
Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target

 ,
, 
 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Autophagy
2.1. Autophagy Types and Selectivity
2.2. Autophagy Process and Machinery
2.2.1. Autophagy Initiation
2.2.2. Autophagosome Formation
2.2.3. Autophagosome Maturation
2.2.4. Autolysosome Formation and Cargo Degradation
2.3. Upstream Autophagy Regulation
2.3.1. PI3K/AKT/mTORC1 Pathway
2.3.2. MAPK Pathway
2.3.3. AMPK
2.3.4. Beclin-1 & Bcl-2
3. Pancreatic Cancer
3.1. Current Treatment Options
3.2. Genetic Landscape of PDAC
3.2.1. KRAS
3.2.2. TP53
3.2.3. SMAD4
3.2.4. CDKN2A
3.2.5. PTEN and BRCA1/2
4. Pancreatic Tumor Microenvironment and Autophagy
4.1. Tumor Microenvironment
4.2. Pancreatic Tumor Microenvironmental Stress
4.2.1. Altered Energy Metabolism
4.2.2. Reactive Oxygen Species (ROS)
4.2.3. Acidosis
4.2.4. Hypoxia and Angiogenesis
4.2.5. Extracellular Matrix and Mechanical Stress
4.3. Supporting Cells of the Tumor Microenvironment
4.3.1. Cancer Associated Fibroblasts and Pancreatic Stellate Cells
4.3.2. Schwann Cells
4.3.3. Endothelial Progenitor Cells
4.3.4. Immune Cell Infiltration
5. Autophagy in Pancreatic Cancer Progression
5.1. Autophagic Regulation in PDAC
5.2. Autophagy Promotes Pancreatic Tumor Progression
5.3. The Role of Autophagy in Pancreatic Cancer Metastasis
5.3.1. Autophagy as a Metastasis Promoter in Pancreatic Cancer
5.3.2. Autophagy as a Metastasis Suppressor in Pancreatic Cancer
6. Inhibiting Autophagic Machinery
6.1. Targeting Late-Stage Autophagy
6.2. Targeting Autophagy Initiation
6.2.1. ULK1 Complex Inhibitors
MRT68921
SBI-0206965
6.2.2. PI3K Class III Complex Inhibitors
Spautin-1
SAR405
6.2.3. New Autophagy Initiation Inhibitors
6.3. Natural Products Targeting Autophagy
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Estimating the world cancer burden: Globocan 2000. Int. J. Cancer 2001, 94, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef]
- Cowgill, S.M.; Muscarella, P. The genetics of pancreatic cancer. Am. J. Surg. 2003, 186, 279–286. [Google Scholar] [CrossRef]
- Gao, H.-L.; Wang, W.-Q.; Yu, X.-J.; Liu, L. Molecular drivers and cells of origin in pancreatic ductal adenocarcinoma and pancreatic neuroendocrine carcinoma. Exp. Hematol. Oncol. 2020, 9, 28. [Google Scholar] [CrossRef]
- De La Cruz, M.S.D.; Young, A.P.; Ruffin, M.T. Diagnosis and management of pancreatic cancer. Am. Fam. Physician 2014, 89, 626–632. [Google Scholar]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Li, H.; Wang, X.; Fang, Y.; Huo, Z.; Lu, X.; Zhan, X.; Deng, X.; Peng, C.; Shen, B. Integrated expression profiles analysis reveals novel predictive biomarker in pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 52571–52583. [Google Scholar] [CrossRef]
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Omary, M.B.; Lugea, A.; Lowe, A.W.; Pandol, S.J. The pancreatic stellate cell: A star on the rise in pancreatic diseases. J. Clin. Investig. 2007, 117, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.-H.; Li, J.; Dong, F.-Y.; Yang, J.-Y.; Liu, D.-J.; Yang, X.-M.; Wang, Y.-H.; Yang, M.-W.; Fu, X.-L.; Zhang, X.-X.; et al. Increased Serotonin Signaling Contributes to the Warburg Effect in Pancreatic Tumor Cells Under Metabolic Stress and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 153, 277–291.e19. [Google Scholar] [CrossRef]
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Farrow, B.; Albo, D.; Berger, D.H. The Role of the Tumor Microenvironment in the Progression of Pancreatic Cancer. J. Surg. Res. 2008, 149, 319–328. [Google Scholar] [CrossRef]
- Hadden, M.; Mittal, A.; Samra, J.; Zreiqat, H.; Sahni, S.; Ramaswamy, Y. Mechanically stressed cancer microenvironment: Role in pancreatic cancer progression. Biochim. Biophys. Acta 2020, 1874, 188418. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Menzies, F.M.; Rubinsztein, D.C. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2009, 584, 1393–1398. [Google Scholar] [CrossRef] [Green Version]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.; Lichtenberg, M.; Luo, S.; et al. Regulation of Mammalian Autophagy in Physiology and Pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [Green Version]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin–proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–689. [Google Scholar] [CrossRef] [Green Version]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [Green Version]
- Nam, H.Y.; Han, M.W.; Chang, H.W.; Kim, S.Y.; Kim, S.W. Prolonged autophagy by MTOR inhibitor leads radioresistant cancer cells into senescence. Autophagy 2013, 9, 1631–1632. [Google Scholar] [CrossRef] [Green Version]
- Nedelsky, N.B.; Todd, P.K.; Taylor, J.P. Autophagy and the ubiquitin-proteasome system: Collaborators in neuroprotection. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2008, 1782, 691–699. [Google Scholar] [CrossRef] [Green Version]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Ding, W.-X.; Ni, H.-M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.-M. Linking of Autophagy to Ubiquitin-Proteasome System Is Important for the Regulation of Endoplasmic Reticulum Stress and Cell Viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, X.; Kang, R.; Zeh, H.; Klionsky, D.J.; Tang, D. Regulation and function of autophagy in pancreatic cancer. Autophagy 2020, 17, 3275–3296. [Google Scholar] [CrossRef]
- Kraft, C.; Reggiori, F.; Peter, M. Selective types of autophagy in yeast. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 1404–1412. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. 2008, 24, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Bejarano, E.; Cuervo, A.M. Chaperone-Mediated Autophagy. Proc. Am. Thorac. Soc. 2010, 7, 29–39. [Google Scholar] [CrossRef]
- Majeski, A.E.; Fred, J. Dice, Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2435–2444. [Google Scholar] [CrossRef]
- Li, W.-W.; Li, J.; Bao, J.-K. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Shpilka, T.; Elazar, Z. Mechanisms of Autophagosome Biogenesis. Curr. Biol. 2012, 22, R29–R34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrpour, M.; Esclatine, A.; Beau, I.; Codogno, P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010, 20, 748–762. [Google Scholar] [CrossRef]
- Reggiori, F.; Komatsu, M.; Finley, K.; Simonsen, A. Autophagy: More Than a Nonselective Pathway. Int. J. Cell Biol. 2012, 2012, 3275–3296. [Google Scholar] [CrossRef]
- Welter, E.; Thumm, M.; Krick, R. Quantification of nonselective bulk autophagy in S. cerevisiae using Pgk1-GFP. Autophagy 2010, 6, 794–797. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, P.; Zhang, Z.; Farré, J.-C.; Li, X.; Wang, R.; Xia, Z.; Subramani, S.; Ma, C. The autophagic degradation of cytosolic pools of peroxisomal proteins by a new selective pathway. Autophagy 2020, 16, 154–166. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, Y.; Kumanomidou, T.; Sou, Y.-S.; Mizushima, T.; Ezaki, J.; Ueno, T.; Kominami, E.; Yamane, T.; Tanaka, K.; Komatsu, M. Structural Basis for Sorting Mechanism of p62 in Selective Autophagy. J. Biol. Chem. 2008, 283, 22847–22857. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Mizushima, N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 2011, 192, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, A.R.; Schandorff, S.; Høyer-Hansen, M.; Nielsen, M.O.; Jaüaüttelaü, M.; Dengjel, J.; Andersen, J.S. Ordered Organelle Degradation during Starvation-induced Autophagy. Mol. Cell. Proteom. 2008, 7, 2419–2428. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Tripathi, D.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [Green Version]
- Goldman, S.J.; Taylor, R.; Zhang, Y.; Jin, S. Autophagy and the degradation of mitochondria. Mitochondrion 2010, 10, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Castrejón-Jiménez, N.S.; Leyva-Paredes, K.; Hernández-González, J.C.; Luna-Herrera, J.; García-Pérez, B.E. The role of autophagy in bacterial infections. Biosci. Trends 2015, 9, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, J.L.; Sumpter, R.; Levine, B.; Hooper, L.V. Intestinal Epithelial Autophagy Is Essential for Host Defense against Invasive Bacteria. Cell Host Microbe 2013, 13, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Luo, G.; Jian, Z.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. Int. J. Mol. Med. 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [Green Version]
