
Inflammatory Myofibroblastic Tumour: State of the Art



Abstract
Simple Summary
Abstract
1. Introduction
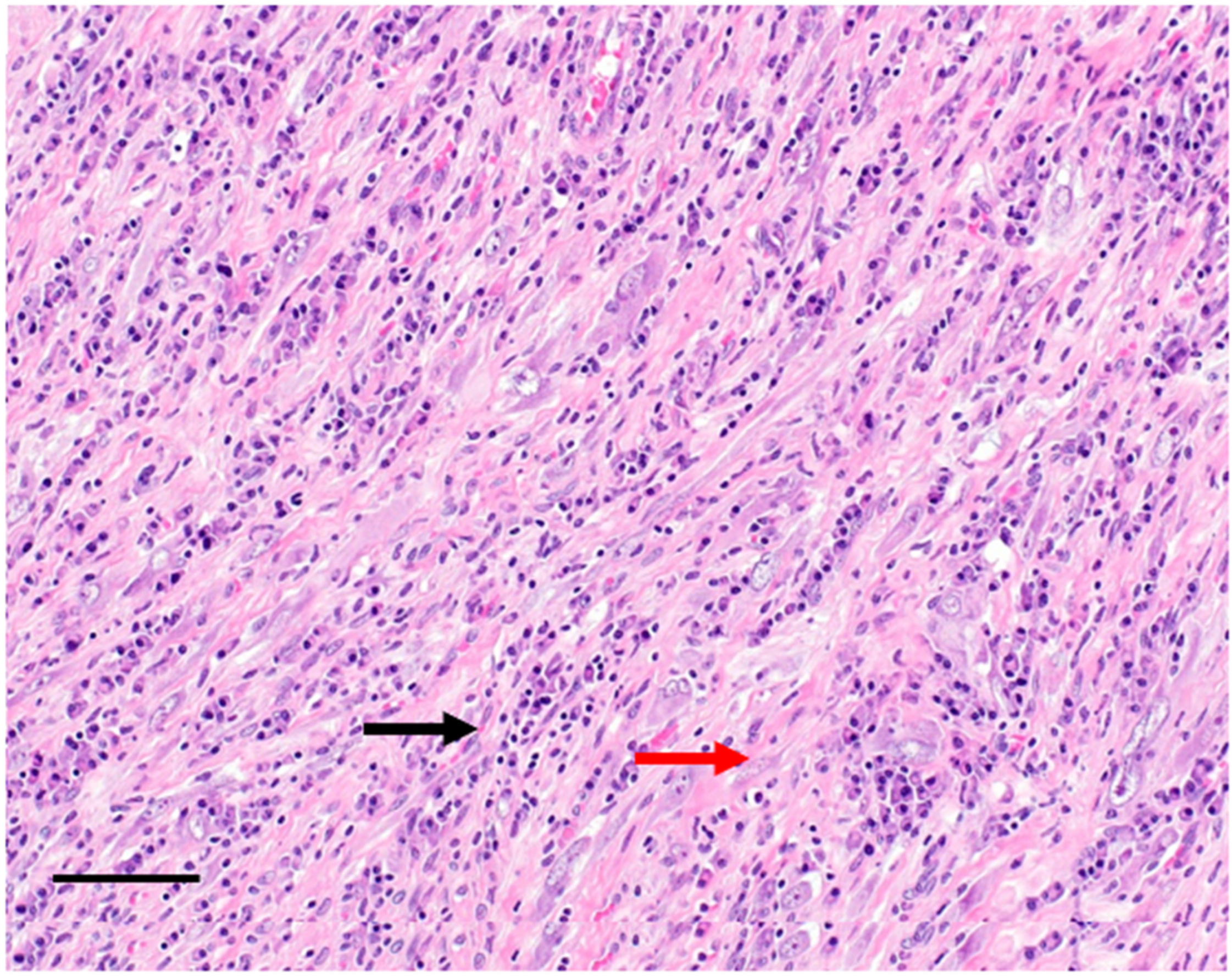
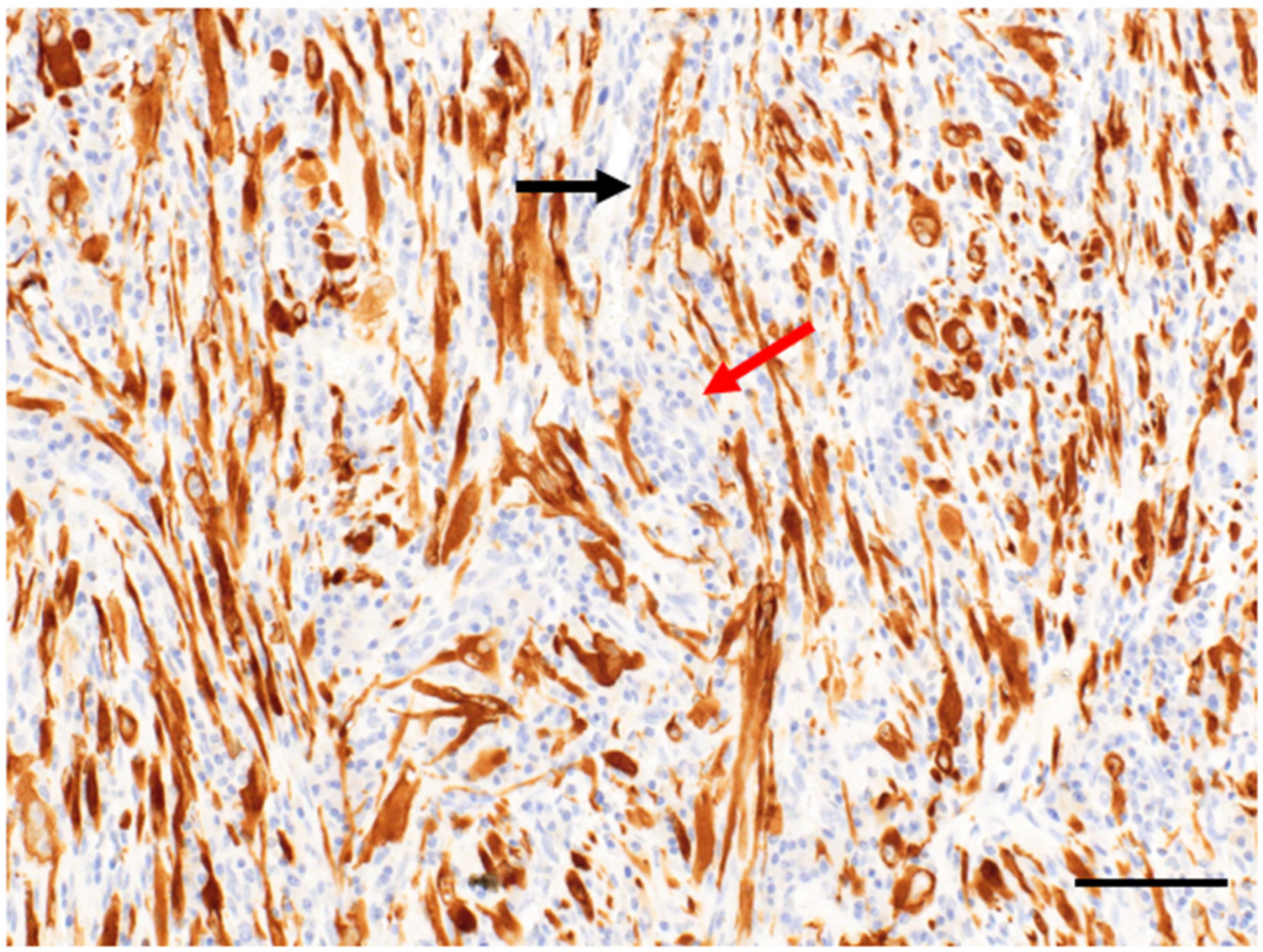
2. Clinicopathological Characteristics
3. Diagnosis
4. Current Treatments
4.1. Management of Localized Disease
4.2. Current Management Options for Advanced Disease
4.2.1. Chemotherapy
4.2.2. Targeted Therapy
4.2.3. Immunotherapy
5. Available Clinical Data on Epithelioid Inflammatory Myofibroblastic Sarcomas
6. Future Directions
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jo, V.Y.; Fletcher, C.D. WHO classification of soft tissue tumours: An update based on the 2013 (4th) edition. Pathology 2014, 46, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Gleason, B.C.; Hornick, J.L. Inflammatory myofibroblastic tumours: Where are we now? J. Clin. Pathol. 2008, 61, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Gupta, A.; Lipson, D.; Otto, G.; Brennan, T.; Chung, C.T.; Borinstein, S.C.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014, 4, 889–895. [Google Scholar] [CrossRef]
- Surabhi, V.R.; Chua, S.; Patel, R.P.; Takahashi, N.; Lalwani, N.; Prasad, S.R. Inflammatory Myofibroblastic Tumors: Current Update. Radiol. Clin. N. Am. 2016, 54, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Panagiotopoulos, N.; Patrini, D.; Gvinianidze, L.; Woo, W.L.; Borg, E.; Lawrence, D. Inflammatory myofibroblastic tumour of the lung: A reactive lesion or a true neoplasm? J. Thorac. Dis. 2015, 7, 908–911. [Google Scholar] [CrossRef]
- Siemion, K.; Reszec-Gielazyn, J.; Kisluk, J.; Roszkowiak, L.; Zak, J.; Korzynska, A. What do we know about inflammatory myofibroblastic tumors?—A systematic review. Adv. Med. Sci. 2022, 67, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.H.; Bohn, O.L.; Beddow, T.D.; McHenry, C.R. Inflammatory myofibroblastic tumor of the small bowel mesentery: An unusual cause of abdominal pain and uveitis. J. Gastrointest. Surg. 2011, 15, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Coffin, C.M.; Watterson, J.; Priest, J.R.; Dehner, L.P. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am. J. Surg. Pathol. 1995, 19, 859–872. [Google Scholar] [CrossRef]
- Palaskar, S.; Koshti, S.; Maralingannavar, M.; Bartake, A. Inflammatory myofibroblastic tumor. Contemp. Clin. Dent. 2011, 2, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef]
- Coffin, C.M.; Hornick, J.L.; Fletcher, C.D. Inflammatory myofibroblastic tumor: Comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am. J. Surg. Pathol. 2007, 31, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Wang, L.; Nascimento, A.G.; Moir, C.R.; Rodeberg, D.A. Pediatric inflammatory myofibroblastic tumor: Anaplastic lymphoma kinase (ALK) expression and prognosis. Pediatr. Blood Cancer 2005, 45, 796–801. [Google Scholar] [CrossRef]
- Gomez-Roman, J.J.; Sanchez-Velasco, P.; Ocejo-Vinyals, G.; Hernandez-Nieto, E.; Leyva-Cobian, F.; Val-Bernal, J.F. Human herpesvirus-8 genes are expressed in pulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). Am. J. Surg. Pathol. 2001, 25, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.W.; Kirstein, M.N.; Valentine, M.B.; Dittmer, K.G.; Shapiro, D.N.; Saltman, D.L.; Look, A.T. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Cheuk, W.; Shimizu, M. Anaplastic lymphoma kinase expression in inflammatory pseudotumors. Am. J. Surg. Pathol. 2001, 25, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.R.; Dehner, L.P.; Collins, M.H.; Ma, Z.; Morris, S.W.; Coffin, C.M.; Hill, D.A. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: A comparative immunohistochemical study. Am. J. Surg. Pathol. 2001, 25, 1364–1371. [Google Scholar] [CrossRef]
