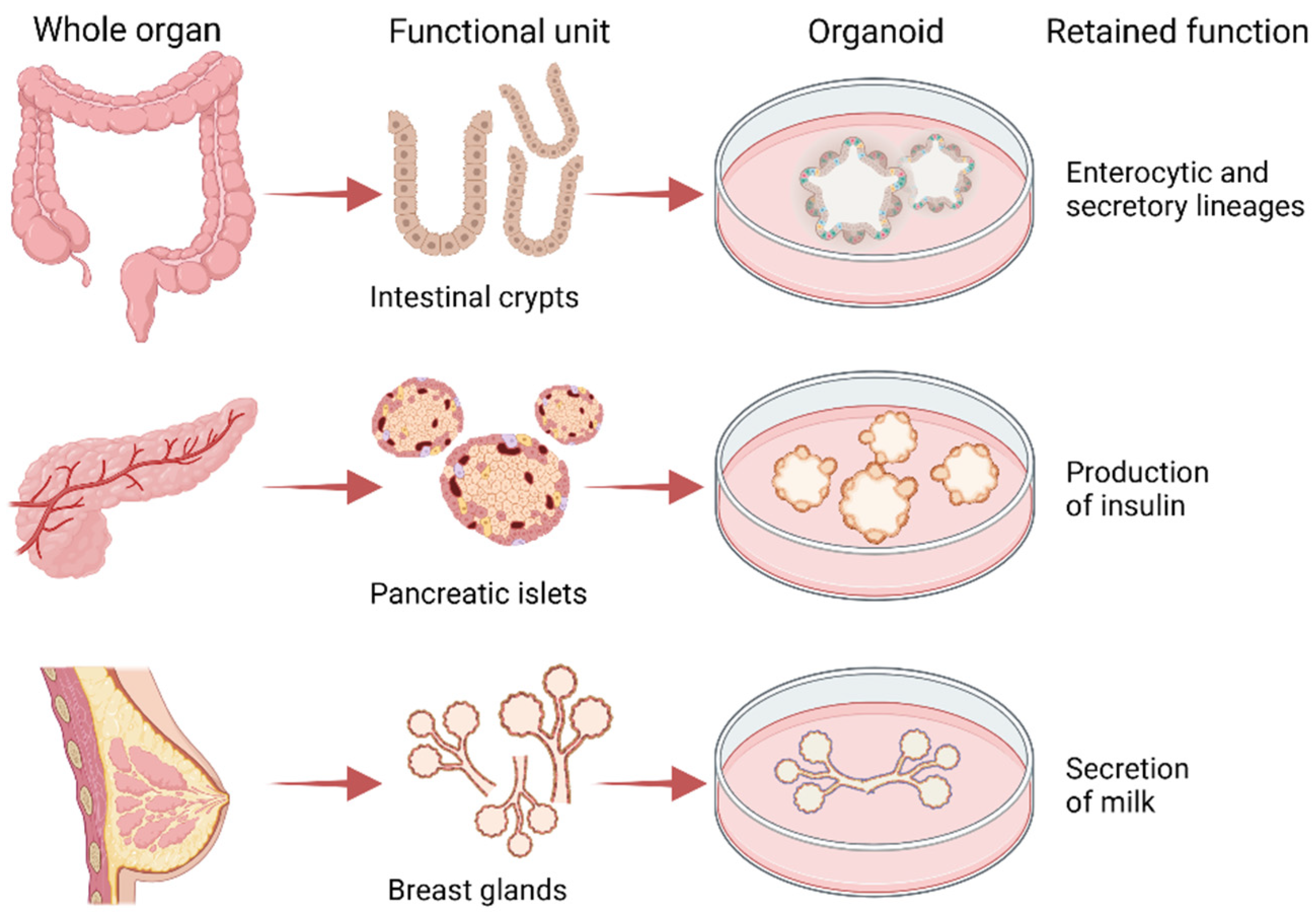
3.2. Patient-Derived Organoids
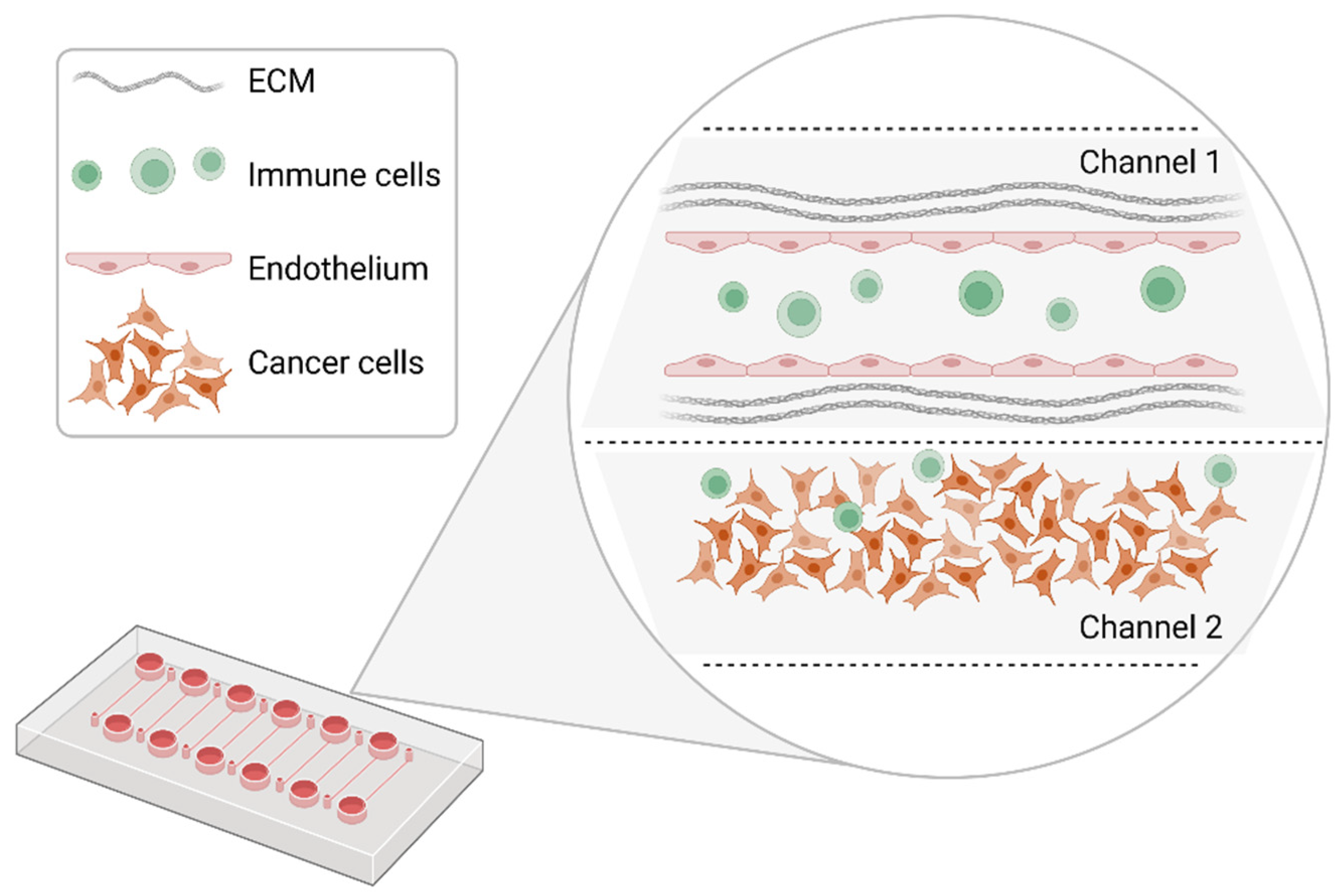
4. Microfluidic-Based Systems and the Organ-on-a-Chip Technology
4.1. Spheroids and Organoids Cultured in Microfluidic Systems
4.2. Organ-on-a-Chip Platforms
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wyld, L.; Audisio, R.A.; Poston, G.J. The evolution of cancer surgery and future perspectives. Nat. Rev. Clin. Oncol. 2015, 12, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Leyland-Jones, B. Pharmacology and pharmacogenetics of chemotherapeutic agents. Cancer Investig. 2009, 27, 482–488. [Google Scholar] [CrossRef]
- Lengauer, C.; Diaz, L.A., Jr.; Saha, S. Cancer drug discovery through collaboration. Nat. Rev. Drug Discov. 2005, 4, 375–380. [Google Scholar] [CrossRef]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug. Discov. 2004, 3, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Ibarrola-Villava, M.; Cervantes, A.; Bardelli, A. Preclinical models for precision oncology. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef]
- DeWeirdt, P.C.; Sangree, A.K.; Hanna, R.E.; Sanson, K.R.; Hegde, M.; Strand, C.; Persky, N.S.; Doench, J.G. Genetic screens in isogenic mammalian cell lines without single cell cloning. Nat. Commun. 2020, 11, 752. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.V.S.; De Sousa Portilho, A.J.; Da Silva, E.L.; De Oliveira Sales, L.; Da Silva Maues, J.H.; De Moraes, M.E.A.; Moreira-Nunes, C.A. Establishment of drug-resistant cell lines as a model in experimental oncology: A Review. Anticancer. Res. 2019, 39, 6443–6455. [Google Scholar] [CrossRef]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Siranosian, B.; Ha, G.; Tang, H.; Oren, Y.; Hinohara, K.; Strathdee, C.A.; Dempster, J.; Lyons, N.J.; Burns, R.; et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018, 560, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Dangles-Marie, V.; Pocard, M.; Richon, S.; Weiswald, L.B.; Assayag, F.; Saulnier, P.; Judde, J.G.; Janneau, J.L.; Auger, N.; Validire, P.; et al. Establishment of human colon cancer cell lines from fresh tumors versus xenografts: Comparison of success rate and cell line features. Cancer Res. 2007, 67, 398–407. [Google Scholar] [CrossRef]
