
Brassinin Promotes the Degradation of Tie2 and FGFR1 in Endothelial Cells and Inhibits Triple-Negative Breast Cancer Angiogenesis

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Chemicals
2.3. Cell Culture
2.4. Water-Soluble Tetrazolium (WST)-1 Assay
2.5. Lactate Dehydrogenase (LDH) Assay
2.6. Bromodeoxyuridine (BrdU) Incorporation Assay
2.7. Transwell Migration Assay
2.8. Tube Formation Assay
2.9. Spheroid Sprouting Assay
2.10. Aortic Ring Assay
2.11. Animal Models
2.12. Histology and Immunohistochemistry
2.13. Western Blotting
2.14. Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.15. Statistics
3. Results
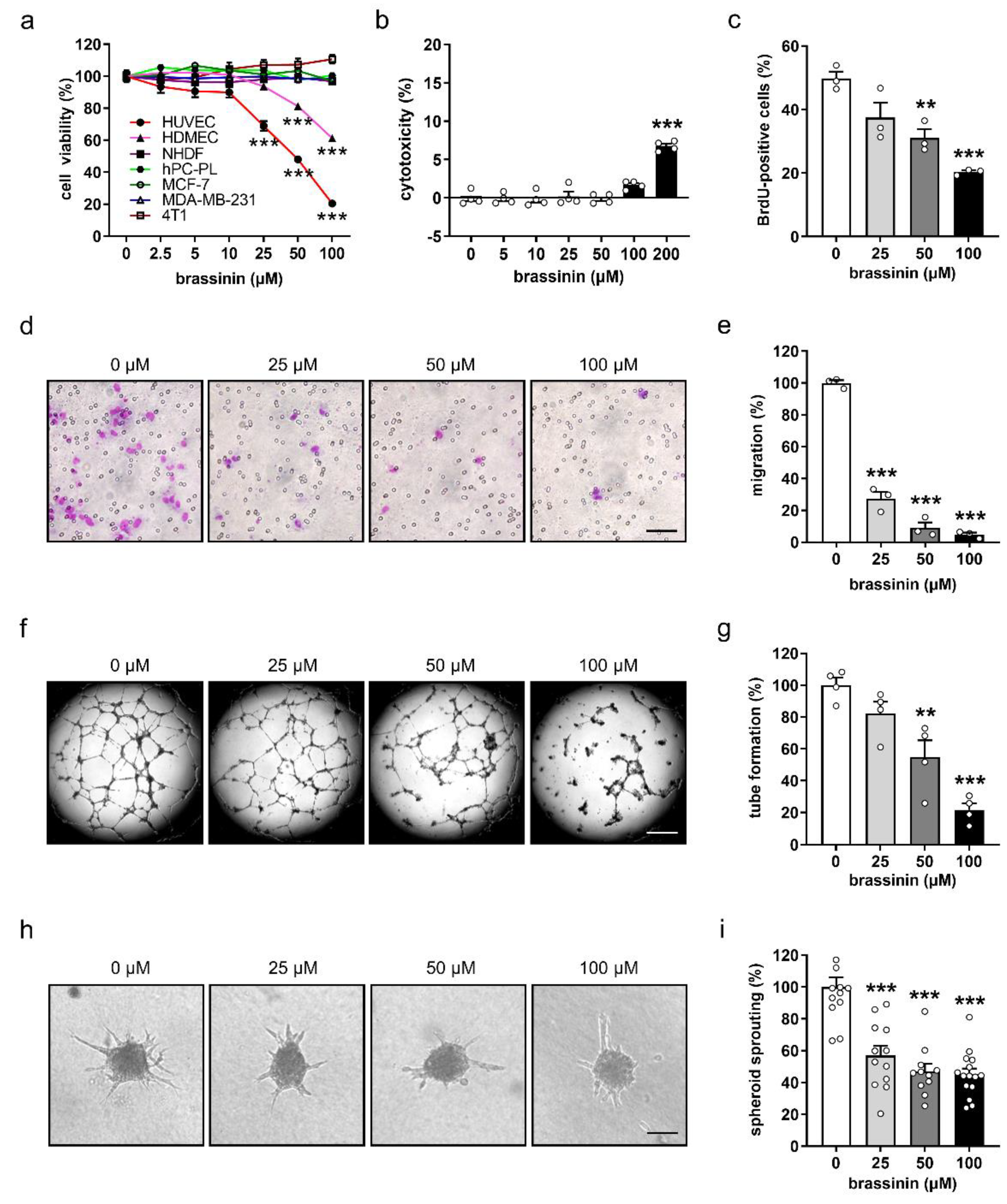
3.1. Brassinin Preferentially Targets ECs
3.2. Brassinin Suppresses the Angiogenic Activity of ECs
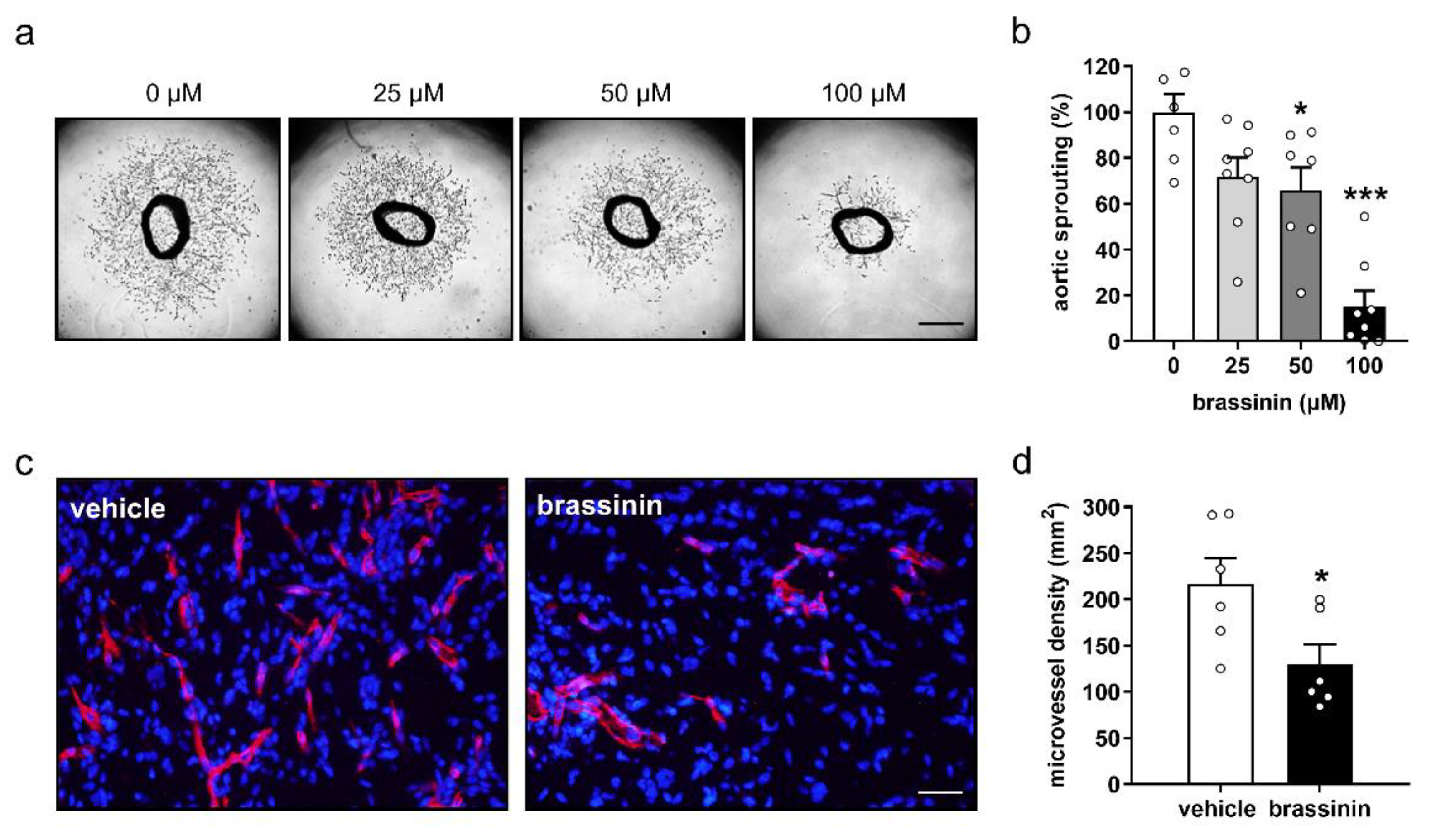
3.3. Brassinin Inhibits Angiogenesis Ex Vivo and In Vivo
3.4. Brassinin Inhibits Tumor Growth and Angiogenesis
3.5. Brassinin Down-Regulates Key Angiogenic Signaling Pathways
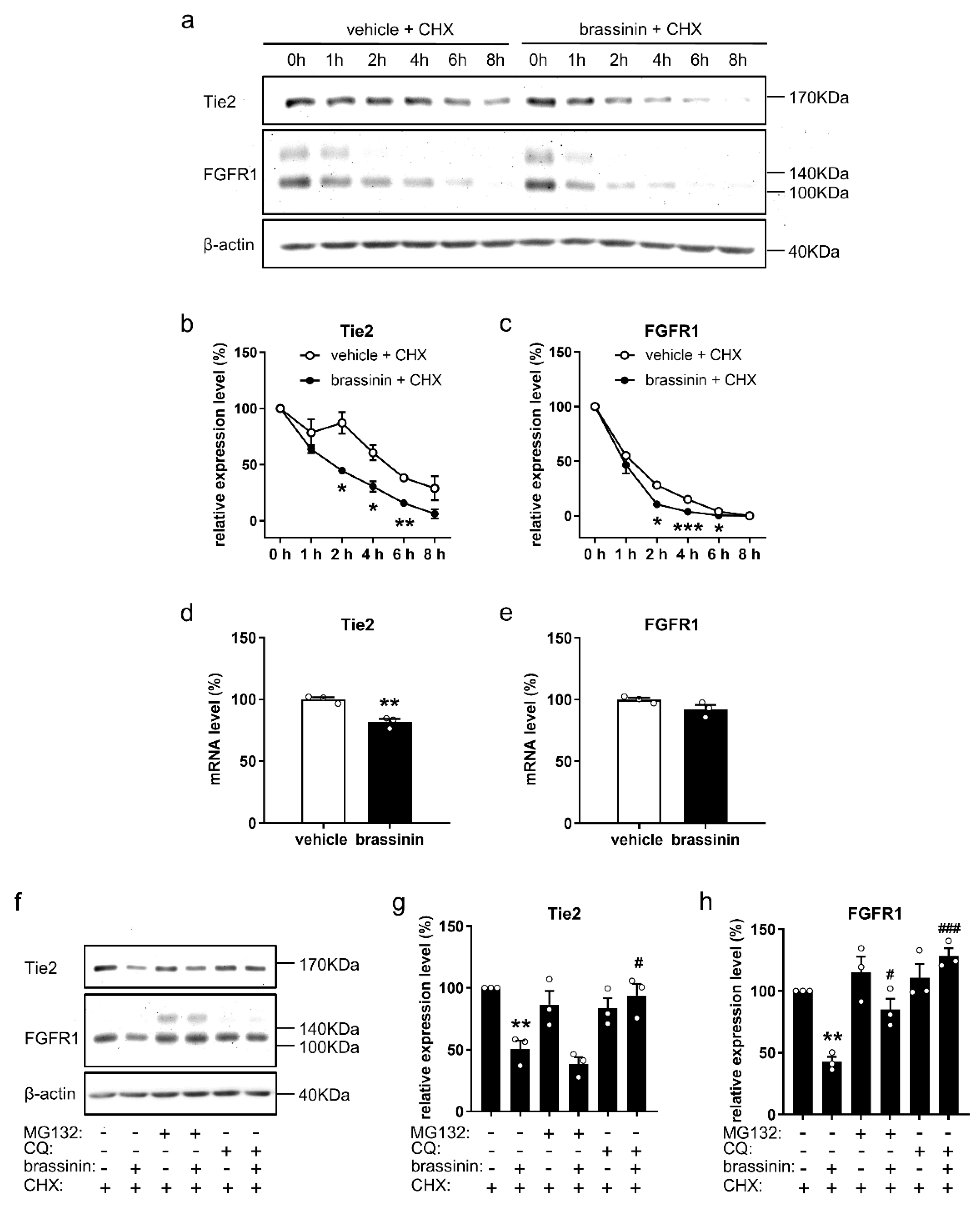
3.6. Brassinin Promotes Tie2 and FGFR1 Degradation
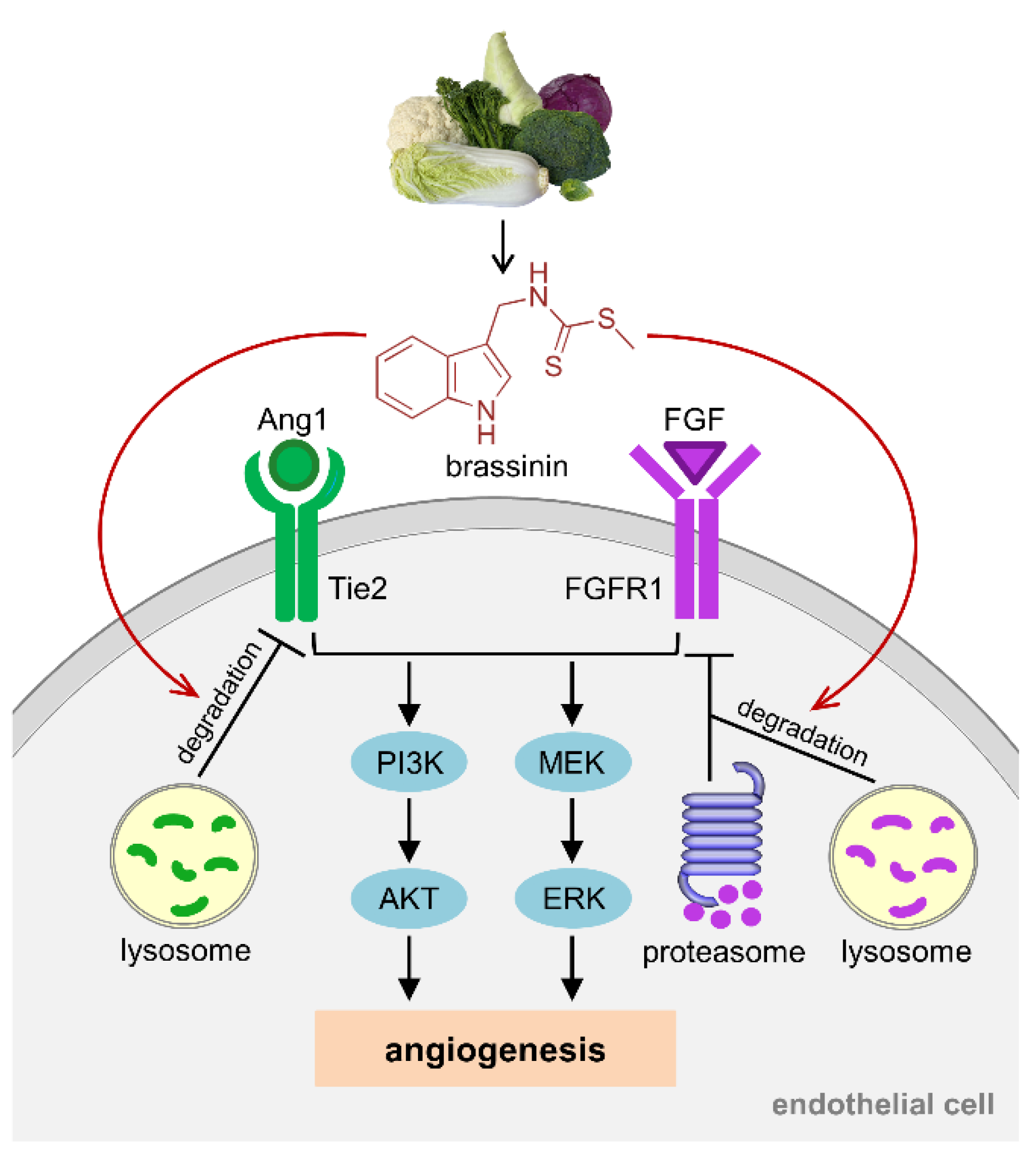
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pedras, M.S.; Okanga, F.I.; Zaharia, I.L.; Khan, A.Q. Phytoalexins from crucifers: Synthesis, biosynthesis, and biotransformation. Phytochemistry 2000, 53, 161–176. [Google Scholar] [CrossRef]
- Kello, M.; Drutovic, D.; Chripkova, M.; Pilatova, M.; Budovska, M.; Kulikova, L.; Urdzik, P.; Mojzis, J. ROS-dependent antiproliferative effect of brassinin derivative homobrassinin in human colorectal cancer Caco2 cells. Molecules 2014, 19, 10877–10897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izutani, Y.; Yogosawa, S.; Sowa, Y.; Sakai, T. Brassinin induces G1 phase arrest through increase of p21 and p27 by inhibition of the phosphatidylinositol 3-kinase signaling pathway in human colon cancer cells. Int. J. Oncol. 