- Marino-Enriquez, A.; Wang, W.L.; Roy, A.; Lopez-Terrada, D.; Lazar, A.J.; Fletcher, C.D.; Coffin, C.M.; Hornick, J.L. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am. J. Surg. Pathol. 2011, 35, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Sobanko, J.F.; Meijer, L.; Nigra, T.P. Epithelioid sarcoma: A review and update. J. Clin. Aesthet. Dermatol. 2009, 2, 49–54. [Google Scholar] [PubMed]
- Yu, L.; Liu, J.; Lao, I.W.; Luo, Z.; Wang, J. Epithelioid inflammatory myofibroblastic sarcoma: A clinicopathological, immunohistochemical and molecular cytogenetic analysis of five additional cases and review of the literature. Diagn. Pathol. 2016, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.T.; Lee, J.C. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: Combination of distinct morphologic, immunohistochemical, and genetic features. Hum. Pathol. 2008, 39, 1854–1858. [Google Scholar] [CrossRef]
- Martorell, M.; Perez-Valles, A.; Gozalbo, F.; Garcia-Garcia, J.A.; Gutierrez, J.; Gaona, J. Solitary fibrous tumor of the thigh with epithelioid features: A case report. Diagn. Pathol. 2007, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Nozaki, Y.; Kohashi, K.; Kinoshita, I.; Oda, Y. Diagnostic utility of pan-Trk immunohistochemistry for inflammatory myofibroblastic tumours. Histopathology 2020, 76, 774–778. [Google Scholar] [CrossRef]
- Elpek, G.O. Inflammatory Myofibroblastic Tumor of the Liver: A Diagnostic Challenge. J. Clin. Transl. Hepatol. 2014, 2, 53–57. [Google Scholar] [CrossRef][Green Version]
- Gronchi, A.; Miah, A.B.; Dei Tos, A.P.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2021, 32, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Cecchetto, G.; Bisogno, G.; Gambini, C.; Calabro, M.L.; Inserra, A.; Boldrini, R.; De Salvo, G.L.; ES, G.d.A.; Dall’igna, P. Inflammatory myofibroblastic tumors in childhood: A report from the Italian Cooperative Group studies. Cancer 2010, 116, 216–226. [Google Scholar] [CrossRef]
- Souid, A.K.; Ziemba, M.C.; Dubansky, A.S.; Mazur, M.; Oliphant, M.; Thomas, F.D.; Ratner, M.; Sadowitz, P.D. Inflammatory myofibroblastic tumor in children. Cancer 1993, 72, 2042–2048. [Google Scholar] [CrossRef]
- Zhu, Z.; Zha, Y.; Wang, W.; Wang, X.; Gao, Y.; Lv, W. Inflammatory Myofibroblastic Tumors in Paranasal Sinus and Nasopharynx: A Clinical Retrospective Study of 13 Cases. Biomed. Res. Int. 2018, 2018, 7928241. [Google Scholar] [CrossRef] [PubMed]
- Biswas, R.; Halder, A.; Gangopadhyay, M.; Biswas, D. Inflammatory myofibroblastic tumor of maxillary sinus successfully treated with radiotherapy and corticosteroid: Report of a rare case. J. Egypt. Natl. Canc. Inst. 2020, 32, 26. [Google Scholar] [CrossRef] [PubMed]
- Baldi, G.G.; Brahmi, M.; Lo Vullo, S.; Cojocaru, E.; Mir, O.; Casanova, M.; Vincenzi, B.; De Pas, T.M.; Grignani, G.; Pantaleo, M.A.; et al. The Activity of Chemotherapy in Inflammatory Myofibroblastic Tumors: A Multicenter, European Retrospective Case Series Analysis. Oncologist 2020, 25, e1777–e1784. [Google Scholar] [CrossRef]
- Kwak, E.L.; Bang, Y.J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.H.; Dezube, B.J.; Janne, P.A.; Costa, D.B.; et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef]