- Mohan, N.; Banik, N.L.; Ray, S.K. Combination of N-(4-hydroxyphenyl) retinamide and apigenin suppressed starvation-induced autophagy and promoted apoptosis in malignant neuroblastoma cells. Neurosci. Lett. 2011, 502, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Sahni, S.; Gillson, J.; Park, K.C.; Chiang, S.; Leck, L.Y.W.; Jansson, P.; Richardson, D.R. NDRG1 suppresses basal and hypoxia-induced autophagy at both the initiation and degradation stages and sensitizes pancreatic cancer cells to lysosomal membrane permeabilization. Biochim. Biophys. Acta (BBA) Gen. Subj. 2020, 1864, 129625. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef]
- Boya, P.; Codogno, P.; Rodriguez-Muela, N. Autophagy in stem cells: Repair, remodelling and metabolic reprogramming. Development 2018, 145, dev146506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Contento, A.L.; Nguyen, P.Q.; Bassham, D.C. Degradation of Oxidized Proteins by Autophagy during Oxidative Stress in Arabidopsis. Plant Physiol. 2007, 143, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, N.; Sasaki, T.; Iemura, S.-I.; Natsume, T.; Hara, T.; Mizushima, N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 2009, 5, 973–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Guan, K.-L. Regulation of the Autophagy Initiating Kinase ULK1 By Nutrients: Roles of Mtorc1 and AMPK. Cell Cycle 2011, 10, 1337–1338. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.-C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.R.; Celano, S.L.; Solitro, A.R.; Gunaydin, H.; Scott, M.; O’Hagan, R.C.; Shumway, S.D.; Fuller, P.; MacKeigan, J.P. A Potent and Selective ULK1 Inhibitor Suppresses Autophagy and Sensitizes Cancer Cells to Nutrient Stress. iScience 2018, 8, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.-Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.-L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Di Bartolomeo, S.; Corazzari, M.; Nazio, F.; Oliverio, S.; Lisi, G.; Antonioli, M.; Pagliarini, V.; Matteoni, S.; Fuoco, C.; Giunta, L.; et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol. 2010, 191, 155–168. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 Antiapoptotic Proteins Inhibit Beclin 1-Dependent Autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A. Autophagy Regulation and Integration with Cell Signaling. Antioxid. Redox Signal. 2012, 17, 756–765. [Google Scholar] [CrossRef]
- Van Humbeeck, C.; Cornelissen, T.; Vandenberghe, W. Ambra1: A Parkin-binding protein involved in mitophagy. Autophagy 2011, 7, 1555–1556. [Google Scholar] [CrossRef] [Green Version]
- Itakura, E.; Kishi, C.; Inoue, K.; Mizushima, N. Beclin 1 Forms Two Distinct Phosphatidylinositol 3-Kinase Complexes with Mammalian Atg14 and UVRAG. Mol. Biol. Cell 2008, 19, 5360–5372. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Schekman, R. The ER-Golgi intermediate compartment feeds the phagophore membrane. Autophagy 2014, 10, 170–172. [Google Scholar] [CrossRef] [Green Version]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schüchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5–Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Otomo, C.; Metlagel, Z.; Takaesu, G.; Otomo, T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat. Struct. Mol. Biol. 2013, 20, 59. [Google Scholar] [CrossRef] [Green Version]
- Giménez-Xavier, P.; Francisco, R.; Platini, F.; Pérez, R.; Ambrosio, S. LC3-I conversion to LC3-II does not necessarily result in complete autophagy. Int. J. Mol. Med. 2008, 22, 781. [Google Scholar]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Høyvarde Clausen, T.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.D.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Kirkin, V.; Lamark, T.; Sou, Y.-S.; Bjørkøy, G.; Nunn, J.L.; Bruun, J.-A.; Shvets, E.; McEwan, D.G.; Clausen, T.H.; Wild, P.; et al. A Role for NBR1 in Autophagosomal Degradation of Ubiquitinated Substrates. Mol. Cell 2009, 33, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.; Ohsumi, Y.; Inagaki, F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett. 2010, 584, 1379–1385. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yamamoto, A.; Hatano, M.; Kobayashi, Y.; Kabeya, Y.; Suzuki, K.; Tokuhisa, T.; Ohsumi, Y.; Yoshimori, T. Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells. J. Cell Biol. 2001, 152, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Jung, C.H.; Seo, M.; Kim, E.K.; Park, J.-M.; Bae, S.S.; Kim, D.-H. mTORC1 Phosphorylates UVRAG to Negatively Regulate Autophagosome and Endosome Maturation. Mol. Cell 2015, 57, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Lee, J.-S.; Inn, K.-S.; Gack, M.U.; Li, Q.; Roberts, E.A.; Vergne, I.; Deretic, V.; Feng, P.; Akazawa, C.; et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat. Cell Biol. 2008, 10, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, Y.; Coppola, D.; Matsushita, N.; Cualing, H.D.; Sun, M.; Sato, Y.; Liang, C.; Jung, J.U.; Cheng, J.Q.; Mul, J.J.; et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat. Cell Biol. 2007, 9, 1142–1151. [Google Scholar] [CrossRef]
- Matsunaga, K.; Saitoh, T.; Tabata, K.; Omori, H.; Satoh, T.; Kurotori, N.; Maejima, I.; Shirahama-Noda, K.; Ichimura, T.; Isobe, T.; et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009, 11, 385–396. [Google Scholar] [CrossRef]
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Davis, T.; Loos, B.; Sishi, B.; Huisamen, B.; Strijdom, H.; Engelbrecht, A.-M. Autophagy is essential for the maintenance of amino acids and ATP levels during acute amino acid starvation in MDAMB231 cells. Cell Biochem. Funct. 2018, 36, 65–79. [Google Scholar] [CrossRef]
- Onodera, J.; Ohsumi, Y. Autophagy Is Required for Maintenance of Amino Acid Levels and Protein Synthesis under Nitrogen Starvation. J. Biol. Chem. 2005, 280, 31582–31586. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, G.P. Autophagy protects proximal tubular cells from injury and apoptosis. Kidney Int. 2012, 82, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.C.; Levine, B. Autophagy in cellular growth control. FEBS Lett. 2010, 584, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Sansal, I.; Sellers, W.R. The Biology and Clinical Relevance of the PTEN Tumor Suppressor Pathway. J. Clin. Oncol. 2004, 22, 2954–2963. [Google Scholar] [CrossRef]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef]
- Vara, J.Á.F.; Casado, E.; De Castro, J.; Cejas, P.; Belda-Iniesta, C.; González-Barón, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Acosta-Jaquez, H.A.; Romeo, Y.; Ekim, B.; Soliman, G.A.; Carriere, A.; Roux, P.P.; Ballif, B.A.; Fingar, D.C. Regulation of mTOR Complex 1 (mTORC1) by Raptor Ser863 and Multisite Phosphorylation. J. Biol. Chem. 2010, 285, 80–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, L.; Wang, X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy 2011, 7, 924–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschläger, M.; Weichhart, T. mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Galbaugh, T.; Cerrito, M.G.; Jose, C.C.; Cutler, M.L. EGF-induced activation of Akt results in mTOR-dependent p70S6 kinase phosphorylation and inhibition of HC11 cell lactogenic differentiation. BMC Cell Biol. 2006, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F.; Gajate, C. Novel therapeutic approaches for pancreatic cancer by combined targeting of RAF→MEK→ERK signaling and autophagy survival response. Ann. Transl. Med. 2019, 7, S153. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Eychene, A. The Raf/MEK/ERK pathway: New concepts of activation. Biol. Cell 2001, 93, 53–62. [Google Scholar] [CrossRef]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Wang, J.; Whiteman, M.W.; Lian, H.; Wang, G.; Singh, A.; Huang, D.; Denmark, T. A Non-canonical MEK/ERK Signaling Pathway Regulates Autophagy via Regulating Beclin 1. J. Biol. Chem. 2009, 284, 21412–21424. [Google Scholar] [CrossRef] [Green Version]