2012, 40, 816–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, T.; Ham, J.; Song, J.; Song, G.; Lim, W. Brassinin Inhibits Proliferation in Human Liver Cancer Cells via Mitochondrial Dysfunction. Cells 2021, 10, 332. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Sethi, G.; Ahn, K.S. Brassinin inhibits STAT3 signaling pathway through modulation of PIAS-3 and SOCS-3 expression and sensitizes human lung cancer xenograft in nude mice to paclitaxel. Oncotarget 2015, 6, 6386–6405. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, T.; Duhadaway, J.B.; Gaspari, P.; Sutanto-Ward, E.; Munn, D.H.; Mellor, A.L.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. A key in vivo antitumor mechanism of action of natural product-based brassinins is inhibition of indoleamine 2,3-dioxygenase. Oncogene 2008, 27, 2851–2857. [Google Scholar] [CrossRef] [Green Version]
- Bielenberg, D.R.; Zetter, B.R. The Contribution of Angiogenesis to the Process of Metastasis. Cancer J. 2015, 21, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Hoeben, A.; Landuyt, B.; Highley, M.S.; Wildiers, H.; Van Oosterom, A.T.; De Bruijn, E.A. Vascular endothelial growth factor and angiogenesis. Pharmacol. Rev. 2004, 56, 549–580. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Haibe, Y.; Kreidieh, M.; El Hajj, H.; Khalifeh, I.; Mukherji, D.; Temraz, S.; Shamseddine, A. Resistance Mechanisms to Anti-angiogenic Therapies in Cancer. Front. Oncol. 2020, 10, 221. [Google Scholar] [CrossRef] [Green Version]
- Elice, F.; Rodeghiero, F. Side effects of anti-angiogenic drugs. Thromb. Res. 2012, 129, S50–S53. [Google Scholar] [CrossRef]