- Xu, P.; Shen, P.; Jin, Y.; Wang, L.; Wu, W. Epithelioid inflammatory myofibroblastic sarcoma of stomach: Diagnostic pitfalls and clinical characteristics. Int. J. Clin. Exp. Pathol. 2019, 12, 1738–1744. [Google Scholar] [PubMed]
- Kozu, Y.; Isaka, M.; Ohde, Y.; Takeuchi, K.; Nakajima, T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen. Thorac. Cardiovasc. Surg. 2014, 62, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Kimbara, S.; Takeda, K.; Fukushima, H.; Inoue, T.; Okada, H.; Shibata, Y.; Katsushima, U.; Tsuya, A.; Tokunaga, S.; Daga, H.; et al. A case report of epithelioid inflammatory myofibroblastic sarcoma with RANBP2-ALK fusion gene treated with the ALK inhibitor, crizotinib. Jpn. J. Clin. Oncol. 2014, 44, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Kurihara-Hosokawa, K.; Kawasaki, I.; Tamai, A.; Yoshida, Y.; Yakushiji, Y.; Ueno, H.; Fukumoto, M.; Fukushima, H.; Inoue, T.; Hosoi, M. Epithelioid inflammatory myofibroblastic sarcoma responsive to surgery and an ALK inhibitor in a patient with panhypopituitarism. Intern. Med. 2014, 53, 2211–2214. [Google Scholar] [CrossRef]
- Sarmiento, D.E.; Clevenger, J.A.; Masters, G.A.; Bauer, T.L.; Nam, B.T. Epithelioid inflammatory myofibroblastic sarcoma: A case report. J. Thoracic. Dis. 2015, 7, E513–E516. [Google Scholar]
- Lee, J.C.; Wu, J.M.; Liau, J.Y.; Huang, H.Y.; Lo, C.Y.; Jan, I.S.; Hornick, J.L.; Qian, X. Cytopathologic features of epithelioid inflammatory myofibroblastic sarcoma with correlation of histopathology, immunohistochemistry, and molecular cytogenetic analysis. Cancer Cytopathol. 2015, 123, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Kan, Y.; Zhao, Y.; He, H.; Kong, L. Epithelioid inflammatory myofibroblastic sarcoma treated with ALK inhibitor: A case report and review of literature. Int. J. Clin. Exp. Pathol. 2015, 8, 15328–15332. [Google Scholar]
- Fang, H.; Langstraat, C.L.; Visscher, D.W.; Folpe, A.L.; Schoolmeester, J.K. Epithelioid Inflammatory Myofibroblastic Sarcoma of the Ovary With RANB2-ALK Fusion: Report of a Case. Int. J. Gynecol. Pathol. 2018, 37, 468–472. [Google Scholar] [CrossRef]
- Schoffski, P.; Sufliarsky, J.; Gelderblom, H.; Blay, J.Y.; Strauss, S.J.; Stacchiotti, S.; Rutkowski, P.; Lindner, L.H.; Leahy, M.G.; Italiano, A.; et al. Crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumours with and without anaplastic lymphoma kinase gene alterations (European Organisation for Research and Treatment of Cancer 90101 CREATE): A multicentre, single-drug, prospective, non-randomised phase 2 trial. Lancet Respir. Med. 2018, 6, 431–441. [Google Scholar] [CrossRef]
- Schoffski, P.; Kubickova, M.; Wozniak, A.; Blay, J.Y.; Strauss, S.J.; Stacchiotti, S.; Switaj, T.; Bucklein, V.; Leahy, M.G.; Italiano, A.; et al. Long-term efficacy update of crizotinib in patients with advanced, inoperable inflammatory myofibroblastic tumour from EORTC trial 90101 CREATE. Eur. J. Cancer 2021, 156, 12–23. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Perol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus Crizotinib in Untreated ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef]
- Hida, T.; Nokihara, H.; Kondo, M.; Kim, Y.H.; Azuma, K.; Seto, T.; Takiguchi, Y.; Nishio, M.; Yoshioka, H.; Imamura, F.; et al. Alectinib versus crizotinib in patients with ALK-positive non-small-cell lung cancer (J-ALEX): An open-label, randomised phase 3 trial. Lancet 2017, 390, 29–39. [Google Scholar] [CrossRef]