- Richter, M.; Piwocka, O.; Musielak, M.; Piotrowski, I.; Suchorska, W.M.; Trzeciak, T. From donor to the lab: A fascinating journey of primary cell lines. Front. Cell Dev. Biol. 2021, 9, 711381. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Mukohara, T.; Shimono, Y.; Funakoshi, Y.; Chayahara, N.; Toyoda, M.; Kiyota, N.; Takao, S.; Kono, S.; Nakatsura, T.; et al. Comparison of 2D- and 3D-culture models as drug-testing platforms in breast cancer. Oncol. Rep. 2015, 33, 1837–1843. [Google Scholar] [CrossRef]
- Ivascu, A.; Kubbies, M. Rapid generation of single-tumor spheroids for high-throughput cell function and toxicity analysis. J. Biomol. Screen 2006, 11, 922–932. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Ackermann, T.; Pfetzer, N.; Sumpton, D.; Mackay, G.; Kalna, G.; Nixon, C.; Blyth, K.; Gottlieb, E.; Tardito, S. Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv. 2019, 5, eaau7314. [Google Scholar] [CrossRef]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-derived spheroids: Relevance to cancer stem cells and clinical applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef]
- Baker, B.M.; Chen, C.S. Deconstructing the third dimension: How 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012, 125, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R., Jr. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef] [PubMed]
- Bialkowska, K.; Komorowski, P.; Bryszewska, M.; Milowska, K. Spheroids as a Type of Three-Dimensional Cell Cultures-Examples of Methods of Preparation and the Most Important Application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef]
- Phelan, M.A.; Gianforcaro, A.L.; Gerstenhaber, J.A.; Lelkes, P.I. An Air Bubble-Isolating Rotating Wall Vessel Bioreactor for Improved Spheroid/Organoid Formation. Tissue Eng. Part C Methods 2019, 25, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Costa, A.; Zaffaroni, N.; Pratesi, G.; Petrangolini, G.; Coradini, D.; Pilotti, S.; Pierotti, M.A.; Daidone, M.G. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005, 65, 5506–5511. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Zhang, S.; Balch, C.; Chan, M.W.; Lai, H.C.; Matei, D.; Schilder, J.M.; Yan, P.S.; Huang, T.H.; Nephew, K.P. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res 2008, 68, 4311–4320. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Richon, S.; Massonnet, G.; Guinebretiere, J.M.; Vacher, S.; Laurendeau, I.; Cottu, P.; Marangoni, E.; Nemati, F.; Validire, P.; et al. A short-term colorectal cancer sphere culture as a relevant tool for human cancer biology investigation. Br. J. Cancer 2013, 108, 1720–1731. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sette, G.; Fecchi, K.; Salvati, V.; Lotti, F.; Pilozzi, E.; Duranti, E.; Biffoni, M.; Pagliuca, A.; Martinetti, D.; Memeo, L.; et al. Mek inhibition results in marked antitumor activity against metastatic melanoma patient-derived melanospheres and in melanosphere-generated xenografts. J. Exp. Clin. Cancer Res. 2013, 32, 91. [Google Scholar] [CrossRef] [PubMed]
- Weiswald, L.B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Barbone, D.; Yang, T.M.; Morgan, J.R.; Gaudino, G.; Broaddus, V.C. Mammalian target of rapamycin contributes to the acquired apoptotic resistance of human mesothelioma multicellular spheroids. J. Biol. Chem. 2008, 283, 13021–13030. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Gencoglu, M.F.; Barney, L.E.; Hall, C.L.; Brooks, E.A.; Schwartz, A.D.; Corbett, D.C.; Stevens, K.R.; Peyton, S.R. Comparative study of multicellular tumor spheroid formation methods and implications for drug screening. ACS Biomater. Sci. Eng. 2018, 4, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Kim, J.S.; Kim, S.H.; Park, Y.K.; Yu, E.; Kim, K.H.; Seo, E.J.; Oh, H.B.; Lee, H.C.; Kim, K.M.; et al. Patient-derived multicellular tumor spheroids towards optimized treatment for patients with hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2018, 37, 109. [Google Scholar] [CrossRef]
- Azharuddin, M.; Roberg, K.; Dhara, A.K.; Jain, M.V.; Darcy, P.; Hinkula, J.; Slater, N.K.H.; Patra, H.K. Dissecting multi drug resistance in head and neck cancer cells using multicellular tumor spheroids. Sci. Rep. 2019, 9, 20066. [Google Scholar] [CrossRef] [PubMed]
- Kiyohara, Y.; Yoshino, K.; Kubota, S.; Okuyama, H.; Endo, H.; Ueda, Y.; Kimura, T.; Kimura, T.; Kamiura, S.; Inoue, M. Drug screening and grouping by sensitivity with a panel of primary cultured cancer spheroids derived from endometrial cancer. Cancer Sci. 2016, 107, 452–460. [Google Scholar] [CrossRef]
- Wilson, K.M.; Mathews-Griner, L.A.; Williamson, T.; Guha, R.; Chen, L.; Shinn, P.; McKnight, C.; Michael, S.; Klumpp-Thomas, C.; Binder, Z.A.; et al. Mutation profiles in glioblastoma 3D oncospheres modulate drug efficacy. SLAS Technol. 2019, 24, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Arnadottir, S.S.; Jeppesen, M.; Lamy, P.; Bramsen, J.B.; Nordentoft, I.; Knudsen, M.; Vang, S.; Madsen, M.R.; Thastrup, O.; Thastrup, J.; et al. Characterization of genetic intratumor heterogeneity in colorectal cancer and matching patient-derived spheroid cultures. Mol. Oncol. 2018, 12, 132–147. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Corro, C.; Novellasdemunt, L.; Li, V.S.W. A brief history of organoids. Am. J. Physiol. Cell Physiol. 2020, 319, C151–C165. [Google Scholar] [CrossRef]