- Ugland, H.; Naderi, S.; Brech, A.; Collas, P.; Blomhoff, H.K. cAMP induces autophagy via a novel pathway involving ERK, cyclin E and Beclin 1. Autophagy 2011, 7, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.-W.; Farkas, A.M.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J.; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]
- Tong, Y.; Huang, H.; Pan, H. Inhibition of MEK/ERK activation attenuates autophagy and potentiates pemetrexed-induced activity against HepG2 hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2015, 456, 86–91. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ross, F.A.; Russell, F.M.; Atrih, A.; Lamont, D.J.; Hardie, D.G. Mechanism of Activation of AMPK by Cordycepin. Cell Chem. Biol. 2020, 27, 214–222.e4. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C. AMPK: Keeping the (power) house in order? Neuronal Signal. 2017, 1, NS20160020. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Göransson, O.; Hardie, D.G.; Alessi, D.R. Activity of LKB1 and AMPK-related kinases in skeletal muscle: Effects of contraction, phenformin, and AICAR. Am. J. Physiol. Metab. 2004, 287, E310–E317. [Google Scholar] [CrossRef] [Green Version]
- Neumann, D. Is TAK1 a direct upstream kinase of AMPK? Int. J. Mol. Sci. 2018, 19, 2412. [Google Scholar] [CrossRef] [Green Version]
- Peng, R.; Chen, Y.; Wei, L.; Li, G. Resistance to FGFR1-targeted therapy leads to autophagy via TAK1/AMPK activation in gastric cancer. Gastric Cancer 2020, 23, 988–1002. [Google Scholar] [CrossRef]
- Dunlop, E.A.; Tee, A. The Kinase Triad, AMPK, mTORC1 and ULK1, Maintains Energy and Nutrient Homoeostasis. Biochem. Soc. Trans. 2013, 41, 939–943. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S. The Antiapoptotic Protein BCL-2 Has Also an Antiautophagy Role Through Beclin 1 Inhibition. In Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging; Elsevier: Amsterdam, The Netherlands, 2016; Volume 9, pp. 165–174. [Google Scholar] [CrossRef]
- Xu, H.-D.; Qin, Z.-H. Beclin 1, Bcl-2 and autophagy. Autophagy Biol. Dis. 2019, 1206, 109–126. [Google Scholar]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-Mediated Phosphorylation of Bcl-2 Regulates Starvation-Induced Autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Wisnoski, N.C.; Townsend, C.M., Jr.; Nealon, W.H.; Freeman, J.L.; Riall, T.S. 672 patients with acinar cell carcinoma of the pancreas: A population-based comparison to pancreatic adenocarcinoma. Surgery 2008, 144, 141–148. [Google Scholar] [CrossRef]
- Klaiber, U.; Hackert, T. Conversion Surgery for Pancreatic Cancer—The Impact of Neoadjuvant Treatment. Front. Oncol. 2020, 9, 1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, Z.V.; Ferrone, C.R. Surgery After Response to Chemotherapy for Locally Advanced Pancreatic Ductal Adenocarcinoma: A Guide for Management. J. Natl. Compr. Cancer Netw. 2021, 19, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Mahipal, A.; Frakes, J.; Hoffe, S.; Kim, R. Management of borderline resectable pancreatic cancer. World J. Gastrointest. Oncol. 2015, 7, 241. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; De La Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Der, C.J.; Cox, A.D. The role of wild type RAS isoforms in cancer. Semin. Cell Dev. Biol. 2016, 58, 60–69. [Google Scholar] [CrossRef] [Green Version]
- Forbes, S.A.; Bindal, N.; Bamford, S.; Cole, C.; Kok, C.Y.; Beare, D.; Jia, M.; Shepherd, R.; Leung, K.; Menzies, A.; et al. COSMIC: Mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2010, 39, D945–D950. [Google Scholar] [CrossRef] [Green Version]
- Voice, J.K.; Klemke, R.L.; Le, A.; Jackson, J.H. Four Human Ras Homologs Differ in Their Abilities to Activate Raf-1, Induce Transformation, and Stimulate Cell Motility. J. Biol. Chem. 1999, 274, 17164–17170. [Google Scholar] [CrossRef] [Green Version]
- Windon, A.L.; Loaiza-Bonilla, A.; Jensen, C.E.; Randall, M.; Morrissette, J.J.D.; Shroff, S.G. A KRAS wild type mutational status confers a survival advantage in pancreatic ductal adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 1. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell signaling network, target genes, biological processes to therapeutic targeting. Crit. Rev. Oncol. 2017, 111, 7–19. [Google Scholar] [CrossRef]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is a Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273. [Google Scholar] [CrossRef]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef] [Green Version]
- Gillson, J.; Ramaswamy, Y.; Singh, G.; Gorfe, A.A.; Pavlakis, N.; Samra, J.; Mittal, A.; Sahni, S. Small Molecule KRAS Inhibitors: The Future for Targeted Pancreatic Cancer Therapy? Cancers 2020, 12, 1341. [Google Scholar] [CrossRef]
- Kennedy, A.L.; Adams, P.D.; Morton, J.P. Ras, PI3K/Akt and senescence: Paradoxes provide clues for pancreatic cancer therapy. Small GTPases 2011, 2, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Elpek, K.G.; Vinjamoori, A.; Zimmerman, S.M.; Chu, G.C.; Yan, H.; Fletcher-Sananikone, E.; Zhang, H.; Liu, Y.; Wang, W.; et al. PTEN is a major tumor suppressor in pancreatic ductal adenocarcinoma and regulates an NF-κB–cytokine network. Cancer Discov. 2011, 1, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Payne, S.N.; Maher, M.E.; Tran, N.H.; Van De Hey, D.R.; Foley, T.M.; Yueh, A.E.; Leystra, A.A.; Pasch, C.A.; Jeffrey, J.J.; Clipson, L.; et al. PIK3CA mutations can initiate pancreatic tumorigenesis and are targetable with PI3K inhibitors. Oncogenesis 2015, 4, e169. [Google Scholar] [CrossRef] [Green Version]
- Kong, B.; Wu, W.; Cheng, T.; Schlitter, A.M.; Qian, C.; Bruns, P.; Jian, Z.; Jäger, C.; Regel, I.; Raulefs, S.; et al. A subset of metastatic pancreatic ductal adenocarcinomas depends quantitatively on oncogenic Kras/Mek/Erk-induced hyperactive mTOR signalling. Gut 2016, 65, 647–657. [Google Scholar] [CrossRef]
- Xiang, J.-F.; Wang, W.-Q.; Liu, L.; Xu, H.-X.; Wu, C.-T.; Yang, J.-X.; Qi, Z.-H.; Wang, Y.-Q.; Xu, J.; Liu, C.; et al. Mutant p53 determines pancreatic cancer poor prognosis to pancreatectomy through upregulation of cavin-1 in patients with preoperative serum CA19-9 ≥ 1000 U/mL. Sci. Rep. 2016, 6, 19222. [Google Scholar] [CrossRef] [Green Version]
- Broz, D.K.; Mello, S.S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [Green Version]
- Pellegata, N.S.; Sessa, F.; Renault, B.; Bonato, M.; Leone, B.E.; Solcia, E.; Ranzani, G. K-ras and p53 gene mutations in pancreatic cancer: Ductal and nonductal tumors progress through different genetic lesions. Cancer Res. 1994, 54, 1556–1560. [Google Scholar]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Tang, J.; Di, J.; Cao, H.; Bai, J.; Zheng, J. p53-mediated autophagic regulation: A prospective strategy for cancer therapy. Cancer Lett. 2015, 363, 101–107. [Google Scholar] [CrossRef]
- Hu, H.-F.; Ye, Z.; Qin, Y.; Xu, X.-W.; Yu, X.-J.; Zhuo, Q.-F.; Ji, S.-R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 Gene Mutations Are Associated with Poor Prognosis in Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef] [Green Version]
