
Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview

 ,
,  ,
,  ,
,  ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pathological and Molecular Features
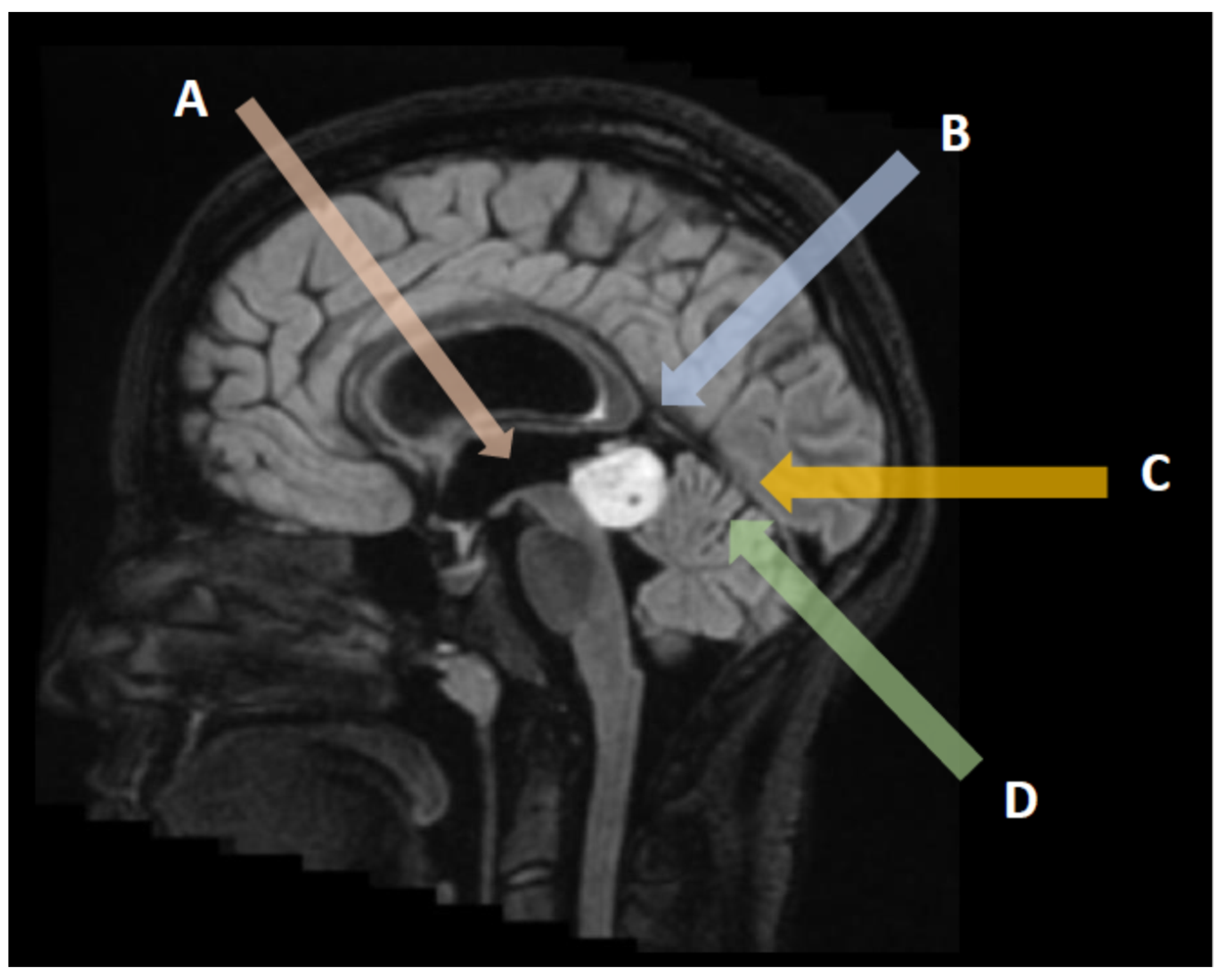
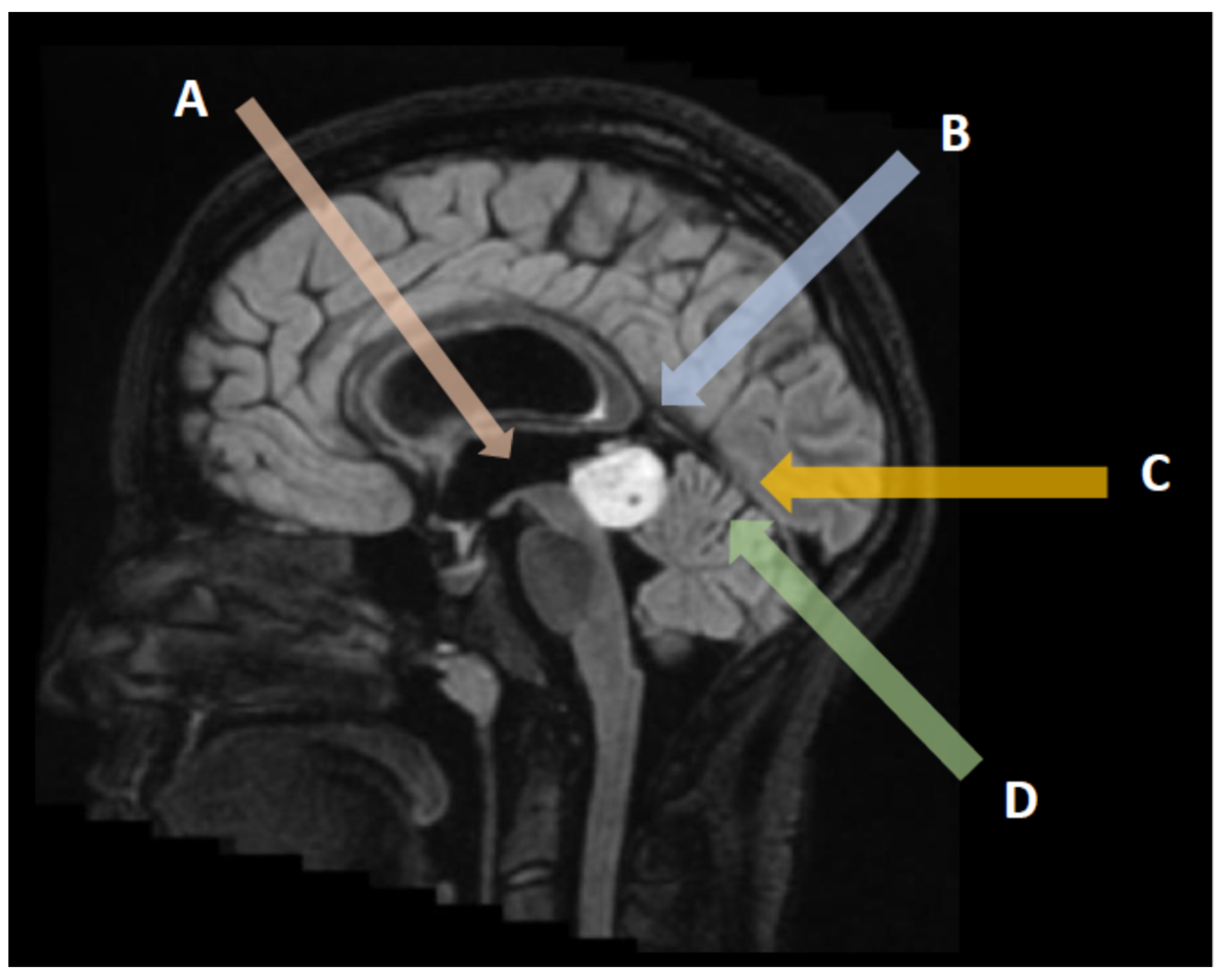
3. Neuroradiological Assessment
3.1. Pineal Parenchymal Tumors
- −
- Pineocytoma generally appears as a well-circumscribed tumor with a maximum dimension of less than 3 cm. Compared to the adjacent brain, the lesion appears iso-/hyperdense on CT, hypo-/isointense on T1, and iso-hyperintense on T2. The tumor tends to be solid and presents strong homogeneous enhancement. Cystic changes may be present which, in some cases, can make it difficult to differentiate it from a pineal cyst; a nodular or thickened wall enhancement helps identify its neoplastic nature [33]. Intralesional hemorrhage may occur, while calcifications are usually peripheral, distinguishing pineocytomas from pineal germinomas that tend to engulf pineal calcification. Pineal apoplexy is reported [34].