- Wang, Z.; Geng, Y.; Yuan, L.Y.; Wang, M.M.; Ye, C.Y.; Sun, L.; Dai, W.P.; Zang, Y.S. Durable Clinical Response to ALK Tyrosine Kinase Inhibitors in Epithelioid Inflammatory Myofibroblastic Sarcoma Harboring PRRC2B-ALK Rearrangement: A Case Report. Front. Oncol. 2022, 12, 761558. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, A.S.; Murphy, S.J.; Harris, F.R.; Robinson, S.I.; Marks, R.S.; Johnson, S.H.; Smadbeck, J.B.; Halling, G.C.; Yi, E.S.; Wigle, D.; et al. Chromoplectic TPM3-ALK rearrangement in a patient with inflammatory myofibroblastic tumor who responded to ceritinib after progression on crizotinib. Ann. Oncol. 2016, 27, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Michels, S.Y.F.; Scheel, A.H.; Wundisch, T.; Heuckmann, J.M.; Menon, R.; Puesken, M.; Kobe, C.; Pasternack, H.; Heydt, C.; Scheffler, M.; et al. ALK(G1269A) mutation as a potential mechanism of acquired resistance to crizotinib in an ALK-rearranged inflammatory myofibroblastic tumor. NPJ Precis. Oncol. 2017, 1, 4. [Google Scholar] [CrossRef]
- Lee, C.J.; Schoffski, P.; Modave, E.; van Wezel, T.; Boeckx, B.; Sufliarsky, J.; Gelderblom, H.; Blay, J.Y.; Debiec-Rychter, M.; Sciot, R.; et al. Comprehensive Molecular Analysis of Inflammatory Myofibroblastic Tumors Reveals Diverse Genomic Landscape and Potential Predictive Markers for Response to Crizotinib. Clin. Cancer Res. 2021, 27, 6737–6748. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Zhao, X.; Huang, C.; Zhou, X.; You, Y.; Zhang, L.; Lu, C.; Yao, F.; Li, S. Double amplifications of CDK4 and MDM2 in a gastric inflammatory myofibroblastic tumor mimicking cancer with local invasion of the spleen and diaphragm. Cancer Biol. Ther. 2018, 19, 967–972. [Google Scholar] [CrossRef]
- Yamamoto, H.; Oda, Y.; Saito, T.; Sakamoto, A.; Miyajima, K.; Tamiya, S.; Tsuneyoshi, M. p53 Mutation and MDM2 amplification in inflammatory myofibroblastic tumours. Histopathology 2003, 42, 431–439. [Google Scholar] [CrossRef]
- Liu, D.; Offin, M.; Harnicar, S.; Li, B.T.; Drilon, A. Entrectinib: An orally available, selective tyrosine kinase inhibitor for the treatment of NTRK, ROS1, and ALK fusion-positive solid tumors. Ther. Clin. Risk Manag. 2018, 14, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.H.; Bentley, H.; Bulusu, V.R.; Anyaegbu, G.; Watkins, J.; Horan, G.; Hatcher, H. Lorlatinib for the treatment of inflammatory myofibroblastic tumour with TPM4-ALK fusion following failure of entrectinib. Anticancer Drugs 2020, 31, 1106–1110. [Google Scholar] [CrossRef]
- Carcamo, B.; Bista, R.; Wilson, H.; Reddy, P.; Pacheco, J. Rapid Response to Lorlatinib in a Patient With TFG-ROS1 Fusion Positive Inflammatory Myofibroblastic Tumor of the Chest Wall Metastatic to the Brain and Refractory to First and Second Generation ROS1 Inhibitors. J. Pediatr. Hematol. Oncol. 2021, 43, e718–e722. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yoshida, A.; Taguchi, K.; Kohashi, K.; Hatanaka, Y.; Yamashita, A.; Mori, D.; Oda, Y. ALK, ROS1 and NTRK3 gene rearrangements in inflammatory myofibroblastic tumours. Histopathology 2016, 69, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Hornick, J.L.; Sholl, L.M.; Dal Cin, P.; Childress, M.A.; Lovly, C.M. Expression of ROS1 predicts ROS1 gene rearrangement in inflammatory myofibroblastic tumors. Mod. Pathol. 2015, 28, 732–739. [Google Scholar] [CrossRef]