- David, C.J.; Huang, Y.-H.; Chen, M.; Su, J.; Zou, Y.; Bardeesy, N.; Iacobuzio-Donahue, C.A.; Massagué, J. TGF-β Tumor Suppression through a Lethal EMT. Cell 2016, 164, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Xu, J.; Meng, Q.; Zhang, B.; Liu, J.; Hua, J.; Zhang, Y.; Shi, S.; Yu, X. TGFB1-induced autophagy affects the pattern of pancreatic cancer progression in distinct ways depending on SMAD4 status. Autophagy 2020, 16, 486–500. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, C.; Chiavarina, B.; Whitaker-Menezes, D.; Pestell, T.G.; Pestell, R.G.; Hulit, J.; Andò, S.; Howell, A.; Martinez-Outschoorn, U.E.; Sotgia, F.; et al. CDK inhibitors (p16/p19/p21) induce senescence and autophagy in cancer-associated fibroblasts,“fueling” tumor growth via paracrine interactions, without an increase in neo-angiogenesis. Cell Cycle 2012, 11, 3599–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, R.R.; Wieben, E.D.; Rabe, K.G.; Pedersen, K.; Wu, Y.; Sicotte, H.; Petersen, G.M. Prevalence of CDKN2A mutations in pancreatic cancer patients: Implications for genetic counseling. Eur. J. Hum. Genet. 2011, 19, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morselli, E.; Galluzzi, L.; Kepp, O.; Vicencio, J.M. Anti-and pro-tumor functions of autophagy. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2009, 1793, 1524–1532. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sawai, H.; Ochi, N.; Matsuo, Y.; Xu, D.; Yasuda, A.; Takahashi, H.; Wakasugi, T.; Takeyama, H. PTEN regulate angiogenesis through PI3K/Akt/VEGF signaling pathway in human pancreatic cancer cells. Mol. Cell. Biochem. 2009, 331, 161. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Stiles, B.L.; Kuralwalla-Martinez, C.; Guo, W.; Gregorian, C.; Wang, Y.; Tian, J.; Magnuson, M.A.; Wu, H. Selective Deletion of Pten in Pancreatic β Cells Leads to Increased Islet Mass and Resistance to STZ-Induced Diabetes. Mol. Cell. Biol. 2006, 26, 2772–2781. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. PTEN loss accelerates KrasG12D-induced pancreatic cancer development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Ehdaie, B.; Ohara, N.; Yoshino, T.; Deng, C.-X. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene 2009, 29, 674–686. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Flossbach, L.; Nixon, C.; Morton, J.; Sansom, O.J.; Ryan, K.M. PTEN deficiency permits the formation of pancreatic cancer in the absence of autophagy. Cell Death Differ. 2017, 24, 1303–1304. [Google Scholar] [CrossRef]
- Aquila, S.; Santoro, M.; Caputo, A.; Panno, M.L.; Pezzi, V.; De Amicis, F. The Tumor Suppressor PTEN as Molecular Switch Node Regulating Cell Metabolism and Autophagy: Implications in Immune System and Tumor Microenvironment. Cells 2020, 9, 1725. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Gress, T.M.; Langer, P. Familial pancreatic cancer—Current knowledge. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 445–453. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Rosen, M.N.; Goodwin, R.A.; Vickers, M.M. BRCA mutated pancreatic cancer: A change is coming. World J. Gastroenterol. 2021, 27, 1943. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Ren, H.; Bakas, N.A.; Vamos, N.; Chaikuad, A. Design, Synthesis, and Characterization of an Orally Active Dual-Specific ULK1/2 Autophagy Inhibitor that Synergizes with the PARP Inhibitor Olaparib for the Treatment of Triple-Negative Breast Cancer. J. Med. Chem. 2020, 63, 14609–14625. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef] [Green Version]
- Duffy, J.P.; Eibl, G.; Reber, H.A.; Hines, O.J. Influence of hypoxia and neoangiogenesis on the growth of pancreatic cancer. Mol. Cancer 2003, 2, 12. [Google Scholar] [CrossRef]
- Martin, J.D.; Fukumura, D.; Duda, D.G.; Boucher, Y.; Jain, R.K. Reengineering the tumor microenvironment to alleviate hypoxia and overcome cancer heterogeneity. Cold Spring Harb. Perspect. Med. 2016, 6, a027094. [Google Scholar] [CrossRef] [Green Version]
- Delrue, L.; Blanckaert, P.; Mertens, D.; Cesmeli, E.; Ceelen, W.P.; Duyck, P. Assessment of Tumor Vascularization in Pancreatic Adenocarcinoma Using 128-Slice Perfusion Computed Tomography Imaging. J. Comput. Assist. Tomogr. 2011, 35, 434–438. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Cheng, T.; Wu, W.; Regel, I.; Raulefs, S.; Friess, H.; Erkan, M.; Esposito, I.; Kleeff, J.; Michalski, C.W. Hypoxia-induced endoplasmic reticulum stress characterizes a necrotic phenotype of pancreatic cancer. Oncotarget 2015, 6, 32154–32160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2014, 4, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forster, J.C.; Harriss-Phillips, W.M.; Douglass, M.; Bezak, E. A review of the development of tumor vasculature and its effects on the tumor microenvironment. Hypoxia 2017, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Zeh, H.J.; Bahary, N.; Boone, B.A.; Singhi, A.D.; Miller-Ocuin, J.L.; Normolle, D.P.; Zureikat, A.H.; Hogg, M.E.; Bartlett, D.L.; Lee, K.K.; et al. A Randomized Phase II Preoperative Study of Autophagy Inhibition with High-Dose Hydroxychloroquine and Gemcitabine/Nab-Paclitaxel in Pancreatic Cancer Patients. Clin. Cancer Res. 2020, 26, 3126–3134. [Google Scholar] [CrossRef] [Green Version]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Bartrons, R.; Caro, J. Hypoxia, glucose metabolism and the Warburg’s effect. J. Bioenerg. Biomembr. 2007, 39, 223–229. [Google Scholar] [CrossRef]
- Komar, G.; Kauhanen, S.; Liukko, K.; Seppänen, M.; Kajander, S.; Ovaska, J.; Nuutila, P.; Minn, H. Decreased Blood Flow with Increased Metabolic Activity: A Novel Sign of Pancreatic Tumor Aggressiveness. Clin. Cancer Res. 2009, 15, 5511–5517. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, H.-X.; Wu, C.-T.; Wang, W.-Q.; Jin, W.; Gao, H.-L.; Li, H.; Zhang, S.-R.; Xu, J.-Z.; Qi, Z.-H.; et al. Angiogenesis in pancreatic cancer: Current research status and clinical implications. Angiogenesis 2019, 22, 15–36. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef] [Green Version]
- Chiche, J.; Brahimi-Horn, M.C.; Pouysségur, J. Tumour hypoxia induces a metabolic shift causing acidosis: A common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. [Google Scholar] [CrossRef] [Green Version]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar] [CrossRef]
- Pouysségur, J.; Dayan, F.; Mazure, N.M. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature 2006, 441, 437–443. [Google Scholar] [CrossRef]
- Hu, M.; Chen, X.; Ma, L.; Ma, Y.; Li, Y.; Song, H.; Xu, J.; Zhou, L.; Li, X.; Jiang, Y.; et al. AMPK Inhibition Suppresses the Malignant Phenotype of Pancreatic Cancer Cells in Part by Attenuating Aerobic Glycolysis. J. Cancer 2019, 10, 1870. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Wu, S.; Li, H.; Duan, Q.; Zhang, Z.; Shen, Q.; Wang, C.; Yin, T. ROS/KRAS/AMPK Signaling Contributes to Gemcitabine-Induced Stem-like Cell Properties in Pancreatic Cancer. Mol. Ther. Oncolytics 2019, 14, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef] [Green Version]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 2019, 29, 285–302.e7. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Ding, H.; Yang, Y.; Yang, L.; Yang, Y.; Fang, M.; Ren, J.; Hu, R.; Wang, C.; Geng, W. BAP1 regulates AMPK-mTOR signalling pathway through deubiquitinating and stabilizing tumour-suppressor LKB1. Biochem. Biophys. Res. Commun. 2020, 529, 1025–1032. [Google Scholar] [CrossRef]
- Reyes-Castellanos, G.; Masoud, R.; Carrier, A. Mitochondrial Metabolism in PDAC: From Better Knowledge to New Targeting Strategies. Biomedicines 2020, 8, 270. [Google Scholar] [CrossRef]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Storz, P. Oxidative Stress in Cancer. In Oxidative Stress and Redox Regulation; Springer: Berlin/Heidelberg, Germany, 2013; pp. 427–447. [Google Scholar]
- Pelicano, H.; Lu, W.; Zhou, Y.; Zhang, W.; Chen, Z.; Hu, Y.; Huang, P. Mitochondrial Dysfunction and Reactive Oxygen Species Imbalance Promote Breast Cancer Cell Motility through a CXCL14-Mediated Mechanism. Cancer Res. 2009, 69, 2375–2383. [Google Scholar] [CrossRef] [Green Version]
- Dansen, T.B.; Wirtz, K.W.A. The Peroxisome in Oxidative Stress. IUBMB Life 2001, 51, 223–230. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [Green Version]
- Moloney, J.N.; Cotter, T.G. ROS Signalling in the Biology of Cancer. In Oxidative Stress and Redox Regulation; Springer: Berlin/Heidelberg, Germany, 2013; pp. 427–447. [Google Scholar]
- Alexander, M.S.; Cullen, J.J. Treating pancreatic cancer: More antioxidants more problems? Expert Rev. Gastroenterol. Hepatol. 2018, 12, 849–851. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Masuda, A.; Sun, M.; Centonze, V.E.; Herman, B. Oxidative stress-induced apoptosis is associated with alterations in mitochondrial caspase activity and Bcl-2-dependent alterations in mitochondrial pH (pHm). Brain Res. Bull. 2004, 62, 497–504. [Google Scholar] [CrossRef]