- −
- Pineoblastomas usually present as large (more than 3 cm), irregular, poorly defined lobulated tumors, often invading the adjacent brain. On CT, they appear hyperdense compared to the adjacent brain due to high cellularity, with frequent necrotic areas and hemorrhagic changes due to their highly malignant nature. As in pineocytomas, calcifications are peripherally dispersed (“blasted calcifications”). A cystic appearance may occur, but the walls are usually thicker and more irregular than in pineal cysts. On MRI, pineoblastomas present heterogeneous signal intensity (hypo- to isointense on T1 and iso- to hyperintense T2) and restricted diffusion. Besides elevated choline and decreased N-acetylaspartate, MR spectroscopy reveals slightly elevated glutamate and taurine peaks (∼3.4 ppm). Contrast enhancement is vivid and CSF seeding is present in 15% of patients at the time of diagnosis and up to 45% during the course of the disease. Consequently, screening of the entire neural axis is necessary both at the time of biopsy diagnosis/imaging suspicion and during follow-up. Obstructive hydrocephalus is common at presentation.
- −
- Pineal parenchymal tumors of intermediate differentiation range from well-circumscribed pineal tumors with pineocytoma-like characteristics to poorly defined, invasive masses with rapid growth and/or low ADC values. They often appear iso-hyperintense on T2, and may present cystic areas and heterogeneous contrast enhancement [34]. As these tumors may spread along the CSF, contrast-enhanced MRI of the entire craniospinal axis is required.
- −
- Papillary tumors of the pineal region also appear on MRI as well-circumscribed T2-hyperintense masses with variable contrast enhancement. Intralesional cysts are common, and their proteinaceous content may result in a heterogeneous T1 signal. Contrast-enhanced MRI of the entire craniospinal axis is recommended because local tumor recurrence and leptomeningeal tumor spread may occur.
3.2. Germ Cell Tumors
- −
- On CT, a pineal germinoma typically appears as a homogeneous, hyperdense mass compared to the adjacent brain. Cysts are present in 20–52% of cases [33,35,36]. Inner calcifications are common and often represent normal pineal calcifications engulfed within the tumor. On MRI, germinomas are usually T1 and T2 isointense to grey matter, DWI comparatively hyperintense, and ADC values are typically higher than in pineoblastomas. Contrast enhancement is vivid. Disseminated disease and subependymal tumor spread along the third ventricle are common at onset (13%).
- −
- Teratomas often manifest as large, multiloculated, lobulated lesions containing cysts and solid components, as well as intralesional areas of fat, calcifications, and fluid lesions [37]. On CT, the majority of teratomas demonstrate at least some highly hypodense or hyperdense components (fat and calcification, respectively). On MRI, teratomas may present T1-hyperintense components due to fat and proteinaceous/lipid-rich fluid and T1-hypointense components due to calcification and blood products. Given the extremely variable histological components, T2-weighted imaging also tends to be heterogeneous, with hypo- or hyperintense intratumoral components. After contrast medium administration, solid components show variable enhancement. Immature teratomas appear more homogeneous, with irregular infiltrating margins and with fewer cysts and calcifications, thus becoming more difficult to differentiate from other pineal tumors. Secondary somatic malignancies are not rare; therefore, surveillance for both secondary malignancy and growing teratoma syndrome is recommended [37]. Mature teratomas are well-circumscribed and often present fat components that help narrow the differential.
- −
- Pineal yolk sac tumors are rare and do not seem to differ from other germ cell tumors. These lesions are usually large, irregular, and frequently infiltrate the adjacent structures; they present variable densities and calcifications. On MRI, the lesions are hypo-isointense on T1 and hyperintense on T2-weighted images. Contrast enhancement is intense and homogeneous [38].
- −
- Embryonal carcinomas usually have large masses that are iso-hypointense on T1 and iso-hyperintense compared to grey matter on T2 [39]. Their margins are lobulated and irregular; invasion of the adjacent structures is suggested by edema along the tumor margins. Cystic areas are common; the solid portions show vivid contrast enhancement.
- −
- Pineal choriocarcinomas are large [40], highly vascular [41] lesions with a propensity to hemorrhage (as do their metastases). The masses are usually ovoid and relatively well-defined, although irregular infiltrating margins have been observed. Most lesions are iso-hypointense compared to the cortex on T1 and markedly heterogeneous on T2. Contrast enhancement is usually marked but heterogeneous. Micro and macrocysts or necrotic areas and mild-to-moderate peritumoral edema are common. Hemorrhages result in signal heterogeneity with blooming on T2* and SWI and with hyperintense foci on T1. On CT, the tumor appears heterogeneously hypodense; calcifications are uncommon.
- −
- Radiologic characteristics and survival outcomes of most frequent pineal parechymal tumors and germ cell tumors are summarized in Table 2.
3.3. Other Neoplasms of the Pineal Region
- −
- Pineal meningiomas are well-circumscribed masses, mostly arising from the contiguous tentorium, the tela choroidea, or the velum interpositum [53]. On CT, lesions are often iso- or hyperdense compared to grey matter; calcifications are detected in 15–20% of cases [34]. The MRI signal is variable. The mass shows intense homogeneous contrast enhancement, typically involving the contiguous dural structures (dural tail).
- −
- Primary pineal melanoma is exceedingly rare and can present as either melanotic or amelanotic MRI patterns. The former is more common, and the mass appears hyperintense on T1 and hypo- or isointense on T2 due to the presence of melanin. The amelanotic MRI pattern occurs less frequently, and the lesion is hypo- or isointense on T1 and hyperintense on T2 (less than 10% of cells contain melanin). Contrast enhancement is typically present [34].
- −
- Ependymomas appear heterogeneous, usually hypo- to isointense on T1 and iso- to hyperintense on T2. Cysts, calcifications, and hemorrhages are common. Contrast enhancement may be present; diffusion restriction is heterogeneous, but the ADC is predominantly high due to relatively low cellularity.
- −
- Pineal gliomas include pilocytic astrocytoma, IDH-mutant astrocytoma and oligodendroglioma, glioblastoma, and choroid plexus papilloma [54], although well-differentiated astrocytomas are the most prevalent [55]. Apart from tumors originating from the pineal glands, gliomas of the pineal region also include tectal, thalamic, and splenial gliomas. Imaging features are therefore rather heterogeneous based on the tumor’s grading and anatomical origin. Intralesional necrosis, calcifications, cysts, and hemorrhages are variably found; contrast enhancement is also variable.
- −
- Pineal metastases should be considered in patients with a history of any malignancy. Although lung cancer is the most frequently implicated, pineal metastases of almost all malignant tumors have been reported [56]. On neuroimaging, metastases are often isodense/isointense to grey matter. Lesion margins may be well-demarcated or may infiltrate the adjacent structures. Vivid contrast enhancement is the rule; necrosis and cysts may be present. Leptomeningeal seeding has been observed in two-thirds of cases; contrast-enhanced MRI of the entire craniospinal axis is therefore required.
3.4. Non-Neoplastic and Benign Pineal Region Lesions
- −
- Pineal cysts are very common in the general population (up to 40% in autopsy series) and almost always asymptomatic. On MRI, benign pineal cysts are usually oval, thin-walled, uni- or multi-loculated, or filled with a proteinaceous or hemorrhagic fluid that usually does not restrict on DWI. On MRI, the cysts are hypo- to isointense on T1 and iso- to hyperintense on T2 and FLAIR compared to grey matter. Common features are a thin rim of calcification (25% of cases) and, above all, a smooth (i.e., non-nodular) thin rim of wall enhancement, usually <2 mm in thickness. Internal enhancement may be observed on delayed images due to seepage of gadolinium into the cyst fluid. There is no invasion of adjacent structures, and minimal or no mass effect. The midbrain aqueduct remains unobstructed [57].