- Fujii, M.; Matano, M.; Toshimitsu, K.; Takano, A.; Mikami, Y.; Nishikori, S.; Sugimoto, S.; Sato, T. Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem. Cell 2018, 23, 787–793.e786. [Google Scholar] [CrossRef]
- Sumbal, J.; Chiche, A.; Charifou, E.; Koledova, Z.; Li, H. Primary Mammary Organoid Model of Lactation and Involution. Front. Cell Dev. Biol. 2020, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Bai, L.; Pan, H.; Feng, H.; Clevers, H.; Zeng, Y.A. Long-Term Expansion of Pancreatic Islet Organoids from Resident Procr(+) Progenitors. Cell 2020, 180, 1198–1211.e1119. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef]
- Wang, Z.; Guo, Y.; Jin, Y.; Zhang, X.; Geng, H.; Xie, G.; Ye, D.; Yu, Y.; Liu, D.; Zhou, D.; et al. Establishment and drug screening of patient-derived extrahepatic biliary tract carcinoma organoids. Cancer Cell Int. 2021, 21, 519. [Google Scholar] [CrossRef]
- Larsen, B.M.; Kannan, M.; Langer, L.F.; Leibowitz, B.D.D.; Bentaieb, A.; Cancino, A.; Dolgalev, I.; Drummond, B.E.; Dry, J.R.; Ho, C.S.; et al. A pan-cancer organoid platform for precision medicine. Cell Rep. 2021, 36, 109429. [Google Scholar] [CrossRef]
- Steele, N.G.; Chakrabarti, J.; Wang, J.; Biesiada, J.; Holokai, L.; Chang, J.; Nowacki, L.M.; Hawkins, J.; Mahe, M.; Sundaram, N.; et al. An Organoid-Based Preclinical Model of Human Gastric Cancer. Cell Mol. Gastroenterol. Hepatol. 2019, 7, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Maenhoudt, N.; Defraye, C.; Boretto, M.; Jan, Z.; Heremans, R.; Boeckx, B.; Hermans, F.; Arijs, I.; Cox, B.; Van Nieuwenhuysen, E.; et al. Developing organoids from ovarian cancer as experimental and preclinical models. Stem. Cell Rep. 2020, 14, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Schutte, M.; Risch, T.; Abdavi-Azar, N.; Boehnke, K.; Schumacher, D.; Keil, M.; Yildiriman, R.; Jandrasits, C.; Borodina, T.; Amstislavskiy, V.; et al. Molecular dissection of colorectal cancer in pre-clinical models identifies biomarkers predicting sensitivity to EGFR inhibitors. Nat. Commun. 2017, 8, 14262. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Holtzinger, A.; Jagan, I.; BeGora, M.; Lohse, I.; Ngai, N.; Nostro, C.; Wang, R.; Muthuswamy, L.B.; Crawford, H.C.; et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med. 2015, 21, 1364–1371. [Google Scholar] [CrossRef]