- Zhao, S.; Zuo, W.J.; Shao, Z.M.; Jiang, Y.Z. Molecular subtypes and precision treatment of triple-negative breast cancer. Ann. Transl. Med. 2020, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Schneeweiss, A.; Denkert, C.; Fasching, P.A.; Fremd, C.; Gluz, O.; Kolberg-Liedtke, C.; Loibl, S.; Luck, H.J. Diagnosis and Therapy of Triple-Negative Breast Cancer (TNBC)—Recommendations for Daily Routine Practice. Geburtshilfe Frauenheilkd 2019, 79, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, V.; Hui, X.; Nalbach, L.; Ampofo, E.; Lipp, P.; Menger, M.D.; Laschke, M.W.; Gu, Y. Linalool inhibits the angiogenic activity of endothelial cells by downregulating intracellular ATP levels and activating TRPM8. Angiogenesis 2021, 24, 613–630. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Ampofo, E.; Menger, M.D.; Laschke, M.W. miR-191 suppresses angiogenesis by activation of NF-kappaB signaling. FASEB J. 2017, 31, 3321–3333. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Becker, V.; Zhao, Y.; Menger, M.D.; Laschke, M.W. miR-370 inhibits the angiogenic activity of endothelial cells by targeting smoothened (SMO) and bone morphogenetic protein (BMP)-2. FASEB J. 2019, 33, 7213–7224. [Google Scholar] [CrossRef]
- Gu, Y.; Scheuer, C.; Feng, D.; Menger, M.D.; Laschke, M.W. Inhibition of angiogenesis: A novel antitumor mechanism of the herbal compound arctigenin. Anticancer Drugs 2013, 24, 781–791. [Google Scholar] [CrossRef]
- Baker, M.; Wayland, H. On-line volume flow rate and velocity profile measurement for blood in microvessels. Microvasc. Res. 1974, 7, 131–143. [Google Scholar] [CrossRef]
- Riva, C.E.; Grunwald, J.E.; Sinclair, S.H.; Petrig, B.L. Blood velocity and volumetric flow rate in human retinal vessels. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1124–1132. [Google Scholar]
- Gu, Y.; Körbel, C.; Scheuer, C.; Nenicu, A.; Menger, M.D.; Laschke, M.W. Tubeimoside-1 suppresses tumor angiogenesis by stimulation of proteasomal VEGFR2 and Tie2 degradation in a non-small cell lung cancer xenograft model. Oncotarget 2016, 7, 5258–5272. [Google Scholar] [CrossRef] [Green Version]