- −
- Cysts of the velum interpositum are a common anatomic variation with dilatation of the normal cistern of the velum interpositum (diameter > 1 cm). The cyst usually assumes a triangular configuration pointing anteriorly and typically incorporates the internal cerebral veins along its lateral walls. On CT and MRI, the cyst’s content has the features of CSF.
- −
- Arachnoid cysts appear as a sharply defined extra-axial fluid collection, which is similar in signal to CSF on all MRI sequences, including DWI. There is no post-contrast enhancement. When severe hydrocephalus is present, arachnoid cysts of the pineal region should be differentiated from ventricular diverticula, as the latter usually disappear with ventricle shunting.
- −
- Epidermoid cysts are non-neoplastic lesions that mostly appear hypodense on CT; focal calcifications may be observed. On MRI, these masses usually present CSF-like signals on T1 and T2, but exhibit a bright signal on DWI [58]. Contrast enhancement is typically absent, although faint, very late peripheral enhancement may be observed.
- −
- Dermoid cysts are usually well-defined rounded midline masses that commonly appear markedly hypodense on CT, hyperintense on T1, and hypointense on fat-saturated T1 due to intracyst lipid content [59]. Calcification may be present in the wall. Typically, dermoid cysts do not enhance after contrast administration, although a thin peripheral rim may be observed. The cyst’s rupture may release lipid droplets into the subarachnoid spaces, aiding in the distinction between lipomas and mature teratomas.
- −
- Pineal lipomas are easily recognized with CT due to the very low density of fat. Calcifications may be present. On MRI, the well-demarcated lesion appears homogeneously hyperintense on T1 and hypointense on fat-suppressed imaging. The typical lack of contrast enhancement helps to differentiate lipomas from mature teratomas.
- −
- Vein of Galen aneurysm malformation is the abnormal dilation of the embryonic precursor to the vein of Galen due to congenital arteriovenous malformations. It is usually observed in neonatal or fetal imaging. This condition is easily identified by MR-angiography and signal void due to blood flow.
- −
- Cavernomas are rare, but usually their MRI characteristics allow a prompt diagnosis in the majority of cases. On T2 images, they usually have a “pop-corn like” appearance surrounded by a hypointense hemosiderin ring. MRI signal intensity depends on whether there has been recent hemorrhage or thrombosis [60].
4. The Role of Surgery
4.1. Management of Hydrocephalus
4.2. Biopsy
4.3. Surgical Removal
4.4. Specific Management of Pineal Cysts
5. The Role of Radiotherapy

{kind=link}
{kind=link}
| Tumor Type | WHO Grade | Treatment Strategy | Extent of Irradiation and Dose Fractionation |
|---|---|---|---|
| Pure germinoma [73,74] | NA | RT in combination with ChT or as an exclusive treatment | Limited disease: WVI 24 Gy and tumor boost 16 Gy; WVI 18 Gy and boost 12 Gy in 1.6–1.8 Gy fractions for patients with complete response; metastatic disease: CSI alone 24 Gy and tumor boost 16 Gy in 1.8 Gy fractions; |
| Non-germinomatous germ cell tumor [76,77,78] | NA | Postoperative RT in combination with ChT | Limited disease: focal RT 54 Gy or CSI 30.4–36 Gy and tumor boost 18–23.4 Gy; Metastatic disease: CSI, 30.4–36 Gy and tumor boost 18–24 Gy in 1.8 Gy fractions; |
| Pineoblastoma [80,81] | 4 | Postoperative RT in combination with ChT | CSI 24–36 Gy and tumor boost 45–54 Gy in 1.8–2 Gy fractions |
| Pineal parenchymal tumor of intermediate differentiation [46] | 2,3 | Postoperative RT alone or in combination with ChT | Focal RT 50.4–54 Gy or CSI 24–36 Gy and tumor boost 45–54 Gy in 1.8–2 Gy fractions; |
| Papillary tumor of the pineal region [48] | 2,3 | Incompletely resected or recurrent tumors | Focal RT 50.4–54 Gy in 1.8–2 Gy fractions; |
| Pineocytoma [42] | 1 | Commonly not indicated. To be evaluated in recurrent inoperable tumors | Focal RT 50.4–54 Gy in 1.8–2 Gy fractions or SRS 15–18 Gy (small residual or recurrent tumors) |
6. The Role of Sytemic Treatment
6.1. Pinealoblastoma
6.2. Germ Cell Tumor
| Pinealoblastoma | ||||||
|---|---|---|---|---|---|---|
| Author | Date | Phase | Trial Name | Patients | Chemotherapy | Results |
| Liu [26] | 2020 | III | SJMB03 | 30 and 12 non-protocol | Risk-adapted IR + HDC (cisplatin/cyclphosphamide/VCR) + ASCR | IR: 5y-PFS & 5y-OS: 100% HR: 5y-PFS: 56%; 5y-OS: 60% |
| Gerber [88] | 2014 | II | HIT2000 | 11 | IR + weekly concomitant VCR + adjuvant (lomustine/cisplatin/VCR) | 5y-PFS & 5y-OS: 64% |
| Cohen [86] | 1995 | III | CCG-921 | 17 | A: IR + weekly concomitant vincristine + adjuvant (VCR/CCNU/prednisone) B: neoadjuvant 8-in-1 (methylprednisolone/VCR/CCNU/procarbazine/hydroxyurea/cisplatin/cytarabine/cyclophosphamide) + IR + adjuvant 8-in-1 | 3y-PFS: 61%; 3y-OS: 73% |
| Gururangan [90] | 2003 | II | 12 | IR + HDC (cyclophosphamide/melphalan or busulfan/melphalan) + ASCR | 4y-PFS: 69%; 4y-OS: 71% | |
| Hinkes [80] | 2007 | IIB | HIT91 | 6 | A: sandwich (ifosfamide/VP16/MTX/cisplatin/cytarabin) + IR + concomitant VCR +/− maintenance (carboplatin/VCR/CCNU) B: IR + concomitant VCR + adjuvant (CCNU/Cisplatin/VCR) | 5y-PFS & 5y-OS: 83% |
| Pizer [87] | 2006 | III | SIOP PNET 3 | 10 | Alternance (VCR/VP16/Carboplatin) and VCR/VP16/cyclophosphamide) + IR | 5y-PFS & 5y-OS: 71% |
| Jakacki [89] | 2015 | I/II | COG 99701 | 23 | IR + concomitant VCR and carboplatin + adjuvant (cyclophosphamide/VCR+/−cisplatin) | 5y-PFS: 62%; 5y-OS: 81% |
| Massimino [101] | 2006 | II | 3 | MTX/VCR/VP16/cyclophosphamide/carboplatin + IR + maintenance (VCR/CCNU) or HDC (thiotepa) + ASCR | CR at the end of trial: 3/3 | |
| Chintagumpala [91] | 2009 | II | 7 | +/−topotecan + IR + HDC (cisplatin/cyclophosphamide/VCR) + ASCR | 5y-PFS: 54%; 5y-OS: 67% | |
| Germ Cell Tumor | ||||||