- Mai, S.; Xiong, G.; Diao, D.; Wang, W.; Zhou, Y.; Cai, R. Case report: Crizotinib is effective in a patient with ROS1-rearranged pulmonary inflammatory myofibroblastic tumor. Lung Cancer 2019, 128, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Comandini, D.; Catalano, F.; Grassi, M.; Pesola, G.; Bertulli, R.; Guadagno, A.; Spina, B.; Mascherini, M.; De Cian, F.; Pistoia, F.; et al. Outstanding Response in a Patient With ROS1-Rearranged Inflammatory Myofibroblastic Tumor of Soft Tissues Treated with Crizotinib: Case Report. Front. Oncol. 2021, 11, 658327. [Google Scholar] [CrossRef] [PubMed]
- Applebaum, H.; Kieran, M.W.; Cripe, T.P.; Coffin, C.M.; Collins, M.H.; Kaipainen, A.; Laforme, A.; Shamberger, R.C. The rationale for nonsteroidal anti-inflammatory drug therapy for inflammatory myofibroblastic tumors: A Children’s Oncology Group study. J. Pediatr. Surg. 2005, 40, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- van der Graaf, W.T.; Blay, J.Y.; Chawla, S.P.; Kim, D.W.; Bui-Nguyen, B.; Casali, P.G.; Schoffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y.; et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Takahashi, A.; Kurosawa, M.; Uemura, M.; Kitazawa, J.; Hayashi, Y. Anaplastic lymphoma kinase-negative uterine inflammatory myofibroblastic tumor containing the ETV6-NTRK3 fusion gene: A case report. J. Int. Med. Res. 2018, 46, 3498–3503. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, T.R.; Duong, A.T.; Gocke, C.D.; Xu, H.; Ogurtsova, A.; Taube, J.M.; Belchis, D.A. PD-L1 expression in inflammatory myofibroblastic tumors. Mod. Pathol. 2018, 31, 1155–1163. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, Z. A case report on epithelioid inflammatory myofibroblastic sarcoma in the abdominal cavity. Int. J. Clin. Exp. Pathol. 2019, 12, 3934–3939. [Google Scholar] [PubMed]
- Quiroga, D.; Liebner, D.A.; Philippon, J.S.; Hoffman, S.; Tan, Y.; Chen, J.L.; Lenobel, S.; Wakely, P.E., Jr.; Pollock, R.; Tinoco, G. Activity of PD1 inhibitor therapy in advanced sarcoma: A single-center retrospective analysis. BMC Cancer 2020, 20, 527. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, L.; Wang, Q.; Chen, J.; Zhang, C.; Tao, R.; Wang, Y. Genetic Testing and Immunotherapy for Intracranial Inflammatory Myofibroblastic Tumor: A Case Report. Oncol. Targets Ther. 2022, 15, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yin, W.H.; Takeuchi, K.; Guan, H.; Huang, Y.H.; Chan, J.K. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: A report of two cases and literature review. Diagn. Pathol. 2013, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Rafee, S.; Elamin, Y.Y.; Joyce, E.; Toner, M.; Flavin, R.; McDermott, R.; Sheehy, N.; Hennessy, B.; O’Byrne, K.; Gleeson, N.; et al. Neoadjuvant crizotinib in advanced inflammatory myofibroblastic tumour with ALK gene rearrangement. Tumori 2015, 101, e35–e39. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Jiang, J.; Tian, X.Y.; Li, Z. Pulmonary epithelioid inflammatory myofibroblastic sarcoma with multiple bone metastases: Case report and review of literature. Diagn. Pathol. 2015, 10, 106. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Jiang, M.; Liang, W.; Chen, F. Incomplete Intestinal Obstruction Caused by a Rare Epithelioid Inflammatory Myofibroblastic Sarcoma of the Colon: A Case Report. Medicine 2015, 94, e2342. [Google Scholar] [CrossRef]