- Labani-Motlagh, A.; Ashja-Mahdavi, M.; Loskog, A. The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 2020, 11, 940. [Google Scholar] [CrossRef] [PubMed]
- Pietras, K.; Ostman, A. Hallmarks of cancer: Interactions with the tumor stroma. Exp. Cell Res. 2010, 316, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Park, J.H.; Kim, K.D.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H.; Ahn, K.S. Brassinin induces apoptosis in PC-3 human prostate cancer cells through the suppression of PI3K/Akt/mTOR/S6K1 signaling cascades. Phytother. Res. 2014, 28, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Lee, J.H.; Ko, J.H.; Jung, S.H.; Sethi, G.; Ahn, K.S. Brassinin Represses Invasive Potential of Lung Carcinoma Cells through Deactivation of PI3K/Akt/mTOR Signaling Cascade. Molecules 2019, 24, 1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosaka, K.; Yang, Y.; Seki, T.; Fischer, C.; Dubey, O.; Fredlund, E.; Hartman, J.; Religa, P.; Morikawa, H.; Ishii, Y.; et al. Pericyte-fibroblast transition promotes tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, E5618–E5627. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, A.L.; Okamoto, O.K. Combined effects of pericytes in the tumor microenvironment. Stem. Cells Int. 2015, 2015, 868475. [Google Scholar] [CrossRef] [Green Version]
- Navarro, R.; Compte, M.; Alvarez-Vallina, L.; Sanz, L. Immune Regulation by Pericytes: Modulating Innate and Adaptive Immunity. Front. Immunol. 2016, 7, 480. [Google Scholar] [CrossRef] [Green Version]
- Truffi, M.; Sorrentino, L.; Corsi, F. Fibroblasts in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1234, 15–29. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Laschke, M.W.; Menger, M.D. The dorsal skinfold chamber: A versatile tool for preclinical research in tissue engineering and regenerative medicine. Eur. Cell Mater. 