| Authors | Date | Phase | Trial Name | Patients | Chemotherapy | Results |
| Calaminus [73] | 2013 | II | SIOP-CNS-GCT-96 | 65 G | carboplatin/VP16 alternating with ifosfamide/VP16 + IR | 5y-PFS: 88%; 5y-OS: 96% |
| Allen [102] | 1994 | II | 11 G | carboplatin + IR | 91% in remission for a median of 25 months | |
| Bartels [95] | 2021 | II | 137 G | carboplatin/VP16 + IR | 3y-PFS: 94% | |
| Kretschmar [103] | 2007 | II | POG9530 | 12 G and 14 NG | cisplatin/VP16 alternating with VCR/cyclophosphamide + IR | G: 66mo-PFS: 92%; NG: 58mo-PFS: 79% |
| Da Silva [104] | 2010 | II | 3rd international CNS GCT | 25 (G and NG) | carboplatin/VP16 alternating with cyclophosphamide/VP16 alone | 6y-PFS: 46%; 6y-OS: 75% |
| Fangusaro [77] | 2019 | II | ACNS1123 | 107 NG | carboplatin/VP16 alternating with ifosfamide/VP16 + IR | 3y-PFS: 88%; 3y-OS: 92% |
| Calaminus [78] | 2017 | II | SIOP-CNS-GCT-96 | 149 NG | cisplatin/VP16/ifosfamide + IR | localized: 5y-PFS: 72%; 5y-OS: 82% metastatic: 5y-PFS: 68%; 5y-OS: 75% |
| Goldman [76] | 2015 | II | ACNS0122 | 102 NG | carboplatin/VP16 alternating ifosfamide/VP16 + IR | 5y-PFS: 84%; 5y-OS: 93% |
7. The Future of Systemic Therapy
Author Contributions
Funding
Conflicts of Interest
References
- Goschzik, T.; Gessi, M.; Denkhaus, D.; Pietsch, T. PTEN Mutations and Activation of the PI3K/Akt/MTOR Signaling Pathway in Papillary Tumors of the Pineal Region. J. Neuropathol. Exp. Neurol. 2014, 73, 747–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheban, B.A.; Rosca, I.A.; Crisan, M. The Morphological and Functional Characteristics of the Pineal Gland. Med. Pharm. Rep. 2019, 92, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Kong, Z.; Wang, Y.; Dai, C.; Yao, Y.; Ma, W.; Wang, Y. Central Nervous System Germ Cell Tumors: A Review of the Literature. J. Child Neurol. 2018, 33, 610–620. [Google Scholar] [CrossRef] [PubMed]
- Palmisciano, P.; Ogasawara, C.; Nwagwu, C.D.; Bin Alamer, O.; Gupta, A.D.; Giantini-Larsen, A.M.; Scalia, G.; Yu, K.; Umana, G.E.; Cohen-Gadol, A.A.; et al. Metastases in the Pineal Region: A Systematic Review of Clinical Features, Management Strategies, and Survival Outcomes. World Neurosurg. 2022, 159, 156–167.e2. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Xiao, M.; Jiang, W.; Yue, C.; Qu, L.; Zhao, G. Paediatric Atypical Teratoid/Rhabdoid Tumour of the Pineal Region Mimicking a Meningioma: A Case Report and Literature Review. Br. J. Neurosurg. 2021, 16, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Baehring, J.; Vives, K.; Duncan, C.; Piepmeier, J.; Bannykyh, S. Tumors of the Posterior Third Ventricle and Pineal Region: Ependymoma and Germinoma. J. Neuro-Oncol. 2004, 70, 273–274. [Google Scholar] [CrossRef] [PubMed]
- Lu, V.M.; Luther, E.M.; Eichberg, D.G.; Morell, A.A.; Shah, A.H.; Komotar, R.J.; Ivan, M.E. Prognosticating Survival of Pineal Parenchymal Tumors of Intermediate Differentiation (PPTID) by Grade. J. Neuro-Oncol. 2021, 155, 165–172. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Dario, A.; Cerati, M.; Taborelli, M.; Finzi, G.; Pozzi, M.; Dorizzi, A. Cytogenetic and Ultrastructural Study of a Pineocytoma Case Report. J. Neuro-Oncol. 2000, 48, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Dahiya, S.; Perry, A. Pineal Tumors. Adv. Anat. Pathol. 2010, 17, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, W.; Lai, X.; Zhou, Y.; Zhou, X.; Li, J.; Liang, Y.; Zhu, X.; Ren, X.; Ding, Y.; et al. CD24 and PRAME Are Novel Grading and Prognostic Indicators for Pineal Parenchymal Tumors of Intermediate Differentiation. Am. J. Surg. Pathol. 2020, 44, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Bi, W.L.; Dubuc, A.M.; Martineau, L.; Ligon, A.H.; Berkowitz, A.L.; Aizer, A.A.; Lee, E.Q.; Ligon, K.L.; Ramkissoon, S.H.; et al. Integrated Genomic Characterization of a Pineal Parenchymal Tumor of Intermediate Differentiation. World Neurosurg. 2016, 85, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Mazor, T.; Lao, R.; Wan, E.; Diallo, A.B.; Hill, N.S.; Thangaraj, N.; Wendelsdorf, K.; Samuel, D.; Kline, C.N.; et al. Recurrent KBTBD4 Small In-Frame Insertions and Absence of DROSHA Deletion or DICER1 Mutation Differentiate Pineal Parenchymal Tumor of Intermediate Differentiation (PPTID) from Pineoblastoma. Acta Neuropathol. 2019, 137, 851–854. [Google Scholar] [CrossRef] [Green Version]
- Martínez, H.; Nagurney, M.; Wang, Z.-X.; Eberhart, C.G.; Heaphy, C.M.; Curtis, M.T.; Rodriguez, F.J. ATRX Mutations in Pineal Parenchymal Tumors of Intermediate Differentiation. J. Neuropathol. Exp. Neurol. 2019, 78, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.G.; Ngo, T.N.M.; Dunn, I.F. Incidence, Prognostic Factors, and Survival Trend in Pineal Gland Tumors: A Population-Based Analysis. Front. Oncol. 2021, 11, 780173. [Google Scholar] [CrossRef] [PubMed]
- Han, S.J.; Clark, A.J.; Ivan, M.E.; Parsa, A.T.; Perry, A. Pathology of Pineal Parenchymal Tumors. Neurosurg. Clin. N. Am. 2011, 22, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Fèvre-Montange, M.; Champier, J.; Szathmari, A.; Wierinckx, A.; Mottolese, C.; Guyotat, J.; Figarella-Branger, D.; Jouvet, A.; Lachuer, J. Microarray Analysis Reveals Differential Gene Expression Patterns in Tumors of the Pineal Region. J. Neuropathol. Exp. Neurol. 2006, 65, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fèvre-Montange, M.; Vasiljevic, A.; Champier, J.; Jouvet, A. Histopathology of Tumors of the Pineal Region. Futur. Oncol. 2010, 6, 791–809. [Google Scholar] [CrossRef]
- Gielen, G.H.; Gessi, M.; Denkhaus, D.; Pietsch, T. CRX/OTX3: A Useful Marker in the Differential Diagnosis of Tumors of the Pineal Region and Indicator of Photoreceptor Differentiation in Medulloblastomas and Atypical Teratoid Rhabdoid Tumors. Appl. Immunohistochem. Mol. Morphol. AIMM 2013, 21, 248–253. [Google Scholar] [CrossRef]
- Manila, A.; Mariangela, N.; Libero, L.; Francesca, G.; Romana, B.F.; Felice, G. Is CRX Protein a Useful Marker in Differential Diagnosis of Tumors of the Pineal Region? Pediatr. Dev. Pathol. 2014, 17, 85–88. [Google Scholar] [CrossRef]