- Pauli, C.; Hopkins, B.D.; Prandi, D.; Shaw, R.; Fedrizzi, T.; Sboner, A.; Sailer, V.; Augello, M.; Puca, L.; Rosati, R.; et al. Personalized in vitro and in vivo cancer models to guide precision medicine. Cancer Discov. 2017, 7, 462–477. [Google Scholar] [CrossRef]
- Verissimo, C.S.; Overmeer, R.M.; Ponsioen, B.; Drost, J.; Mertens, S.; Verlaan-Klink, I.; Gerwen, B.V.; van der Ven, M.; Wetering, M.V.; Egan, D.A.; et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife 2016, 5, e18489. [Google Scholar] [CrossRef] [PubMed]
- Tiriac, H.; Belleau, P.; Engle, D.D.; Plenker, D.; Deschenes, A.; Somerville, T.D.D.; Froeling, F.E.M.; Burkhart, R.A.; Denroche, R.E.; Jang, G.H.; et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov. 2018, 8, 1112–1129. [Google Scholar] [CrossRef]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Muramatsu, T.; Kanai, Y.; Ojima, H.; Sukeda, A.; Hiraoka, N.; Arai, E.; Sugiyama, Y.; Matsuzaki, J.; Uchida, R.; et al. Establishment of patient-derived organoids and drug screening for biliary tract carcinoma. Cell Rep. 2019, 27, 1265–1276.e1264. [Google Scholar] [CrossRef] [PubMed]
- Puca, L.; Bareja, R.; Prandi, D.; Shaw, R.; Benelli, M.; Karthaus, W.R.; Hess, J.; Sigouros, M.; Donoghue, A.; Kossai, M.; et al. Patient derived organoids to model rare prostatte canccerr phenotypes. Nat. Commun. 2018, 9, 2404. [Google Scholar] [CrossRef]
- Kim, M.; Mun, H.; Sung, C.O.; Cho, E.J.; Jeon, H.J.; Chun, S.M.; Jung, D.J.; Shin, T.H.; Jeong, G.S.; Kim, D.K.; et al. Patient-derived lung cancer organoids as in vitro cancer models for therapeutic screening. Nat. Commun. 2019, 10, 3991. [Google Scholar] [CrossRef] [PubMed]
- Bjornson-Hooper, Z.B.; Fragiadakis, G.K.; Spitzer, M.H.; Chen, H.; Madhireddy, D.; Hu, K.; Lundsten, K.; McIlwain, D.R.; Nolan, G.P. A comprehensive atlas of immunological differences between humans, mice, and non-human primates. Front. Immunol. 2022, 13, 867015. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Ping, J.; Chen, H.; Jiao, L.; Zheng, S.; Han, Z.G.; Hao, P.; Huang, J. A comparative analysis of liver transcriptome suggests divergent liver function among human, mouse and rat. Genomics 2010, 96, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Kwapiszewska, K.; Michalczuk, A.; Rybka, M.; Kwapiszewski, R.; Brzozka, Z. A microfluidic-based platform for tumour spheroid culture, monitoring and drug screening. Lab Chip 2014, 14, 2096–2104. [Google Scholar] [CrossRef]