- Wu, H.; Meng, Y.H.; Lu, P.; Ning, H.Y.; Hong, L.; Kang, X.L.; Duan, M.G. Epithelioid inflammatory myofibroblastic sarcoma in abdominal cavity: A case report and review of literature. Int. J. Clin. Exp. Pathol. 2015, 8, 4213–4219. [Google Scholar]
- Lee, J.C.; Li, C.F.; Huang, H.Y.; Zhu, M.J.; Marino-Enriquez, A.; Lee, C.T.; Ou, W.B.; Hornick, J.L.; Fletcher, J.A. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: Novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J. Pathol. 2017, 241, 316–323. [Google Scholar] [CrossRef]
- Jiang, Q.; Tong, H.X.; Hou, Y.Y.; Zhang, Y.; Li, J.L.; Zhou, Y.H.; Xu, J.; Wang, J.Y.; Lu, W.Q. Identification of EML4-ALK as an alternative fusion gene in epithelioid inflammatory myofibroblastic sarcoma. Orphanet. J. Rare Dis. 2017, 12, 97. [Google Scholar] [CrossRef]
- Fang, N.; Yang, Q.J.; Deng, Y.T.; Feng, X.; Xia, H.S.; Zhang, Y.G.; Wang, M.W.; Wu, D.; Zhou, H.; Guo, F. Epithelioid inflammatory myofibroblastic sarcoma of small bowel mesentery: Report of a case. Zhonghua Bing Li Xue Za Zhi 2017, 46, 201–202. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Gao, Y.; Zhao, H.; Li, B.; Xue, W.; Wang, D. Clinicopathological analysis of epithelioid inflammatory myofibroblastic sarcoma. Oncol. Lett. 2018, 15, 9317–9326. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Li, H.; Peng, K.; Yu, Y.; Chen, L.; Fang, Y.; Sun, Y.; Hou, Y.; Liu, T. ALK-G1269A mutation in epithelioid inflammatory myofibroblastic sarcoma after progression on crizotinib: A case report. Oncol. Lett. 2019, 17, 2370–2376. [Google Scholar] [CrossRef] [PubMed]
- Hallin, M.; Thway, K. Epithelioid Inflammatory Myofibroblastic Sarcoma. Int. J. Surg. Pathol. 2019, 27, 69–71. [Google Scholar] [CrossRef]
- Liu, D.; Luo, R.; Tang, H.; Li, T. Sigmoid epithelioid inflammatory myofibroblastic sarcoma with high white blood cell count: A case report. Asian J. Surg. 2020, 43, 838–839. [Google Scholar] [CrossRef] [PubMed]
- Kopelevich, A.; Holzman, S.A.; La, J.; Lai, H.A.; Torno, L.; Stephany, H.A. Crizotinib-associated Renal Cyst Formation in a Pediatric Patient with ALK+ Epithelioid Inflammatory Myofibroblastic Sarcoma. Urology 2021, 149, 222–224. [Google Scholar] [CrossRef]
- Zilla, M.L.; Khoshnoodi, P.; Bailey, N.G.; Herradura, A.; Lee, S.J.; John, I. Epithelioid inflammatory myofibroblastic sarcomas are not exclusive to ventral cavity sites. Histopathology 2022, 80, 610–612. [Google Scholar] [CrossRef]
- Chopra, S.; Maloney, N.; Wang, W.L. Epithelioid inflammatory myofibroblastic sarcoma with VCL-ALK fusion of central nervous system: Case report and brief review of the literature. Brain Tumor Pathol. 2022, 39, 35–42. [Google Scholar] [CrossRef]
- Gadeyne, L.; Creytens, D.; Dekeyser, S.; Van der Meulen, J.; Haspeslagh, M. Primary Cutaneous Epithelioid Inflammatory Myofibroblastic Sarcoma Harboring RANBP2-ALK Fusion: Report of an Exceptional Case. Am. J. Dermatopathol. 2022, 44, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.; Ramalingam, P.; Euscher, E.D.; Reques Llanos, A.; Garcia, A.; Malpica, A. Uterine Inflammatory Myofibroblastic Neoplasms With Aggressive Behavior, Including an Epithelioid Inflammatory Myofibroblastic Sarcoma: A Clinicopathologic Study of 9 Cases. Am. J. Surg. Pathol. 2022, 46, 105–117. [Google Scholar] [CrossRef]
- Ma, Z.; Hill, D.A.; Collins, M.H.; Morris, S.W.; Sumegi, J.; Zhou, M.; Zuppan, C.; Bridge, J.A. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer 2003, 37, 98–105. [Google Scholar] [CrossRef]