2016, 32, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Schrors, B.; Boegel, S.; Albrecht, C.; Bukur, T.; Bukur, V.; Holtstrater, C.; Ritzel, C.; Manninen, K.; Tadmor, A.D.; Vormehr, M.; et al. Multi-Omics Characterization of the 4T1 Murine Mammary Gland Tumor Model. Front. Oncol. 2020, 10, 1195. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Liu, D.; Fueyo, J.; Gomez-Manzano, C. Tie2: A journey from normal angiogenesis to cancer and beyond. Histol. Histopathol. 2008, 23, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Polverini, P.; Dewhirst, M.; Shan, S.; Rao, P.S.; Peters, K. Inhibition of tumor angiogenesis using a soluble receptor establishes a role for Tie2 in pathologic vascular growth. J. Clin. Investig. 1997, 100, 2072–2078. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.; Buxton, J.A.; Acheson, A.; Radziejewski, C.; Maisonpierre, P.C.; Yancopoulos, G.D.; Channon, K.M.; Hale, L.P.; Dewhirst, M.W.; George, S.E.; et al. Antiangiogenic gene therapy targeting the endothelium-specific receptor tyrosine kinase Tie2. Proc. Natl. Acad. Sci. USA 1998, 95, 8829–8834. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Sugimachi, K.; Yamashita Yi, Y.; Ohga, T.; Shirabe, K.; Shimada, M.; Wands, J.R.; Sugimachi, K. Tie2 vascular endothelial receptor expression and function in hepatocellular carcinoma. Hepatology 2002, 35, 861–867. [Google Scholar] [CrossRef]
- Siemeister, G.; Schirner, M.; Weindel, K.; Reusch, P.; Menrad, A.; Marme, D.; Martiny-Baron, G. Two independent mechanisms essential for tumor angiogenesis: Inhibition of human melanoma xenograft growth by interfering with either the vascular endothelial growth factor receptor pathway or the Tie-2 pathway. Cancer Res. 1999, 59, 3185–3191. [Google Scholar]