- Pfaff, E.; Aichmüller, C.; Sill, M.; Stichel, D.; Snuderl, M.; Karajannis, M.A.; Schuhmann, M.U.; Schittenhelm, J.; Hasselblatt, M.; Thomas, C.; et al. Molecular Subgrouping of Primary Pineal Parenchymal Tumors Reveals Distinct Subtypes Correlated with Clinical Parameters and Genetic Alterations. Acta Neuropathol. 2020, 139, 243–257. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Li, B.K.; Pfaff, E.; Gudenas, B.; Vasiljevic, A.; Orr, B.A.; Dufour, C.; Snuderl, M.; Karajannis, M.A.; Rosenblum, M.K.; et al. Clinical and Molecular Heterogeneity of Pineal Parenchymal Tumors: A Consensus Study. Acta Neuropathol. 2021, 141, 771–785. [Google Scholar] [CrossRef]
- Li, B.K.; Vasiljevic, A.; Dufour, C.; Yao, F.; Ho, B.L.B.; Lu, M.; Hwang, E.I.; Gururangan, S.; Hansford, J.R.; Fouladi, M.; et al. Pineoblastoma Segregates into Molecular Sub-Groups with Distinct Clinico-Pathologic Features: A Rare Brain Tumor Consortium Registry Study. Acta Neuropathol. 2020, 139, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Kannan, K.; Pfaff, E.; Wang, S.; Stafford, J.M.; Serrano, J.; Heguy, A.; Ray, K.; Faustin, A.; Aminova, O.; et al. Recurrent Homozygous Deletion of DROSHA and Microduplication of PDE4DIP in Pineoblastoma. Nat. Commun. 2018, 9, 2868. [Google Scholar] [CrossRef] [Green Version]
- de Kock, L.; Sabbaghian, N.; Druker, H.; Weber, E.; Hamel, N.; Miller, S.; Choong, C.S.; Gottardo, N.G.; Kees, U.R.; Rednam, S.P.; et al. Germ-Line and Somatic DICER1 Mutations in Pineoblastoma. Acta Neuropathol. 2014, 128, 583–595. [Google Scholar] [CrossRef]
- Liu, A.P.Y.; Gudenas, B.; Lin, T.; Orr, B.A.; Klimo, P.; Kumar, R.; Bouffet, E.; Gururangan, S.; Crawford, J.R.; Kellie, S.J.; et al. Risk-Adapted Therapy and Biological Heterogeneity in Pineoblastoma: Integrated Clinico-Pathological Analysis from the Prospective, Multi-Center SJMB03 and SJYC07 Trials. Acta Neuropathol. 2020, 139, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, N.; Goda, N.; Yashige, K.; Kitagawa, M.; Yamazaki, T.; Nobusawa, S.; Yokoo, H. Desmoplastic Myxoid Tumor, SMARCB1-Mutant: A New Variant of SMARCB1-Deficient Tumor of the Central Nervous System Preferentially Arising in the Pineal Region. Virchows Arch. 2021, 479, 835–839. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Wefers, A.; Bens, S.; Nemes, K.; Agaimy, A.; Oyen, F.; Vogelgesang, S.; Rodriguez, F.J.; Brett, F.M.; McLendon, R.; et al. Desmoplastic Myxoid Tumor, SMARCB1-Mutant: Clinical, Histopathological and Molecular Characterization of a Pineal Region Tumor Encountered in Adolescents and Adults. Acta Neuropathol. 2020, 139, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Heim, S.; Sill, M.; Jones, D.T.W.; Vasiljevic, A.; Jouvet, A.; Fèvre-Montange, M.; Wesseling, P.; Beschorner, R.; Mittelbronn, M.; Kohlhof, P.; et al. Papillary Tumor of the Pineal Region: A Distinct Molecular Entity. Brain Pathol. 2016, 26, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Coy, S.; Dubuc, A.M.; Dahiya, S.; Ligon, K.L.; Vasiljevic, A.; Santagata, S. Nuclear CRX and FOXJ1 Expression Differentiates Non-Germ Cell Pineal Region Tumors and Supports the Ependymal Differentiation of Papillary Tumor of the Pineal Region. Am. J. Surg. Pathol. 2017, 41, 1410–1421. [Google Scholar] [CrossRef]
- Heim, S.; Beschorner, R.; Mittelbronn, M.; Keyvani, K.; Riemenschneider, M.J.; Vajtai, I.; Hartmann, C.; Acker, T.; Blümcke, I.; Paulus, W.; et al. Increased Mitotic and Proliferative Activity Are Associated with Worse Prognosis in Papillary Tumors of the Pineal Region. Am. J. Surg. Pathol. 2014, 38, 106–110. [Google Scholar] [CrossRef]
- Takami, H.; Fukuoka, K.; Fukushima, S.; Nakamura, T.; Mukasa, A.; Saito, N.; Yanagisawa, T.; Nakamura, H.; Sugiyama, K.; Kanamori, M.; et al. Integrated Clinical, Histopathological, and Molecular Data Analysis of 190 Central Nervous System Germ Cell Tumors from the IGCT Consortium. Neuro-Oncology 2019, 21, 1565–1577. [Google Scholar] [CrossRef]
- Smith, A.B.; Rushing, E.J.; Smirniotopoulos, J.G. From the Archives of the AFIP: Lesions of the Pineal Region: Radiologic-Pathologic Correlation. RadioGraphics 2010, 30, 2001–2020. [Google Scholar] [CrossRef]
- Fang, A.S.; Meyers, S.P. Magnetic Resonance Imaging of Pineal Region Tumours. Insights Imaging 2013, 4, 369–382. [Google Scholar] [CrossRef] [Green Version]
- Osborn, A.G.; Preece, M.T. Intracranial Cysts: Radiologic-Pathologic Correlation and Imaging Approach. Radiology 2006, 239, 650–664. [Google Scholar] [CrossRef]
- Moon, W.K.; Chang, K.H.; Han, M.H.; Kim, I.O. Intracranial Germinomas: Correlation of Imaging Findings with Tumor Response to Radiation Therapy. Am. J. Roentgenol. 1999, 172, 713–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruno, F.; Arrigoni, F.; Maggialetti, N.; Natella, R.; Reginelli, A.; Di Cesare, E.; Brunese, L.; Giovagnoni, A.; Masciocchi, C.; Splendiani, A.; et al. Neuroimaging in Emergency: A Review of Possible Role of Pineal Gland Disease. Gland Surg. 2019, 8, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Davaus, T.; Gasparetto, E.L.; Carvalho Neto, A.D.; Jung, J.E.; Bleggi-Torres, L.F. Pineal Yolk Sac Tumor: Correlation between Neuroimaging and Pathological Findings. Arq. Neuro-Psiquiatria 2007, 65, 283–285. [Google Scholar] [CrossRef]
- Liang, L.; Korogi, Y.; Sugahara, T.; Ikushima, I.; Shigematsu, Y.; Okuda, T.; Takahashi, M.; Kochi, M.; Ushio, Y. MRI of Intracranial Germ-Cell Tumours. Neuroradiology 2002, 44, 382–388. [Google Scholar] [CrossRef]
- Lv, X.-F.; Qiu, Y.-W.; Zhang, X.-L.; Han, L.-J.; Qiu, S.-J.; Xiong, W.; Wen, G.; Zhang, Y.-Z.; Zhang, J. Primary Intracranial Choriocarcinoma: MR Imaging Findings. Am. J. Neuroradiol. 2010, 31, 1994–1998. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, F. Available online: https://Radiopaedia.org (accessed on 26 July 2022).