- Rottgers, S.; Gombert, M.; Teigler-Schlegel, A.; Busch, K.; Gamerdinger, U.; Slany, R.; Harbott, J.; Borkhardt, A. ALK fusion genes in children with atypical myeloproliferative leukemia. Leukemia 2010, 24, 1197–1200. [Google Scholar] [CrossRef]
- Sasaki, T.; Okuda, K.; Zheng, W.; Butrynski, J.; Capelletti, M.; Wang, L.; Gray, N.S.; Wilner, K.; Christensen, J.G.; Demetri, G.; et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010, 70, 10038–10043. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Farris, N.; Sampson, M. Single-agent rituximab for treatment of multifocal and multiple relapsed pulmonary inflammatory myofibroblastic tumor in an adolescent patient. Pediatr. Blood Cancer 2021, 68, e29131. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author, Year | Number of Cases | Age Range | M:F | Location | Treatment | Recurrence/ Metastasis | Dead of Disease | ALK IHC Pattern | Fusion Partner |
|---|---|---|---|---|---|---|---|---|---|
| Butrynski J et al., 2010 [32] | 1 | 44 | M | Intraabdominal | SE, HPP, CT Imatinib ALKi (Crizotinib) | Yes | - | Nuclear membrane | RANBP2–ALK |
| Mariño-Enríquez A et al., 2011 [17] | 11 | 6–63 | 10:1 | Intraabdominal | SE (2) SE + CT (5) SE +CT+RT (2) SE + CT +ALKi (1) (Experimental ALK inhibitor) NA (1) | 10 | 5 out of 8 in which follow-up available | Nuclear membrane 9 out of 11 Cytoplasmic with perinuclear attenuation in 2 of 11 | 9 ALK translocation RANBP2–ALK in 3 of the 9 |
| Li J et al., 2013 [65] | 2 | 19 and 39 | 1:1 | Pelvic cavity | SE SE + CT | 2 | 1 | Nuclear membrane | RANBP2–ALK |
| Kozu et al., 2014 [34] | 1 | 57 | M | Pleural cavity | CT + ALKi (ALK inhibitor no precision) | Yes | SE | Cytoplasmic pattern with perinuclear accentuation | RANBP2–ALK |
| Kimbara S et al., 2014 [35] | 1 | 22 | M | Intraabdominal | SE + CT + ALKi (Crizotinib) | Yes | No F/U after 10 months | Nuclear membrane | RANBP2-ALK |
| Kurihara-Hosokawa K et al., 2014 [36] | 1 | 22 | M | Intraabdominal | SE + ALKi (Crizotinib) | Yes | No | Nuclear membrane | RANBP2-ALK |
| Rafee S et al., 2015 [66] | 1 | 55 | F | Intraabdominal | CT ALKi (Crizotinib) SE | Yes | Nuclear membranous staining | Only ALK FISH done, fusion parnters unknown | |
| Fu X et al., 2015 [67] | 1 | 21 | M | Lung | SE ALKi (Crizotinib) | Bone metastasis | No F/U after 3 months | Cytoplasmic | Only ALK FISH done, fusion parnters unknown |
| Bai Y et al. 2015 [68] | 1 | 65 | M | Intraabdominal | Yes | No F/U | No mentioned | No mentioned | |
| Wu H et al., 2015 [69] | 1 | 47 | F | Intraabdominal | Yes | Yes | Nuclear membrane | RANBP2-ALK | |
| Sarmiento et al., 2015 [37] | 1 | 71 | M | Pleural | SE + ALKi (Crizotinib, switch for 2nd line) | No | Alive | ALK positive—pattern not mentioned | Only ALK FISH done—fusion partners unknown |
| Lee JC et al., 2015 [70] | 5 | 16–76 | 3:2 | Liver (1) Lung (1) Intraabdominal (3) | SE (3) SE + CT (1) SE + CT + RTH + ALKi (1) (Crizotinib) | 4 DOD within 12 months 1 Alive at 33 months | Cytoplasmic (2) Nuclear (3) | RANBP2-ALK fusion in the 2 other cases | |
| Liu Q et al., 2015 [39] | 1 | 22 | M | Intraabdominal | SE + ALKi (Crizotinib) | Yes | Alive | Nuclear membrane | RANBP2-ALK |
| Yu L et al., 2016 [19] | 5 | 15–58 | 2:3 | Intraabdominal | 37: SE 55: SE, SE, CT 22: SE, recurrence, ALKi (Crizotinib) 58: SE, CT 15: SE | Yes 3 out of 5 | 37: No recurrene, alive 55: | Nuclear membrane pattern in 4 cases Cytoplasmic staining with perinuclear accentuation fashion in 1case | 5 tumors showed ALK gene rearrangement |