- Gunay, G.; Kirit, H.A.; Kamatar, A.; Baghdasaryan, O.; Hamsici, S.; Acar, H. The effects of size and shape of the ovarian cancer spheroids on the drug resistance and migration. Gynecol. Oncol. 2020, 159, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Fang, G.; Lu, H.; Law, A.; Gallego-Ortega, D.; Jin, D.; Lin, G. Gradient-sized control of tumor spheroids on a single chip. Lab Chip 2019, 19, 4093–4103. [Google Scholar] [CrossRef] [PubMed]
- Patra, B.; Peng, C.C.; Liao, W.H.; Lee, C.H.; Tung, Y.C. Drug testing and flow cytometry analysis on a large number of uniform sized tumor spheroids using a microfluidic device. Sci. Rep. 2016, 6, 21061. [Google Scholar] [CrossRef]
- Jung, D.J.; Shin, T.H.; Kim, M.; Sung, C.O.; Jang, S.J.; Jeong, G.S. A one-stop microfluidic-based lung cancer organoid culture platform for testing drug sensitivity. Lab Chip 2019, 19, 2854–2865. [Google Scholar] [CrossRef]
- Mazzocchi, A.R.; Rajan, S.A.P.; Votanopoulos, K.I.; Hall, A.R.; Skardal, A. In vitro patient-derived 3D mesothelioma tumor organoids facilitate patient-centric therapeutic screening. Sci. Rep. 2018, 8, 2886. [Google Scholar] [CrossRef]
- Meijer, T.G.; Naipal, K.A.; Jager, A.; van Gent, D.C. Ex vivo tumor culture systems for functional drug testing and therapy response prediction. Future Sci. OA 2017, 3, FSO190. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, M.; Peant, B.; Lateef, M.A.; Rousset, N.; Kendall-Dupont, J.; Carmona, E.; Monet, F.; Saad, F.; Provencher, D.; Mes-Masson, A.M.; et al. Micro-dissected tumor tissues on chip: An ex vivo method for drug testing and personalized therapy. Lab Chip 2016, 16, 312–325. [Google Scholar] [CrossRef]
- Ko, J.; Ahn, J.; Kim, S.; Lee, Y.; Lee, J.; Park, D.; Jeon, N.L. Tumor spheroid-on-a-chip: A standardized microfluidic culture platform for investigating tumor angiogenesis. Lab Chip 2019, 19, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Nashimoto, Y.; Okada, R.; Hanada, S.; Arima, Y.; Nishiyama, K.; Miura, T.; Yokokawa, R. Vascularized cancer on a chip: The effect of perfusion on growth and drug delivery of tumor spheroid. Biomaterials 2020, 229, 119547. [Google Scholar] [CrossRef]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Williamson, A.; Singh, S.; Fernekorn, U.; Schober, A. The future of the patient-specific Body-on-a-chip. Lab Chip 2013, 13, 3471–3480. [Google Scholar] [CrossRef]