- Clark, A.J.; Ivan, M.E.; Sughrue, M.E.; Yang, I.; Aranda, D.; Han, S.J.; Kane, A.J.; Parsa, A.T. Tumor Control after Surgery and Radiotherapy for Pineocytoma. J. Neurosurg. 2010, 113, 319–324. [Google Scholar] [CrossRef]
- Zaccagna, F.; Brown, F.S.; Allinson, K.S.J.; Devadass, A.; Kapadia, A.; Massoud, T.F.; Matys, T. In and around the Pineal Gland: A Neuroimaging Review. Clin. Radiol. 2022, 77, e107–e119. [Google Scholar] [CrossRef]
- Favero, G.; Bonomini, F.; Rezzani, R. Pineal Gland Tumors: A Review. Cancers 2021, 13, 1547. [Google Scholar] [CrossRef]
- Tate, M.; Sughrue, M.E.; Rutkowski, M.J.; Kane, A.J.; Aranda, D.; McClinton, L.; McClinton, L.; Barani, I.J.; Parsa, A.T. The Long-Term Postsurgical Prognosis of Patients with Pineoblastoma. Cancer 2012, 118, 173–179. [Google Scholar] [CrossRef]
- Mallick, S.; Benson, R.; Rath, G.K. Patterns of Care and Survival Outcomes in Patients with Pineal Parenchymal Tumor of Intermediate Differentiation: An Individual Patient Data Analysis. Radiother. Oncol. 2016, 121, 204–208. [Google Scholar] [CrossRef] [Green Version]
- Poulgrain, K.; Gurgo, R.; Winter, C.; Ong, B.; Lau, Q. Papillary Tumour of the Pineal Region. J. Clin. Neurosci. 2011, 18, 1007–1017. [Google Scholar] [CrossRef]
- Yamaki, V.N.; Solla, D.J.F.; Ribeiro, R.R.; da Silva, S.A.; Teixeira, M.J.; Figueiredo, E.G. Papillary Tumor of the Pineal Region: Systematic Review and Analysis of Prognostic Factors. Neurosurgery 2019, 85, E420–E429. [Google Scholar] [CrossRef]
- Bamberg, M.; Kortmann, R.D.; Calaminus, G.; Becker, G.; Meisner, C.; Harms, D.; Göbel, U. Radiation Therapy for Intracranial Germinoma: Results of the German Cooperative Prospective Trials MAKEI 83/86/89. J. Clin. Oncol. 1999, 17, 2585–2592. [Google Scholar] [CrossRef]
- Rogers, S.J.; Mosleh-Shirazi, M.A.; Saran, F.H. Radiotherapy of Localised Intracranial Germinoma: Time to Sever Historical Ties? Lancet Oncol. 2005, 6, 509–519. [Google Scholar] [CrossRef]
- Calaminus, G.; Bamberg, M.; Baranzelli, M.C.; Benoit, Y.; di Montezemolo, L.C.; Fossati-Bellani, F.; Jürgens, H.; Kühl, H.J.; Lenard, H.G.; Curto, M.L. Intracranial Germ Cell Tumors: A Comprehensive Update of the European Data. Neuropediatrics 1994, 25, 26–32. [Google Scholar] [CrossRef]
- Losa, F.; García del Muro, J.; Germà, J.R. Primary germ cell tumors of the central nervous system. Neurol. Barc. Spain 1997, 12, 249–254. [Google Scholar]
- Yu, L.; Orazmyradov, B.; Qi, S.; Song, Y.; Fang, L. Reinvestigation of the Origins of Pineal Meningiomas Based on Its Related Veins and Arachnoid Membranes. BMC Neurol. 2020, 20, 200. [Google Scholar] [CrossRef] [PubMed]
- Stowe, H.B.; Miller, C.R.; Wu, J.; Randazzo, D.M.; Ju, A.W. Pineal Region Glioblastoma, a Case Report and Literature Review. Front. Oncol. 2017, 7, 123. [Google Scholar] [CrossRef] [Green Version]
- Hirato, J.; Nakazato, Y. Pathology of Pineal Region Tumors. J. Neurooncol. 2001, 54, 239–249. [Google Scholar] [CrossRef]
- Lassman, A.B.; Bruce, J.N.; Fetell, M.R. Metastases to the Pineal Gland. Neurology 2006, 67, 1303–1304. [Google Scholar] [CrossRef]
- Newton, H.B. (Ed.) Handbook of Neuro-Oncology Neuroimaging; Academic Press: Cambridge, MA, USA, 2016; ISBN 978-0-12-800945-1. [Google Scholar]
- Hassani, F.D.; Bouchaouch, A.; El Fatemi, N.; Gana, R.; El Abbadi, N.; Maaqili, M.R. Pineal Epidermoid Cyst: Case Report and Review of the Literature. Pan Afr. Med. J. 2014, 18, 259. [Google Scholar] [CrossRef]
- Dawes, L.; Knipe, H. Intracranial Dermoid Cyst. Available online: https://radiopaedia.org/articles/intracranial-dermoid-cyst-1?lang=us (accessed on 26 July 2022). [CrossRef]
- Kim, D.-S.; Shim, K.-W.; Kim, T.-G.; Chang, J.-H.; Park, Y.-G.; Choi, J.-U. Pineal Cavernous Malformations: Report of Two Cases. Yonsei Med. J. 2005, 46, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Yamini, B.; Refai, D.; Rubin, C.M.; Frim, D.M. Initial Endoscopic Management of Pineal Region Tumors and Associated Hydrocephalus: Clinical Series and Literature Review. J. Neurosurg. 2004, 100, 437–441. [Google Scholar] [CrossRef]
- Morgenstern, P.F.; Souweidane, M.M. Pineal Region Tumors: Simultaneous Endoscopic Third Ventriculostomy and Tumor Biopsy. World Neurosurg. 2013, 79, S18.e9–S18.e13. [Google Scholar] [CrossRef]
- Balossier, A.; Blond, S.; Touzet, G.; Lefranc, M.; de Saint-Denis, T.; Maurage, C.-A.; Reyns, N. Endoscopic versus Stereotactic Procedure for Pineal Tumour Biopsies: Comparative Review of the Literature and Learning from a 25-Year Experience. Neurochirurgie. 2015, 61, 146–154. [Google Scholar] [CrossRef]
- Masina, R.; Ansaripour, A.; Beneš, V.; Berhouma, M.; Choque-Velasquez, J.; Eide, P.K.; Fedorko, S.; Fleck, S.; Hernesniemi, J.; Koziarski, A.; et al. Surgical Treatment of Symptomatic Pineal Cysts without Hydrocephalus-Meta-Analysis of the Published Literature. Acta Neurochir. 2022, 164, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Sonabend, A.M.; Bowden, S.; Bruce, J.N. Microsurgical Resection of Pineal Region Tumors. J. Neurooncol. 2016, 130, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Berhouma, M.; Ni, H.; Vallee, B. The Endoscopic Intraventricular Management of Pineal Cysts: A Minimally Invasive Modus Operandi. Acta Neurochir. 2013, 155, 1901–1905. [Google Scholar] [CrossRef] [PubMed]
- Berhouma, M.; Jemel, H.; Ksira, I.; Khaldi, M. Transcortical Approach to a Huge Pineal Mature Teratoma. Pediatr. Neurosurg. 2008, 44, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Little, K.M.; Friedman, A.H.; Fukushima, T. Surgical Approaches to Pineal Region Tumors. J. Neurooncol. 2001, 54, 287–299. [Google Scholar] [CrossRef]
- Shepard, M.J.; Haider, A.S.; Prabhu, S.S.; Sawaya, R.; DeMonte, F.; McCutcheon, I.E.; Weinberg, J.S.; Ferguson, S.D.; Suki, D.; Fuller, G.N.; et al. Long Term Outcomes Following Surgery for Pineal Region Tumors. J. Neurooncol. 2022, 156, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Scaringi, C.; Agolli, L.; Minniti, G. Technical Advances in Radiation Therapy for Brain Tumors. Anticancer Res. 2018, 38, 6041–6045. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; DeWees, T.; Shinohara, E.T.; Perkins, S.M. Long-Term Outcomes and Late Effects for Childhood and Young Adulthood Intracranial Germinomas. Neuro-Oncology 2015, 17, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alemany, M.; Velasco, R.; Simó, M.; Bruna, J. Late Effects of Cancer Treatment: Consequences for Long-Term Brain Cancer Survivors. Neuro-Oncol. Pract. 2021, 8, 18–30. [Google Scholar] [CrossRef]