- Sosa, V.; Moliné, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; Lleonart, M.E. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef] [PubMed]
- Lisanti, M.P.; Martinez-Outschoorn, U.E.; Lin, Z.; Pavlides, S.; Whitaker-Menezes, D.; Pestell, R.G.; Howell, A.; Sotgia, F. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: The seed and soil also needs “fertilizer”. Cell Cycle 2011, 10, 2440–2449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Wang, K.; Chen, Y.; Chen, H.; Nice, E.C.; Huang, C. Redox regulation in tumor cell epithelial–mesenchymal transition: Molecular basis and therapeutic strategy. Signal Transduct. Target. Ther. 2017, 2, 17036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Chen, Y.; Gibson, S.B. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell. Signal. 2013, 25, 50–65. [Google Scholar] [CrossRef]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Wang, X.; Contino, G.; Liesa, M.; Sahin, E.; Ying, H.; Bause, A.; Li, Y.; Stommel, J.M.; Dell’Antonio, G.; et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011, 25, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Fu, Z.; Chenghong, P.; Kuang, J.; Feng, H.; Chen, L.; Liang, J.; Shen, X.; Yuen, S.; Peng, C.; Shen, B.; et al. CQ sensitizes human pancreatic cancer cells to gemcitabine through the lysosomal apoptotic pathway via reactive oxygen species. Mol. Oncol. 2018, 12, 529–544. [Google Scholar] [CrossRef]
- Yang, S.; Kimmelman, A.C. A critical role for autophagy in pancreatic cancer. Autophagy 2011, 7, 912–913. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Lagadic-Gossmann, D.; Huc, L.; Lecureur, V. Alterations of intracellular pH homeostasis in apoptosis: Origins and roles. Cell Death Differ. 2004, 11, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity Generated by the Tumor Microenvironment Drives Local Invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [Green Version]
- Annese, T.; Tamma, R.; Ruggieri, S.; Ribatti, D. Angiogenesis in Pancreatic Cancer: Pre-Clinical and Clinical Studies. Cancers 2019, 11, 381. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, Q.-Q.; Pan, G.-X.; Jia, G.-R.; Li, X.; Wang, C.; Zhang, L.-M.; Zuo, C.-J. Retracted Article: ASIC1a involves acidic microenvironment-induced activation and autophagy of pancreatic stellate cells. RSC Adv. 2018, 8, 30950–30956. [Google Scholar] [CrossRef] [Green Version]
- Rabiee, S.; Tavakol, S.; Barati, M.; Joghataei, M.T. Autophagic, apoptotic, and necrotic cancer cell fates triggered by acidic pH microenvironment. J. Cell. Physiol. 2018, 234, 12061–12069. [Google Scholar] [CrossRef]
- Wojtkowiak, J.W.; Rothberg, J.M.; Kumar, V.; Schramm, K.J.; Haller, E.; Proemsey, J.B.; Lloyd, M.C.; Sloane, B.F.; Gillies, R.J. Chronic Autophagy Is a Cellular Adaptation to Tumor Acidic pH Microenvironments. Cancer Res. 2012, 72, 3938–3947. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Zhou, H.-Y.; Deng, S.-C.; Deng, S.-J.; He, C.; Li, X.; Chen, J.-Y.; Jin, Y.; Hu, Z.-L.; Wang, F. ASIC1 and ASIC3 contribute to acidity-induced EMT of pancreatic cancer through activating Ca2+/RhoA pathway. Cell Death Dis. 2017, 8, e2806. [Google Scholar] [CrossRef]
- Endo, S.; Nakata, K.; Ohuchida, K.; Takesue, S.; Nakayama, H.; Abe, T.; Koikawa, K.; Okumura, T.; Sada, M.; Horioka, K.; et al. Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated with Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 2017, 152, 1492–1506.e24. [Google Scholar] [CrossRef] [Green Version]
- Moir, J.A.; Mann, J.; White, S.A. The role of pancreatic stellate cells in pancreatic cancer. Surg. Oncol. 2015, 24, 232–238. [Google Scholar] [CrossRef]
- Spector, I.; Honig, H.; Kawada, N.; Nagler, A.; Genin, O.; Pines, M. Inhibition of Pancreatic Stellate Cell Activation by Halofuginone Prevents Pancreatic Xenograft Tumor Development. Pancreas 2010, 39, 1008–1015. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Demir, R.; Yaba, A.; Huppertz, B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2009, 112, 203–214. [Google Scholar] [CrossRef]
- Bačić, I.; Karlo, R.; Šoštarić Zadro, A.; Zadro, Z.; Skitarelić, N.; Antabak, A. Tumor angiogenesis as an important prognostic factor in advanced non-small cell lung cancer (Stage IIIA). Oncol. Lett. 2018, 15, 2335–2339. [Google Scholar]
- Uzzan, B.; Nicolas, P.; Cucherat, M.; Perret, G.-P. Microvessel density as a prognostic factor in women with breast cancer: A systematic review of the literature and meta-analysis. Cancer Res. 2004, 64, 2941–2955. [Google Scholar] [CrossRef] [Green Version]
- Kraby, M.R.; Opdahl, S.; Akslen, L.A.; Bofin, A.M. Quantifying tumour vascularity in non-luminal breast cancers. J. Clin. Pathol. 2017, 70, 766–774. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. HIF-1, O2, and the 3 PHDs: How animal cells signal hypoxia to the nucleus. Cell 2001, 107, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Dai, L.; Ma, Y.; Wang, J.; Liu, Z. Implications of HIF-1α in the tumorigenesis and progression of pancreatic cancer. Cancer Cell Int. 2020, 20, 273. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhu, D.-M.; Zhou, X.-G.; Yin, N.; Zhang, Y.; Zhang, Z.-X.; Li, D.-C.; Zhou, J. HIF-2α promotes the formation of vasculogenic mimicry in pancreatic cancer by regulating the binding of Twist1 to the VE-cadherin promoter. Oncotarget 2017, 8, 47801. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.-Y.; Zhang, Q.; Bai, X.-L.; Pankaj, P.; Hu, Q.-D.; Liang, T.-B. Hypoxia-inducible factor 1α expression and its clinical significance in pancreatic cancer: A meta-analysis. Pancreatology 2014, 14, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Akakura, N.; Kobayashi, M.; Horiuchi, I.; Suzuki, A.; Wang, J.; Chen, J.; Niizeki, H.; Kawamura, K.-i.; Hosokawa, M.; Masahiro Asaka, M. Constitutive expression of hypoxia-inducible factor-1α renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001, 61, 6548–6554. [Google Scholar]
- Marquez, R.T.; Xu, L. Bcl-2:Beclin 1 complex: Multiple, mechanisms regulating autophagy/apoptosis toggle switch. Am. J. Cancer Res. 2012, 2, 214. [Google Scholar]
- Madhu, V.; Boneski, P.G.; Silagi, E.; Qiu, Y.; Kurland, I.; Guntur, A.R.; Shapiro, I.M.; Risbud, M.V. Hypoxic Regulation of Mitochondrial Metabolism and Mitophagy in Nucleus Pulposus Cells Is Dependent on HIF-1α–BNIP3 Axis. J. Bone Miner. Res. 2020, 35, 1504–1524. [Google Scholar] [CrossRef]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial Autophagy Is an HIF-1-dependent Adaptive Metabolic Response to Hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [Green Version]
- Giaccia, A.J.; Simon, M.C.; Johnson, R. The biology of hypoxia: The role of oxygen sensing in development, normal function, and disease. Genes Dev. 2004, 18, 2183–2194. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Zhang, Q.; Luo, W. Angiogenesis inhibitors as therapeutic agents in cancer: Challenges and future directions. Eur. J. Pharmacol. 2016, 793, 76–81. [Google Scholar] [CrossRef]
- Gui, L.; Liu, B.; Lv, G. Hypoxia induces autophagy in cardiomyocytes via a hypoxia-inducible factor 1-dependent mechanism. Exp. Ther. Med. 2016, 11, 2233–2239. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef]
- Meng, H.; Zhao, Y.; Dong, J.; Xue, M.; Lin, Y.-S.; Ji, Z.; Mai, W.X.; Zhang, H.; Chang, C.H.; Brinker, C.J.; et al. Two-Wave Nanotherapy to Target the Stroma and Optimize Gemcitabine Delivery to a Human Pancreatic Cancer Model in Mice. ACS Nano 2013, 7, 10048–10065. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Tao, Y.; He, M.; Deng, M.; Guo, R.; Sheng, Q.; Wang, X.; Ren, K.; Li, T.; He, X. Co-delivery of autophagy inhibitor and gemcitabine using a pH-activatable core-shell nanobomb inhibits pancreatic cancer progression and metastasis. Theranostics 2021, 11, 8692. [Google Scholar] [CrossRef]
- Phillips, P.; McCarroll, J.; Park, S.; Wu, M.-J.; Pirola, R.C.; Korsten, M.A.; Wilson, J.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef]