| Jiang et al., 2017 [71] | 1 | 45 | M | Intraabdominal | SE + ALKIi adjuvant (Crizotinib –stop for severe vomiting + elevation AST et ALT) ALki for mestatatic disease (Crizotinib) –tumor lyse syndrome, DOD | Metastasis to liver, spleen, small intestine et al. | Yes | Cytoplasmic | EML4-ALK |
| Lee et al., 2017 [70] | 9 | 7 months-76 | 6:3 | Intraabdominal (n = 7) Lung (n = 1) Liver (n = 1) | SE (9) 2 treated with ALKi (Crizotinib) | Yes 9 out of 9 | 6 | 4 Nuclear membrane (n = 4) and 5 cytoplasmic staining (n = 5; 4 with perinuclear accentuation). | RANBP2-ALK (3) RRBP1-ALK (5) No mentionned (1) |
| Fang et al., 2017 [72] | 1 | 52 | F | Small bowel | ? | ? | Yes (8 months) | ? | ALK done, fusion partners unknown |
| Du X et al., 2018 [73] | 1 | 26 | M | Intraabdominal | SE + CT | Yes | Yes | Cell nuclei | RANBP2-ALK |
| Xu X et al., 2019 [74] | 1 | 28 | M | Intraabdominal | ALKi (Crizotinib then Brigatinib) | Yes | No | RANBP2-ALK | |
| Hallin M et al., 2018 [75] | 1 | - | - | - | - | - | - | - | ALK-unknown |
| Xu P et al., 2019 [33] | 1 | 35 | F | Gastric | SE | No | No (limited F/U) | Cytoplasmic | N/A |
| Zhang S et al., 2019 [62] | 1 | 46 | F | Intraabdominal | SE ALKi at progression (Crizotinib), no response and then multi-targeting tyrosine kinase inhibitor (Anlotinib) | Yes | Yes (16 months) | Yes | 2p23 ALK gene rearrangement |
| Liu D et al., 2020 [76] | 1 | N/A | N/A | Sigmoid colon | SE + ALKi (Crizotinib) | Yes | N/A | Perinuclear | RANBP2–ALK |
| Kopelevich A et al., 2020 [77] | 1 | 17 | M | Renal | SE + ALKi (Crizotinib, and then Alectinib) | Yes | N/A | N/A | RANBP2–ALK |
| Zilla et al., 2021 [78] | 1 | 80 | M | Right groin | SE | ? | No G/U | Nuclear membrane | RANBP2-ALK |
| Chopra S et al., 2021 [79] | 1 | 72 | F | Brain | SE At progression ALKi (Alectinib) | Yes | At 4 months | Cytoplasmic | VCL-ALK |
| Gadeyne L et al., 2021 [80] | 1 | 27 | F | Cutaneous | SE | No | No | Very clear cytoplasmic staining with perinuclear accentuation | RANBP2-ALK |
| Collins K et al., 2022 [81] | 1 | 43 | F | Uterus | SE + CT | Yes | No | Nuclear membrane | RANBP2-ALK |
| Wang S et al., 2022 [45] | 1 | 42 | F | Intraabdominal | SE Crizotinib At progression (PD after 5 months) Alectinib At progression (PD after 5 months) Ceritinib At progression (PD after 6 months) Lorlatnib (SD after 5 months) | Yes | No | PRRC2B-ALK | |
| Current case | 1 | 39 | F | Intraabdominal | ALKi (Alectinib) | Yes | Nuclear membrane | RANBP2-ALK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gros, L.; Dei Tos, A.P.; Jones, R.L.; Digklia, A. Inflammatory Myofibroblastic Tumour: State of the Art. Cancers 2022, 14, 3662. https://doi.org/10.3390/cancers14153662
Gros L, Dei Tos AP, Jones RL, Digklia A. Inflammatory Myofibroblastic Tumour: State of the Art. Cancers. 2022; 14(15):3662. https://doi.org/10.3390/cancers14153662
Chicago/Turabian StyleGros, Louis, Angelo Paolo Dei Tos, Robin L. Jones, and Antonia Digklia. 2022. "Inflammatory Myofibroblastic Tumour: State of the Art" Cancers 14, no. 15: 3662. https://doi.org/10.3390/cancers14153662
APA StyleGros, L., Dei Tos, A. P., Jones, R. L., & Digklia, A. (2022). Inflammatory Myofibroblastic Tumour: State of the Art. Cancers, 14(15), 3662. https://doi.org/10.3390/cancers14153662