- Calaminus, G.; Kortmann, R.; Worch, J.; Nicholson, J.C.; Alapetite, C.; Garrè, M.L.; Patte, C.; Ricardi, U.; Saran, F.; Frappaz, D. SIOP CNS GCT 96: Final Report of Outcome of a Prospective, Multinational Nonrandomized Trial for Children and Adults with Intracranial Germinoma, Comparing Craniospinal Irradiation Alone with Chemotherapy Followed by Focal Primary Site Irradiation for Patients with Localized Disease. Neuro-Oncology 2013, 15, 788–796. [Google Scholar] [CrossRef]
- Bartels, U.; Fangusaro, J.; Shaw, D.; Bhatia, A.; Omar-Thomas, A.; Wu, S.; MacDonald, S.; Murphy, E.; Souweidane, M.; Fouladi, M.; et al. GCT-41. Response-based radiation therapy in patients with newly diagnosed central nervous system localized germinoma: A children’s oncology group (cog) prospective phase 2 clinical trial. Neuro-Oncology 2020, 22, iii336. [Google Scholar] [CrossRef]
- Cheng, S.; Kilday, J.-P.; Laperriere, N.; Janzen, L.; Drake, J.; Bouffet, E.; Bartels, U. Outcomes of Children with Central Nervous System Germinoma Treated with Multi-Agent Chemotherapy Followed by Reduced Radiation. J. Neurooncol. 2016, 127, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.; Bouffet, E.; Fisher, P.G.; Allen, J.C.; Robertson, P.L.; Chuba, P.J.; Donahue, B.; Kretschmar, C.S.; Zhou, T.; Buxton, A.B.; et al. Phase II Trial Assessing the Ability of Neoadjuvant Chemotherapy With or Without Second-Look Surgery to Eliminate Measurable Disease for Nongerminomatous Germ Cell Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2015, 33, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Fangusaro, J.; Wu, S.; MacDonald, S.; Murphy, E.; Shaw, D.; Bartels, U.; Khatua, S.; Souweidane, M.; Lu, H.-M.; Morris, D.; et al. Phase II Trial of Response-Based Radiation Therapy for Patients With Localized CNS Nongerminomatous Germ Cell Tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2019, 37, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Calaminus, G.; Frappaz, D.; Kortmann, R.D.; Krefeld, B.; Saran, F.; Pietsch, T.; Vasiljevic, A.; Garre, M.L.; Ricardi, U.; Mann, J.R.; et al. Outcome of Patients with Intracranial Non-Germinomatous Germ Cell Tumors-Lessons from the SIOP-CNS-GCT-96 Trial. Neuro-Oncology 2017, 19, 1661–1672. [Google Scholar] [CrossRef] [Green Version]
- Tamrazi, B.; Nelson, M.; Blüml, S. Pineal Region Masses in Pediatric Patients. Neuroimaging Clin. N. Am. 2017, 27, 85–97. [Google Scholar] [CrossRef]
- Hinkes, B.G.; von Hoff, K.; Deinlein, F.; Warmuth-Metz, M.; Soerensen, N.; Timmermann, B.; Mittler, U.; Urban, C.; Bode, U.; Pietsch, T.; et al. Childhood Pineoblastoma: Experiences from the Prospective Multicenter Trials HIT-SKK87, HIT-SKK92 and HIT91. J. Neurooncol. 2007, 81, 217–223. [Google Scholar] [CrossRef]
- Mynarek, M.; Pizer, B.; Dufour, C.; van Vuurden, D.; Garami, M.; Massimino, M.; Fangusaro, J.; Davidson, T.; Gil-da-Costa, M.J.; Sterba, J.; et al. Evaluation of Age-Dependent Treatment Strategies for Children and Young Adults with Pineoblastoma: Analysis of Pooled European Society for Paediatric Oncology (SIOP-E) and US Head Start Data. Neuro-Oncology 2017, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- de Rojas, T.; Bautista, F.; Flores, M.; Igual, L.; Rubio, R.; Bardón, E.; Navarro, L.; Murillo, L.; Hladun, R.; Cañete, A.; et al. Management and Outcome of Children and Adolescents with Non-Medulloblastoma CNS Embryonal Tumors in Spain: Room for Improvement in Standards of Care. J. Neurooncol. 2018, 137, 205–213. [Google Scholar] [CrossRef]
- Jing, Y.; Deng, W.; Zhang, H.; Jiang, Y.; Dong, Z.; Fan, F.; Sun, P. Development and Validation of a Prognostic Nomogram to Predict Cancer-Specific Survival in Adult Patients With Pineoblastoma. Front. Oncol. 2020, 10, 1021. [Google Scholar] [CrossRef]
- Murray, M.J.; Bailey, S.; Heinemann, K.; Mann, J.; Göbel, U.K.; Saran, F.; Hale, J.P.; Calaminus, G.; Nicholson, J.C. Treatment and Outcomes of UK and German Patients with Relapsed Intracranial Germ Cell Tumors Following Uniform First-Line Therapy. Int. J. Cancer 2017, 141, 621–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callec, L.; Lardy-Cleaud, A.; Guerrini-Rousseau, L.; Alapetite, C.; Vignon, L.; Chastagner, P.; Frappaz, D.; Faure-Conter, C. Relapsing Intracranial Germ Cell Tumours Warrant Retreatment. Eur. J. Cancer 2020, 136, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.H.; Zeltzer, P.M.; Boyett, J.M.; Geyer, J.R.; Allen, J.C.; Finlay, J.L.; McGuire-Cullen, P.; Milstein, J.M.; Rorke, L.B.; Stanley, P. Prognostic Factors and Treatment Results for Supratentorial Primitive Neuroectodermal Tumors in Children Using Radiation and Chemotherapy: A Childrens Cancer Group Randomized Trial. J. Clin. Oncol. 1995, 13, 1687–1696. [Google Scholar] [CrossRef]
- Pizer, B.L.; Weston, C.L.; Robinson, K.J.; Ellison, D.W.; Ironside, J.; Saran, F.; Lashford, L.S.; Tait, D.; Lucraft, H.; Walker, D.A.; et al. Analysis of Patients with Supratentorial Primitive Neuro-Ectodermal Tumours Entered into the SIOP/UKCCSG PNET 3 Study. Eur. J. Cancer 2006, 42, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Gerber, N.U.; von Hoff, K.; Resch, A.; Ottensmeier, H.; Kwiecien, R.; Faldum, A.; Matuschek, C.; Hornung, D.; Bremer, M.; Benesch, M.; et al. Treatment of Children with Central Nervous System Primitive Neuroectodermal Tumors/Pinealoblastomas in the Prospective Multicentric Trial HIT 2000 Using Hyperfractionated Radiation Therapy Followed by Maintenance Chemotherapy. Int. J. Radiat. Oncol. 2014, 89, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Jakacki, R.I.; Burger, P.C.; Kocak, M.; Boyett, J.M.; Goldwein, J.; Mehta, M.; Packer, R.J.; Tarbell, N.J.; Pollack, I.F. Outcome and Prognostic Factors for Children with Supratentorial Primitive Neuroectodermal Tumors Treated with Carboplatin during Radiotherapy: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 776–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gururangan, S.; McLaughlin, C.; Quinn, J.; Rich, J.; Reardon, D.; Halperin, E.C.; Herndon, J.; Fuchs, H.; George, T.; Provenzale, J.; et al. High-Dose Chemotherapy with Autologous Stem-Cell Rescue in Children and Adults with Newly Diagnosed Pineoblastomas. J. Clin. Oncol. 2003, 21, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Chintagumpala, M.; Hassall, T.; Palmer, S.; Ashley, D.; Wallace, D.; Kasow, K.; Merchant, T.E.; Krasin, M.J.; Dauser, R.; Boop, F.; et al. A Pilot Study of Risk-Adapted Radiotherapy and Chemotherapy in Patients with Supratentorial PNET. Neuro-Oncology 2009, 11, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.-S.; Hale, G.A.; Gronewold, L.; Tong, X.; Laningham, F.H.; Gilger, E.A.; Srivastava, D.K.; Kun, L.E.; Gajjar, A.; Fouladi, M. High-Dose Chemotherapy with Autologous Stem Cell Rescue for Children with Recurrent Malignant Brain Tumors. Cancer 2008, 112, 1345–1353. [Google Scholar] [CrossRef]