- Weniger, M.; Honselmann, K.; Liss, A. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, 316. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; DelGiorno, K.E.; Greenberg, P.D.; Hingorani, S.R. Stromal reengineering to treat pancreas cancer. Carcinogenesis 2014, 35, 1451–1460. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.-Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Wei, S.; Yang, J. Forcing through Tumor Metastasis: The Interplay between Tissue Rigidity and Epithelial–Mesenchymal Transition. Trends Cell Biol. 2016, 26, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Gkretsi, V.; Stylianopoulos, T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018, 8, 145. [Google Scholar] [CrossRef]
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018, 359, 1408–1411. [Google Scholar] [CrossRef] [Green Version]
- Hupfer, A.; Brichkina, A.; Koeniger, A.; Keber, C.; Denkert, C.; Pfefferle, P.; Helmprobst, F.; Pagenstecher, A.; Visekruna, A.; Lauth, M. Matrix stiffness drives stromal autophagy and promotes formation of a protumorigenic niche. Proc. Natl. Acad. Sci. USA 2021, 118, e2105367118. [Google Scholar] [CrossRef]
- Bays, J.; Campbell, H.; Heidema, C.; Sebbagh, M.; DeMali, K.A. Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat. Cell Biol. 2017, 19, 724–731. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Ohuchida, K.; Fei, S.; Zheng, B.; Guan, W.; Feng, H.; Kibe, S.; Ando, Y.; Koikawa, K.; Abe, T.; et al. Inhibition of ERK1/2 in cancer-associated pancreatic stellate cells suppresses cancer–stromal interaction and metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 221. [Google Scholar] [CrossRef]
- Galdiero, M.R.; Bonavita, E.; Barajon, I.; Garlanda, C.; Mantovani, A.; Jaillon, S. Tumor associated macrophages and neutrophils in cancer. Immunobiology 2013, 218, 1402–1410. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y.; et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef] [Green Version]
- Vonlaufen, A.; Phillips, P.A.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Pancreatic Stellate Cells and Pancreatic Cancer Cells: An Unholy Alliance. Cancer Res. 2008, 68, 7707–7710. [Google Scholar] [CrossRef] [Green Version]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog Promotes Desmoplasia in Pancreatic Cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachem, M.G.; Schneider, E.; Groß, H.; Weidenbach, H.; Schmid, R.M.; Menke, A.; Siech, M.; Beger, H.; Grünert, A.; Adler, G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998, 115, 421–432. [Google Scholar] [CrossRef]
- Berna, M.J.; Seiz, O.; Nast, J.F.; Benten, D.; Bläker, M.; Koch, J.; Lohse, A.W.; Pace, A. CCK1 and CCK2 Receptors Are Expressed on Pancreatic Stellate Cells and Induce Collagen Production. J. Biol. Chem. 2010, 285, 38905–38914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordes, C.; Sawitza, I.; Gotze, S.; Herebian, D.; Haussinger, D. Stellate Cells Are Mesenchymal Stem Cells. Eur. J. Med. Res. 2014, 19, S6. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz-López, K.G.; Castro-Muñoz, L.J.; Reyes-Hernández, D.O.; García-Carrancá, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.; Radhakrishnan, P. Tumor-stromal crosstalk in pancreatic cancer and tissue fibrosis. Mol. Cancer 2019, 18, 14. [Google Scholar] [CrossRef]
- Jakubowska, K.; Pryczynicz, A.; Januszewska, J.; Sidorkiewicz, I.; Kemona, A.; Niewiński, A.; Lewczuk, Ł.; Kędra, B.; Guzińska-Ustymowicz, K. Expressions of Matrix Metalloproteinases 2, 7, and 9 in Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Dis. Markers 2016, 2016, 9895721. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhang, H.; Ma, Q.; Huang, R.; Lu, J.; Liang, X.; Liu, X.; Zhang, Z.; Yu, L.; Pang, J.; et al. YAP1-mediated pancreatic stellate cell activation inhibits pancreatic cancer cell proliferation. Cancer Lett. 2019, 462, 51–60. [Google Scholar] [CrossRef]
- Sun, T.; Peng, H.; Mao, W.; Ma, L.; Liu, H.; Mai, J.; Jiao, L. Autophagy-mediated negative feedback attenuates the oncogenic activity of YAP in pancreatic cancer. Int. J. Biol. Sci. 2021, 17, 3634. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; He, R.; Yanh, W.; Zhang, Y.; Yuan, Q.; Wang, J.; Liu, Y.; Chen, S.; Zhang, S.; Zhang, W.; et al. Autophagic Schwann cells promote perineural invasion mediated by the NGF/ATG7 paracrine pathway in pancreatic cancer. J. Exp. Clin. Cancer Res. 2022, 41, 48. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, H.-Q.; Li, J.; Liu, X.-L. Endothelial progenitor cells promote tumor growth and progression by enhancing new vessel formation. Oncol. Lett. 2016, 12, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; He, P.; Xu, D.; Li, J.; Peng, X.; Tang, Y. Acidic stress induces apoptosis and inhibits angiogenesis in human bone marrow-derived endothelial progenitor cells. Oncol. Lett. 2017, 14, 5695–5702. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Xue, J.; Jaffee, E.M.; Habtezion, A. Role of Immune Cells and Immune-Based Therapies in Pancreatitis and Pancreatic Ductal Adenocarcinoma. Gastroenterology 2013, 144, 1230–1240. [Google Scholar] [CrossRef] [Green Version]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef] [Green Version]
- Mielgo, A.; Schmid, M.C. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013, 46, 131. [Google Scholar] [CrossRef] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- Jiang, H.; Courau, T.; Borison, J.; Ritchie, A.J.; Mayer, A.T.; Krummel, M.F.; Collisson, E.A. Activating Immune Recognition in Pancreatic Ductal Adenocarcinoma via Autophagy Inhibition, MEK Blockade, and CD40 Agonism. Gastroenterology 2022, 162, 590–603.e14. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Edwards, L.J.; Evavold, B.D.; Zhu, C. Kinetics of MHC-CD8 interaction at the T cell membrane. J. Immunol. 2007, 179, 7653–7662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I. Nature 2020, 581, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Garcia, M.N.; Iovanna, J.L. Autophagy in pancreatic cancer. Int. J. Cell Biol. 2012, 2012, 760498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopp, J.L.; Dubois, C.L.; Schaeffer, D.F.; Samani, A.; Taghizadeh, F.; Cowan, R.W.; Rhim, A.D.; Stiles, B.L.; Valasek, M.; Sander, M. Loss of Pten and Activation of Kras Synergistically Induce Formation of Intraductal Papillary Mucinous Neoplasia from Pancreatic Ductal Cells in Mice. Gastroenterology 2018, 154, 1509–1523.e5. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Ciccarese, F.; Zulato, E.; Indraccolo, S. LKB1/AMPK pathway and drug response in cancer: A therapeutic perspective. Oxidative Med. Cell. Longev. 2019, 2019, 8730816. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 2064. [Google Scholar] [CrossRef]
- Zhu, J.; Chen, Y.; Jin, Y.; Yu, Y.; Zhang, X.; Zhou, J. Gemcitabine induces apoptosis and autophagy via the AMPK/mTOR signaling pathway in pancreatic cancer cells. Biotechnol. Appl. Biochem. 2018, 65, 665–671. [Google Scholar] [CrossRef]
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.-W.; Choi, J.; Lee, S.-Y.; Sung, S.; Yoo, H.J.; Kang, M.-J.; Cheong, H.; Son, J. Autophagy is required for PDAC glutamine metabolism. Sci. Rep. 2016, 6, 37594. [Google Scholar] [CrossRef]
- Nguyen, A.V.; Trompetto, B.; Tan, X.H.M.; Scott, M.B.; Hu, K.H.-H.; Deeds, E.; Butte, M.J.; Chiou, P.Y.; Rowat, A.C. Differential Contributions of Actin and Myosin to the Physical Phenotypes and Invasion of Pancreatic Cancer Cells. Cell. Mol. Bioeng. 2020, 13, 27–44. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Sohal, D.P.S.; Kennedy, E.B.; Khorana, A.; Copur, M.S.; Crane, C.H.; Garrido-Laguna, I.; Krishnamurthi, S.; Moravek, C.; O’Reilly, E.M.; Philip, P.A.; et al. Metastatic Pancreatic Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2018, 36, 2545–2556. [Google Scholar] [CrossRef]
- Hosein, A.N.; Dougan, S.K.; Aguirre, A.J.; Maitra, A. Translational advances in pancreatic ductal adenocarcinoma therapy. Nat. Cancer 2022, 3, 272–286. [Google Scholar] [CrossRef]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a therapeutic target in pancreatic cancer. Br. J. Cancer 2020, 124, 333–344. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.-G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef]