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Martinez-Ledesma, E.; Zhang, C.; Gao, F.; Zheng, S.; Ding, J.; Wu, S.; Nguyen, N.; Clifford, S.C.; Wen, P.Y.; et al. Tie2-FGFR1 Interaction Induces Adaptive PI3K Inhibitor Resistance by Upregulating Aurora A/PLK1/CDK1 Signaling in Glioblastoma. Cancer Res. 2019, 79, 5088–5101. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Wang, X.; Liu, Y.; Singhal, M.; Gürkaşlar, C.; Valls, A.F.; Lei, Y.; Hu, W.; Schermann, G.; Adler, H.; et al. STAT3-YAP/TAZ signaling in endothelial cells promotes tumor angiogenesis. Sci. Signal. 2021, 14, eabj8393. [Google Scholar] [CrossRef]
- He, J.; Wang, L.; Zhang, C.; Shen, W.; Zhang, Y.; Liu, T.; Hu, H.; Xie, X.; Luo, F. Interleukin-9 promotes tumorigenesis through augmenting angiogenesis innon-small cell lung cancer. Int. Immunopharmacol. 2019, 75, 105766. [Google Scholar] [CrossRef] [PubMed]
- Dumont, D.J.; Yamaguchi, T.P.; Conlon, R.A.; Rossant, J.; Breitman, M.L. tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene 1992, 7, 1471–1480. [Google Scholar] [PubMed]
- Teichert, M.; Milde, L.; Holm, A.; Stanicek, L.; Gengenbacher, N.; Savant, S.; Ruckdeschel, T.; Hasanov, Z.; Srivastava, K.; Hu, J.; et al. Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat. Commun. 2017, 8, 16106. [Google Scholar] [CrossRef]
- Nakamura, K.; Arimura, K.; Nishimura, A.; Tachibana, M.; Yoshikawa, Y.; Makihara, N.; Wakisaka, Y.; Kuroda, J.; Kamouchi, M.; Ooboshi, H.; et al. Possible involvement of basic FGF in the upregulation of PDGFRbeta in pericytes after ischemic stroke. Brain Res. 2016, 1630, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.E. Differential expression of the fibroblast growth factor receptor (FGFR) multigene family in normal human adult tissues. J. Histochem. Cytochem. 1997, 45, 1005–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.; Pearson, A.; Sharpe, R.; Lambros, M.; Geyer, F.; Lopez-Garcia, M.A.; Natrajan, R.; Marchio, C.; Iorns, E.; Mackay, A.; et al. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010, 70, 2085–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Bogdanovic, E.; Nguyen, V.P.; Dumont, D.J. Activation of Tie2 by angiopoietin-1 and angiopoietin-2 results in their release and receptor internalization. J. Cell Sci. 2006, 119, 3551–3560. [Google Scholar] [CrossRef] [Green Version]
- Wehrle, C.; Van Slyke, P.; Dumont, D.J. Angiopoietin-1-induced ubiquitylation of Tie2 by c-Cbl is required for internalization and degradation. Biochem. J. 2009, 423, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Haugsten, E.M.; Malecki, J.; Bjorklund, S.M.; Olsnes, S.; Wesche, J. Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol. Biol. Cell 2008, 19, 3390–3403. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, Y.; Becker, V.; Qiu, M.; Tang, T.; Ampofo, E.; Menger, M.D.; Laschke, M.W. Brassinin Promotes the Degradation of Tie2 and FGFR1 in Endothelial Cells and Inhibits Triple-Negative Breast Cancer Angiogenesis. Cancers 2022, 14, 3540. https://doi.org/10.3390/cancers14143540
Gu Y, Becker V, Qiu M, Tang T, Ampofo E, Menger MD, Laschke MW. Brassinin Promotes the Degradation of Tie2 and FGFR1 in Endothelial Cells and Inhibits Triple-Negative Breast Cancer Angiogenesis. Cancers. 2022; 14(14):3540. https://doi.org/10.3390/cancers14143540
Chicago/Turabian StyleGu, Yuan, Vivien Becker, Moqin Qiu, Tianci Tang, Emmanuel Ampofo, Michael D. Menger, and Matthias W. Laschke. 2022. "Brassinin Promotes the Degradation of Tie2 and FGFR1 in Endothelial Cells and Inhibits Triple-Negative Breast Cancer Angiogenesis" Cancers 14, no. 14: 3540. https://doi.org/10.3390/cancers14143540
APA StyleGu, Y., Becker, V., Qiu, M., Tang, T., Ampofo, E., Menger, M. D., & Laschke, M. W. (2022). Brassinin Promotes the Degradation of Tie2 and FGFR1 in Endothelial Cells and Inhibits Triple-Negative Breast Cancer Angiogenesis. Cancers, 14(14), 3540. https://doi.org/10.3390/cancers14143540