- Graham, M.L.; Herndon, J.E.; Casey, J.R.; Chaffee, S.; Ciocci, G.H.; Krischer, J.P.; Kurtzberg, J.; Laughlin, M.J.; Longee, D.C.; Olson, J.F.; et al. High-Dose Chemotherapy with Autologous Stem-Cell Rescue in Patients with Recurrent and High-Risk Pediatric Brain Tumors. J. Clin. Oncol. 1997, 15, 1814–1823. [Google Scholar] [CrossRef]
- Broniscer, A.; Nicolaides, T.P.; Dunkel, I.J.; Gardner, S.L.; Johnson, J.; Allen, J.C.; Sposto, R.; Finlay, J.L. High-Dose Chemotherapy with Autologous Stem-Cell Rescue in the Treatment of Patients with Recurrent Non-Cerebellar Primitive Neuroectodermal Tumors. Pediatr. Blood Cancer 2004, 42, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Bartels, U.; Onar-Thomas, A.; Patel, S.K.; Shaw, D.; Fangusaro, J.; Dhall, G.; Souweidane, M.; Bhatia, A.; Embry, L.; Trask, C.L.; et al. Phase II Trial of Response-Based Radiation Therapy for Patients with Localized Germinoma: A Children’s Oncology Group Study. Neuro-Oncology 2021, 24, 974–983. [Google Scholar] [CrossRef]
- Modak, S.; Gardner, S.; Dunkel, I.J.; Balmaceda, C.; Rosenblum, M.K.; Miller, D.C.; Halpern, S.; Finlay, J.L. Thiotepa-Based High-Dose Chemotherapy with Autologous Stem-Cell Rescue in Patients with Recurrent or Progressive CNS Germ Cell Tumors. J. Clin. Oncol. 2004, 22, 1934–1943. [Google Scholar] [CrossRef]
- Baek, H.J.; Park, H.J.; Sung, K.W.; Lee, S.H.; Han, J.W.; Koh, K.N.; Im, H.J.; Kang, H.J.; Park, K.D. Myeloablative Chemotherapy and Autologous Stem Cell Transplantation in Patients with Relapsed or Progressed Central Nervous System Germ Cell Tumors: Results of Korean Society of Pediatric Neuro-Oncology (KSPNO) S-053 Study. J. Neurooncol. 2013, 114, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Kubota, H.; Umeda, K.; Kagehiro, K.; Tanaka, K.; Daifu, T.; Hamabata, T.; Nodomi, S.; Kato, I.; Hiramatsu, H.; Arakawa, Y.; et al. High-Dose Chemotherapy with Autologous Stem Cell Transplantation Spares Re-Irradiation for Recurrent Intracranial Germinoma. Pediatr. Blood Cancer 2018, 65, e27104. [Google Scholar] [CrossRef]
- Finlay, J.L.; Goldman, S.; Wong, M.C.; Cairo, M.; Garvin, J.; August, C.; Cohen, B.H.; Stanley, P.; Zimmerman, R.A.; Bostrom, B.; et al. Pilot Study of High-Dose Thiotepa and Etoposide with Autologous Bone Marrow Rescue in Children and Young Adults with Recurrent CNS Tumors. The Children’s Cancer Group. J. Clin. Oncol. 1996, 14, 2495–2503. [Google Scholar] [CrossRef]
- Perez-Somarriba, M.; Moreno-Tejero, M.L.; Rozas, M.I.; Pelaez, I.; Madero, L.; Lassaletta, A. Gemcitabine, Paclitaxel, and Oxaliplatin (GEMPOX) in the Treatment of Relapsed/Refractory Intracranial Nongerminomatous Germ Cell Tumors. Pediatr. Blood Cancer 2020, 67, e28089. [Google Scholar] [CrossRef] [PubMed]
- Massimino, M.; Gandola, L.; Spreafico, F.; Luksch, R.; Collini, P.; Giangaspero, F.; Simonetti, F.; Casanova, M.; Cefalo, G.; Pignoli, E.; et al. Supratentorial Primitive Neuroectodermal Tumors (S-PNET) in Children: A Prospective Experience with Adjuvant Intensive Chemotherapy and Hyperfractionated Accelerated Radiotherapy. Int. J. Radiat. Oncol. 2006, 64, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.C.; DaRosso, R.C.; Donahue, B.; Nirenberg, A. A Phase II Trial of Preirradiation Carboplatin in Newly Diagnosed Germinoma of the Central Nervous System. Cancer 1994, 74, 940–944. [Google Scholar] [CrossRef]
- Kretschmar, C.; Kleinberg, L.; Greenberg, M.; Burger, P.; Holmes, E.; Wharam, M. Pre-Radiation Chemotherapy with Response-Based Radiation Therapy in Children with Central Nervous System Germ Cell Tumors: A Report from the Children’s Oncology Group. Pediatr. Blood Cancer 2007, 48, 285–291. [Google Scholar] [CrossRef] [Green Version]
- da Silva, N.S.; Cappellano, A.M.; Diez, B.; Cavalheiro, S.; Gardner, S.; Wisoff, J.; Kellie, S.; Parker, R.; Garvin, J.; Finlay, J. Primary Chemotherapy for Intracranial Germ Cell Tumors: Results of the Third International CNS Germ Cell Tumor Study. Pediatr. Blood Cancer 2010, 54, 377–383. [Google Scholar] [CrossRef] [PubMed]

| Histotypes | WHO Grade | Frequent Age at Diagnosis | Mitotic Count (mitosis/10HPF) | Ki67/Mib1 LI (%) | Most Frequent Molecular Alterations |
|---|---|---|---|---|---|
| PC | 1 | adult | <0–1 | 1–2 | n.s. |
| PPTID | 2 | adult | <5 | 6–10 | KBTBD4 (insertion); ATRX loss of function |
| 3 | 5 | 10–20 | |||
| PB (subtypes) | |||||
| PB-miRNA1 | 4 | older children or young adult | High mitotic count | 20–25% and up to 50 or more | DICER1, DROSHA, DGCR8 (loss of function) |
| PB-miRNA2 | DICER1, DROSHA (loss of function) | ||||
| PB-MYC/FOXR2 | Infant or young children | FOXR2 overexpression; MYC activation | |||
| PB-RB1 | RB1 loss of function | ||||
| DMT-SMARCB1-mut | TBD | young adult | <0–1 | 3 | SMARCB1 loss of function |
| PTPR | 2 | young adult | <2–3 | 2–3 | PTEN mutation; PI3K alteration |
| 3 | ≥3 | ≥10 | |||
| Histotype | Radiological Characteristics | PFS | OS |
|---|---|---|---|
| Pineal parenchymal tumors | |||
| Pineocytoma |
| 5ys PFS 97% [42] | 5 ys OS > 85% [42,43,44] |
| Pineoblastoma |
| - | 5ys OS < 60% [43,44,45] |
| Pineal parenchymal tumors of intermediate differentiation |
| 5ys PFS 74.1% [12] | 5 ys OS 84.1% [46] |
| Papillary tumors |
| PFS 5ys 34.5% [47] | 5ys OS < 75% [44,48] |
| Germ cell tumors | |||
| Germinoma |
| 5ys PFS 91% [49] | 10ys OS 90% [50] 5ys OS > 90% [43,44,49] |
| Teratoma |
| 5 ys PFS 20–45% [51] | 5ys OS 30–100% [52] 5ys OS for immature teratoma: 30–70% [43] 5ys OS for mature teratoma: 90–100% [43] |
| Pineal yolk sac tumors |
| - | 5ys OS 60–70% [43] |
| Embryonal carcinoma |
| - | 5ys OS 60–70% [43] |
| Pineal choriocarcinoma |
| - | 5ys OS 45–70% [43] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardi, G.; Poliani, P.L.; Manara, R.; Berhouma, M.; Minniti, G.; Tabouret, E.; Razis, E.; Cerretti, G.; Zagonel, V.; Weller, M.; et al. Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview. Cancers 2022, 14, 3646. https://doi.org/10.3390/cancers14153646
Lombardi G, Poliani PL, Manara R, Berhouma M, Minniti G, Tabouret E, Razis E, Cerretti G, Zagonel V, Weller M, et al. Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview. Cancers. 2022; 14(15):3646. https://doi.org/10.3390/cancers14153646
Chicago/Turabian StyleLombardi, Giuseppe, Pietro Luigi Poliani, Renzo Manara, Moncef Berhouma, Giuseppe Minniti, Emeline Tabouret, Evangelia Razis, Giulia Cerretti, Vittorina Zagonel, Michael Weller, and et al. 2022. "Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview" Cancers 14, no. 15: 3646. https://doi.org/10.3390/cancers14153646
APA StyleLombardi, G., Poliani, P. L., Manara, R., Berhouma, M., Minniti, G., Tabouret, E., Razis, E., Cerretti, G., Zagonel, V., Weller, M., & Idbaih, A. (2022). Diagnosis and Treatment of Pineal Region Tumors in Adults: A EURACAN Overview. Cancers, 14(15), 3646. https://doi.org/10.3390/cancers14153646