- Wang, Y.; Yin, S.; Zhang, L.; Shi, K.; Tang, J.; Zhang, Z.; He, Q. A tumor-activatable particle with antimetastatic potential in breast cancer via inhibiting the autophagy-dependent disassembly of focal adhesion. Biomaterials 2018, 168, 1–9. [Google Scholar] [CrossRef]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Sahni, S.; Bae, D.-H.; Lane, D.; Kovacevic, Z.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. The Metastasis Suppressor, N-myc Downstream-regulated Gene 1 (NDRG1), Inhibits Stress-induced Autophagy in Cancer Cells. J. Biol. Chem. 2014, 289, 9692–9709. [Google Scholar] [CrossRef] [Green Version]
- Merlot, A.; Porter, G.; Sahni, S.; Lim, E.; Peres, P.; Richardson, D. The metastasis suppressor, NDRG1, differentially modulates the endoplasmic reticulum stress response. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 2094–2110. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Philipson, E.; Engström, C.; Naredi, P.; Bourghardt Fagman, J. High expression of p62/SQSTM1 predicts shorter survival for patients with pancreatic cancer. BMC Cancer 2022, 22, 347. [Google Scholar] [CrossRef]
- Todoric, J.; Antonucci, L.; Di Caro, G.; Li, N.; Wu, X.; Lytle, N.K.; Dhar, D.; Banerjee, S.; Fagman, J.B.; Browne, C.D.; et al. Stress-Activated NRF2-MDM2 Cascade Controls Neoplastic Progression in Pancreas. Cancer Cell 2017, 32, 824–839.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.-F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/β/NF-κB activation by IL-1α and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, A.; Majumder, K.; Giri, B.; Dauer, P.; Dudeja, V.; Roy, S.; Banerjee, S.; Saluja, A.K. Inhibition of NF-kappa B pathway leads to deregulation of epithelial–mesenchymal transition and neural invasion in pancreatic cancer. Lab. Investig. 2016, 96, 1268–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolcet, X.; Llobet, D.; Pallares, J.; Matias-Guiu, X. NF-kB in development and progression of human cancer. Virchows Arch. 2005, 446, 475–482. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Sergeant, G.; Vankelecom, H.; Gremeaux, L.; Topal, B. Role of cancer stem cells in pancreatic ductal adenocarcinoma. Nat. Rev. Clin. Oncol. 2009, 6, 580–586. [Google Scholar] [CrossRef]
- Rausch, V.; Liu, L.; Apel, A.; Rettig, T.; Gladkich, J.; Labsch, S.; Kallifatidis, G.; Kaczorowski, A.; Groth, A.; Gross, W.; et al. Autophagy mediates survival of pancreatic tumour-initiating cells in a hypoxic microenvironment. J. Pathol. 2012, 227, 325–335. [Google Scholar] [CrossRef]
- Kizaka-Kondoh, S.; Itasaka, S.; Zeng, L.; Tanaka, S.; Zhao, T.; Takahashi, Y.; Shibuya, K.; Hirota, K.; Semenza, G.L.; Hiraoka, M. Selective Killing of Hypoxia-Inducible Factor-1–Active Cells Improves Survival in a Mouse Model of Invasive and Metastatic Pancreatic Cancer. Clin. Cancer Res. 2009, 15, 3433–3441. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-C.; Wang, H.-C.; Hou, Y.-C.; Tung, H.-L.; Chiu, T.-J.; Shan, Y.-S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [Green Version]
- Akar, U.; Ozpolat, B.; Mehta, K.; Fok, J.; Kondo, Y.; Lopez-Berestein, G. Tissue Transglutaminase Inhibits Autophagy in Pancreatic Cancer Cells. Mol. Cancer Res. 2007, 5, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Görgülü, K.; Diakopoulos, K.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.-F.; Ciecielski, K.J.; Aksoy, E.K.; Ruess, D.A.; Berninger, A.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203–217.e20. [Google Scholar] [CrossRef] [Green Version]
- Yayon, A.; Cabantchik, Z.I.; Ginsburg, H. Susceptibility of human malaria parasites to chloroquine is pH dependent. Proc. Natl. Acad. Sci. USA 1985, 82, 2784–2788. [Google Scholar] [CrossRef] [Green Version]
- Geser, A.; Brubaker, G.; Draper, C.C. Effect of a malaria suppression program on the incidence of African Burkitt’s lymphoma. Am. J. Epidemiol. 1989, 129, 740–752. [Google Scholar] [CrossRef]
- Gonzalez-Noriega, A.; Grubb, J.H.; Talkad, V.; Sly, W.S. Chloroquine inhibits lysosomal enzyme pinocytosis and enhances lysosomal enzyme secretion by impairing receptor recycling. J. Cell Biol. 1980, 85, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Boya, P.; Gonzalez-Polo, R.-A.; Poncet, D.; Andreau, K.; La Vieira, H.; Roumier, T.; Perfettini, J.-L.; Kroemer, G. Mitochondrial membrane permeabilization is a critical step of lysosome-initiated apoptosis induced by hydroxychloroquine. Oncogene 2003, 22, 3927–3936. [Google Scholar] [CrossRef] [Green Version]
- Browning, D.J. Pharmacology of Chloroquine and Hydroxychloroquine. In Hydroxychloroquine and Chloroquine Retinopathy; Springer: Berlin/Heidelberg, Germany, 2014; pp. 35–63. [Google Scholar] [CrossRef]
- Al-Bari, M.A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Perez, J.; Roustit, M.; Lepelley, M.; Revol, B.; Cracowski, J.-L.; Khouri, C. Reported Adverse Drug Reactions Associated with the Use of Hydroxychloroquine and Chloroquine during the COVID-19 Pandemic. Ann. Intern. Med. 2021, 174, 878–880. [Google Scholar] [CrossRef]
- Yazal, T.; Bailleul, J.; Ruan, Y.; Sung, D.; Chu, F.-I.; Palomera, D.; Dao, A.; Sehgal, A.; Gurunathan, V.; Aryan, L.; et al. Radiosensitizing Pancreatic Cancer via Effective Autophagy Inhibition. Mol. Cancer Ther. 2022, 21, 79–88. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and Pharmacodynamic Study of Autophagy Inhibition Using Hydroxychloroquine in Patients with Metastatic Pancreatic Adenocarcinoma. Oncologist 2014, 19, 637. [Google Scholar] [CrossRef] [Green Version]
- Stalnecker, C.A.; Coleman, M.F.; Bryant, K.L. Susceptibility to autophagy inhibition is enhanced by dual IGF1R and MAPK/ERK inhibition in pancreatic cancer. Autophagy 2022, 18, 1–3. [Google Scholar] [CrossRef]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.-I.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 Association with the ULK1–Atg13–FIP200 Complex Required for Autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- AlMasri, S.S.; Zenati, M.S.; Desilva, A.; Nassour, I.; Boone, B.A.; Singhi, A.D.; Bartlett, D.L.; Liotta, L.A.; Espina, V.; Loughran, P.; et al. Encouraging long-term survival following autophagy inhibition using neoadjuvant hydroxychloroquine and gemcitabine for high-risk patients with resectable pancreatic carcinoma. Cancer Med. 2021, 10, 7233–7241. [Google Scholar] [CrossRef]
- Petherick, K.J.; Conway, O.J.L.; Mpamhanga, C.; Osborne, S.A.; Kamal, A.; Saxty, B.; Ganley, I.G. Pharmacological inhibition of ULK1 kinase blocks mammalian target of rapamycin (mTOR)-dependent autophagy. J. Biol. Chem. 2015, 290, 11376–11383. [Google Scholar] [CrossRef] [Green Version]
- Singha, B.; Laski, J.; Valdés, Y.R.; Liu, E.; DiMattia, G.D.; Shepherd, T.G. Inhibiting ULK1 kinase decreases autophagy and cell viability in high-grade serous ovarian cancer spheroids. Am. J. Cancer Res. 2020, 10, 1384. [Google Scholar]
- Su, H.; Yang, F.; Fu, R.; Li, X.; French, R.; Mose, E.; Pu, X.; Trinh, B.; Kumar, A.; Liu, J.; et al. Cancer cells escape autophagy inhibition via NRF2-induced macropinocytosis. Cancer Cell 2021, 39, 678–693.e11. [Google Scholar] [CrossRef]
- Chen, Y.; Xie, X.; Wang, C.; Hu, Y.; Zhang, H.; Zhang, L.; Tu, S.; He, Y.; Li, Y. Dual targeting of NUAK1 and ULK1 using the multitargeted inhibitor MRT68921 exerts potent antitumor activities. Cell Death Dis. 2020, 11, 712. [Google Scholar] [CrossRef]
- Lu, J.; Zhu, L.; Zheng, L.-P.; Cui, Q.; Zhu, H.-H.; Zhao, H.; Shen, Z.-J.; Dong, H.-Y.; Chen, S.-S.; Wu, W.-Z.; et al. Overexpression of ULK1 Represents a Potential Diagnostic Marker for Clear Cell Renal Carcinoma and the Antitumor Effects of SBI-0206965. eBioMedicine 2018, 34, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Hu, P.; Yang, Z.; Xue, C.; Gong, J.; Sun, S.; Shi, L.; Zhang, S.; Li, Z.; Yang, C.; et al. SBI0206965, a novel inhibitor of Ulk1, suppresses non-small cell lung cancer cell growth by modulating both autophagy and apoptosis pathways. Oncol. Rep. 2017, 37, 3449–3458. [Google Scholar] [CrossRef] [Green Version]