- Lu, S.; Cuzzucoli, F.; Jiang, J.; Liang, L.G.; Wang, Y.; Kong, M.; Zhao, X.; Cui, W.; Li, J.; Wang, S. Development of a biomimetic liver tumor-on-a-chip model based on decellularized liver matrix for toxicity testing. Lab Chip 2018, 18, 3379–3392. [Google Scholar] [CrossRef]
- Phan, D.T.T.; Wang, X.; Craver, B.M.; Sobrino, A.; Zhao, D.; Chen, J.C.; Lee, L.Y.N.; George, S.C.; Lee, A.P.; Hughes, C.C.W. A vascularized and perfused organ-on-a-chip platform for large-scale drug screening applications. Lab Chip 2017, 17, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Smith, A.M.; Garson, C.J.; Hassani, I.; Seeto, W.J.; Pant, K.; Arnold, R.D.; Prabhakarpandian, B.; Lipke, E.A. A microvascularized tumor-mimetic platform for assessing anti-cancer drug efficacy. Sci. Rep. 2018, 8, 3171. [Google Scholar] [CrossRef]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.T.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife 2020, 9, e52253. [Google Scholar] [CrossRef] [PubMed]
- Walter, F.R.; Valkai, S.; Kincses, A.; Petneházi, A.; Czeller, T.; Veszelka, S.; Ormos, P.; Deli, M.A.; Dér, A. A versatile lab-on-a-chip tool for modeling biological barriers. Sens. Actuators B Chem. 2016, 222, 1209–1219. [Google Scholar] [CrossRef]
- Skardal, A.; Aleman, J.; Forsythe, S.; Rajan, S.; Murphy, S.; Devarasetty, M.; Pourhabibi Zarandi, N.; Nzou, G.; Wicks, R.; Sadri-Ardekani, H.; et al. Drug compound screening in single and integrated multi-organoid body-on-a-chip systems. Biofabrication 2020, 12, 025017. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foglizzo, V.; Cocco, E.; Marchiò, S. Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology. Cancers 2022, 14, 3692. https://doi.org/10.3390/cancers14153692
Foglizzo V, Cocco E, Marchiò S. Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology. Cancers. 2022; 14(15):3692. https://doi.org/10.3390/cancers14153692
Chicago/Turabian StyleFoglizzo, Valentina, Emiliano Cocco, and Serena Marchiò. 2022. "Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology" Cancers 14, no. 15: 3692. https://doi.org/10.3390/cancers14153692
APA StyleFoglizzo, V., Cocco, E., & Marchiò, S. (2022). Advanced Cellular Models for Preclinical Drug Testing: From 2D Cultures to Organ-on-a-Chip Technology. Cancers, 14(15), 3692. https://doi.org/10.3390/cancers14153692