- Dite, T.A.; Langendorf, C.; Hoque, A.; Galic, S.; Rebello, R.J.; Ovens, A.J.; Lindqvist, L.M.; Ngoei, K.R.; Ling, N.; Furic, L.; et al. AMP-activated protein kinase selectively inhibited by the type II inhibitor SBI-0206965. J. Biol. Chem. 2018, 293, 8874–8885. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, J.R.; Madsen, A.B.; Persson, K.W.; Henríquez-Olguín, C.; Li, Z.; Jensen, T.E. The ULK1/2 and AMPK inhibitor SBI-0206965 blocks AICAR and insulin-stimulated glucose transport. Int. J. Mol. Sci. 2020, 21, 2344. [Google Scholar] [CrossRef] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wei, M.; Xu, J.; Hua, J.; Liang, C.; Meng, Q.; Zhang, Y.; Liu, J.; Zhang, B.; Yu, X.; et al. PARP inhibitors in pancreatic cancer: Molecular mechanisms and clinical applications. Mol. Cancer 2020, 19, 49. [Google Scholar] [CrossRef] [PubMed]
- Pardo, R.; Ré, A.L.; Archange, C.; Ropolo, A.; Papademetrio, D.L.; Gonzalez, C.D.; Alvarez, E.M.; Iovanna, J.L.; Vaccaro, M.I. Gemcitabine Induces the VMP1 -Mediated Autophagy Pathway to Promote Apoptotic Death in Human Pancreatic Cancer Cells. Pancreatology 2010, 10, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, H.; Kim, M.; Xu, L.; Li, Y.; Zhang, L.; Cai, Y.; Norberg, H.V.; Zhang, T.; Furuya, T.; et al. Beclin1 Controls the Levels of p53 by Regulating the Deubiquitination Activity of USP10 and USP13. Cell 2011, 147, 223–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Luo, K.; Zhang, L.; Cheville, J.C.; Lou, Z. USP10 Regulates p53 Localization and Stability by Deubiquitinating p53. Cell 2010, 140, 384–396. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Shao, S.; Li, S.; Qin, Y.; Wang, X.; Yang, Y.; Bai, H.; Zhou, L.; Zhao, C.; Wang, C. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 2014, 44, 1661–1668. [Google Scholar] [CrossRef] [Green Version]
- Schott, C.; Ludwig, L.; Mutsaers, A.J.; Foster, R.A.; Wood, G.A. The autophagy inhibitor spautin-1, either alone or combined with doxorubicin, decreases cell survival and colony formation in canine appendicular osteosarcoma cells. PLoS ONE 2018, 13, e0206427. [Google Scholar] [CrossRef]
- Yuan, T.; Chen, Z.; Yan, F.; Qian, M.; Luo, H.; Ye, S.; Cao, J.; Ying, M.; Dai, X.; Gai, R.; et al. Deubiquitinating enzyme USP10 promotes hepatocellular carcinoma metastasis through deubiquitinating and stabilizing Smad4 protein. Mol. Oncol. 2020, 14, 197–210. [Google Scholar] [CrossRef]
- Xiao, J.; Feng, X.; Huang, X.-Y.; Huang, Z.; Huang, Y.; Li, C.; Li, G.; Nong, S.; Wu, R.; Huang, Y.; et al. Spautin-1 Ameliorates Acute Pancreatitis via Inhibiting Impaired Autophagy and Alleviating Calcium Overload. Mol. Med. 2016, 22, 643–652. [Google Scholar] [CrossRef]
- Kaushal, J.B.; Bhatia, R.; Kanchan, R.K.; Raut, P.; Mallapragada, S.; Ly, Q.P.; Batra, S.K.; Rachagani, S. Repurposing Niclosamide for Targeting Pancreatic Cancer by Inhibiting Hh/Gli Non-Canonical Axis of Gsk3β. Cancers 2021, 13, 3105. [Google Scholar] [CrossRef]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.-F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013. [Google Scholar] [CrossRef]
- Zhou, P.; Li, Y.; Li, B.; Zhang, M.; Xu, C.; Liu, F.; Bian, L.; Liu, Y.; Yao, Y.; Li, D. Autophagy inhibition enhances celecoxib-induced apoptosis in osteosarcoma. Cell Cycle 2018, 17, 997–1006. [Google Scholar] [CrossRef]
- Young, C.D.; Arteaga, C.L.; Cook, R.S. Dual inhibition of Type I and Type III PI3 kinases increases tumor cell apoptosis in HER2+ breast cancers. Breast Cancer Res. 2015, 17, 148. [Google Scholar] [CrossRef] [Green Version]
- Schluetermann, D.; Skowron, M.; Berleth, N.; Böhler, P.; Deitersen, J.; Stuhldreier, F.; Wallot-Hieke, N.; Wu, W.; Peter, C.; Hoffmann, M.J. Targeting Urothelial Carcinoma Cells by Combining Cisplatin with a Specific Inhibitor of the Autophagy-Inducing Class III Ptdins3k Complex. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 160.e1–160.e13. [Google Scholar] [CrossRef]
- Noman, M.Z.; Berchem, G.; Parpal, S.; Hasmim, M.; Janji, B. Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti–PD-1/PD-L1 immunotherapy. Sci. Adv. 2020, 6, eaax7881. [Google Scholar] [CrossRef]
- Filippakis, H.; Belaid, A.; Siroky, B.; Wu, C.; Alesi, N.; Hougard, T.; Nijmeh, J.; Lam, H.C.; Henske, E.P. Vps34-mediated macropinocytosis in Tuberous Sclerosis Complex 2-deficient cells supports tumorigenesis. Sci. Rep. 2018, 8, 14161. [Google Scholar] [CrossRef]
- Zachari, M.; Rainard, J.M.; Pandarakalam, G.C.; Robinson, L.; Gillespie, J.; Rajamanickam, M.; Hamon, V.; Morrison, A.; Ganley, I.G.; McElroy, S.P. The identification and characterisation of autophagy inhibitors from the published kinase inhibitor sets. Biochem. J. 2020, 477, 801–814. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Jiang, L.; Zhang, Z.; He, Y.; Teng, Y.; Li, J.; Yuan, S.; Pan, Y.; Liang, H.; Yang, H.; et al. Pancreatic cancer cell apoptosis is induced by a proteoglycan extracted from Ganoderma lucidum. Oncol. Lett. 2020, 21, 34. [Google Scholar] [CrossRef]
- He, R.; Shi, X.; Zhou, M.; Zhao, Y.; Pan, S.; Zhao, C.; Guo, X.; Wang, M.; Li, X.; Qin, R. Alantolactone induces apoptosis and improves chemosensitivity of pancreatic cancer cells by impairment of autophagy-lysosome pathway via targeting TFEB. Toxicol. Appl. Pharmacol. 2018, 356, 159–171. [Google Scholar] [CrossRef]
- Zheng, H.; Yang, L.; Kang, Y.; Chen, M.; Lin, S.; Xiang, Y.; Li, C.; Dai, X.; Huang, X.; Liang, G.; et al. Alantolactone sensitizes human pancreatic cancer cells to EGFR inhibitors through the inhibition of STAT3 signaling. Mol. Carcinog. 2019, 58, 565–576. [Google Scholar] [CrossRef]
- Bao, S.; Zheng, H.; Ye, J.; Huang, H.; Zhou, B.; Yao, Q.; Lin, G.; Zhang, H.; Kou, L.; Chen, R. Dual Targeting EGFR and STAT3 With Erlotinib and Alantolactone Co-Loaded PLGA Nanoparticles for Pancreatic Cancer Treatment. Front. Pharmacol. 2021, 12, 625084. [Google Scholar] [CrossRef]
- Zhu, Y.; Bu, S. Curcumin Induces Autophagy, Apoptosis, and Cell Cycle Arrest in Human Pancreatic Cancer Cells. Evid. -Based Complement. Altern. Med. 2017, 2017, 5787218. [Google Scholar] [CrossRef]
- Ali, S.; Ahmad, A.; Banerjee, S.; Padhye, S.; Dominiak, K.; Schaffert, J.M.; Wang, Z.; Philip, P.A.; Sarkar, F.H. Gemcitabine Sensitivity Can Be Induced in Pancreatic Cancer Cells through Modulation of miR-200 and miR-21 Expression by Curcumin or Its Analogue CDF. Cancer Res. 2010, 70, 3606–3617. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-H.; Chen, S.-Y.; Lu, C.-C.; Lin, J.-A.; Yen, G.-C. Ursolic acid promotes apoptosis, autophagy, and chemosensitivity in gemcitabine-resistant human pancreatic cancer cells. Phytother. Res. 2020, 34, 2053–2066. [Google Scholar] [CrossRef]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin; Ahmad, S. The Receptor for Advanced Glycation End Products: A Fuel to Pancreatic Cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Guzmán, E.A.; Pitts, T.P.; Diaz, M.C.; Wright, A.E. The marine natural product Scalarin inhibits the receptor for advanced glycation end products (RAGE) and autophagy in the PANC-1 and MIA PaCa-2 pancreatic cancer cell lines. Investig. New Drugs 2019, 37, 262–270. [Google Scholar] [CrossRef]
- Xie, G.; Sun, L.; Li, Y.; Chen, B.; Wang, C. Periplocin inhibits the growth of pancreatic cancer by inducing apoptosis via AMPK-mTOR signaling. Cancer Med. 2021, 10, 325–336. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy Is Critical for Pancreatic Tumor Growth and Progression in Tumors with p53 Alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Xu, X.; Zhou, S.; Chen, Y.; Ding, G.; Cao, L. Fisetin induces autophagy in pancreatic cancer cells via endoplasmic reticulum stress- and mitochondrial stress-dependent pathways. Cell Death Dis. 2019, 10, 317. [Google Scholar] [CrossRef]
- Murtaza, I.; Adhami, V.M.; Hafeez, B.B.; Saleem, M.; Mukhtar, H. Fisetin, a natural flavonoid, targets chemoresistant human pancreatic cancer AsPC-1 cells through DR3-mediated inhibition of NF-κB. Int. J. Cancer 2009, 125, 2465–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
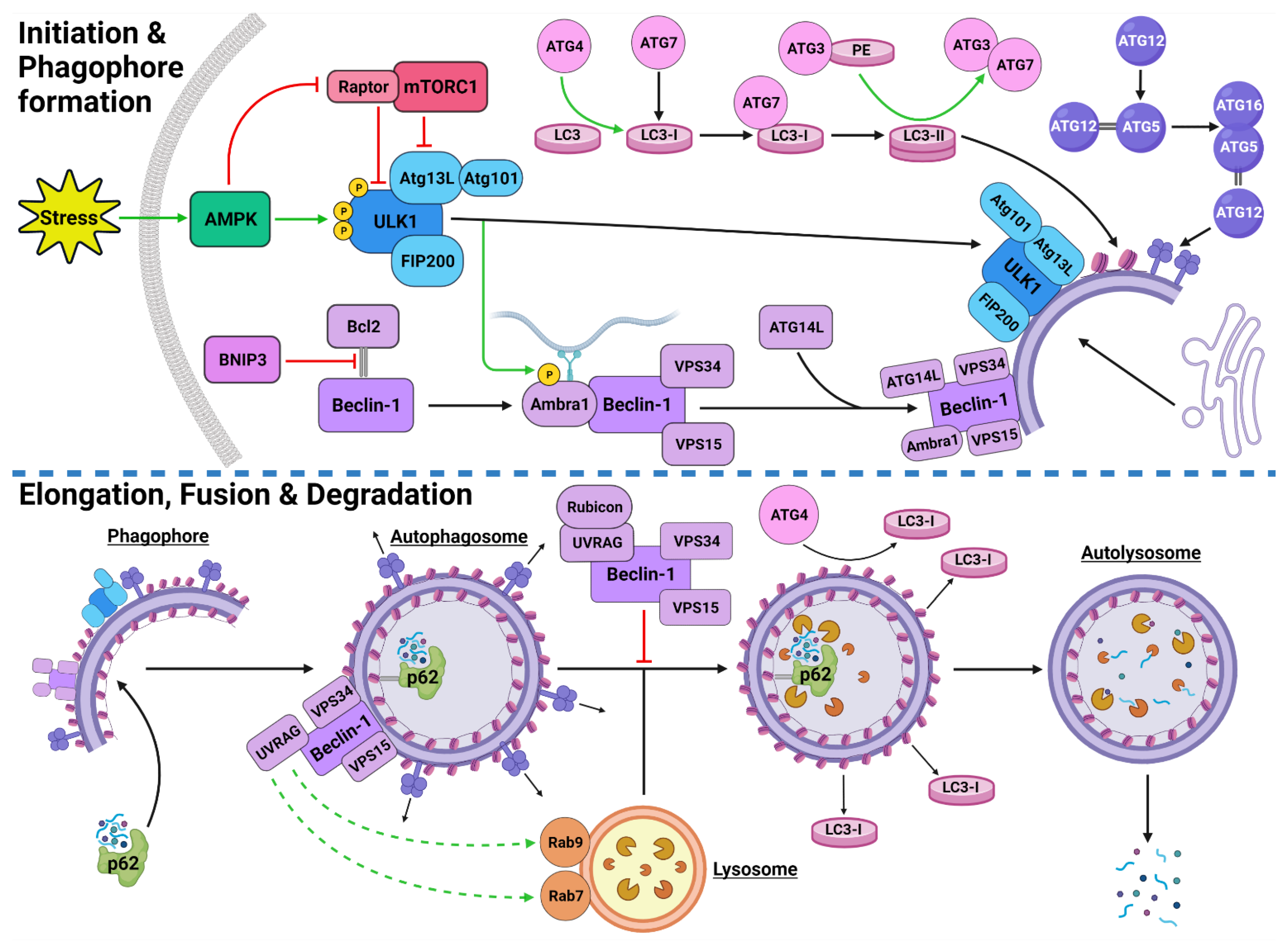

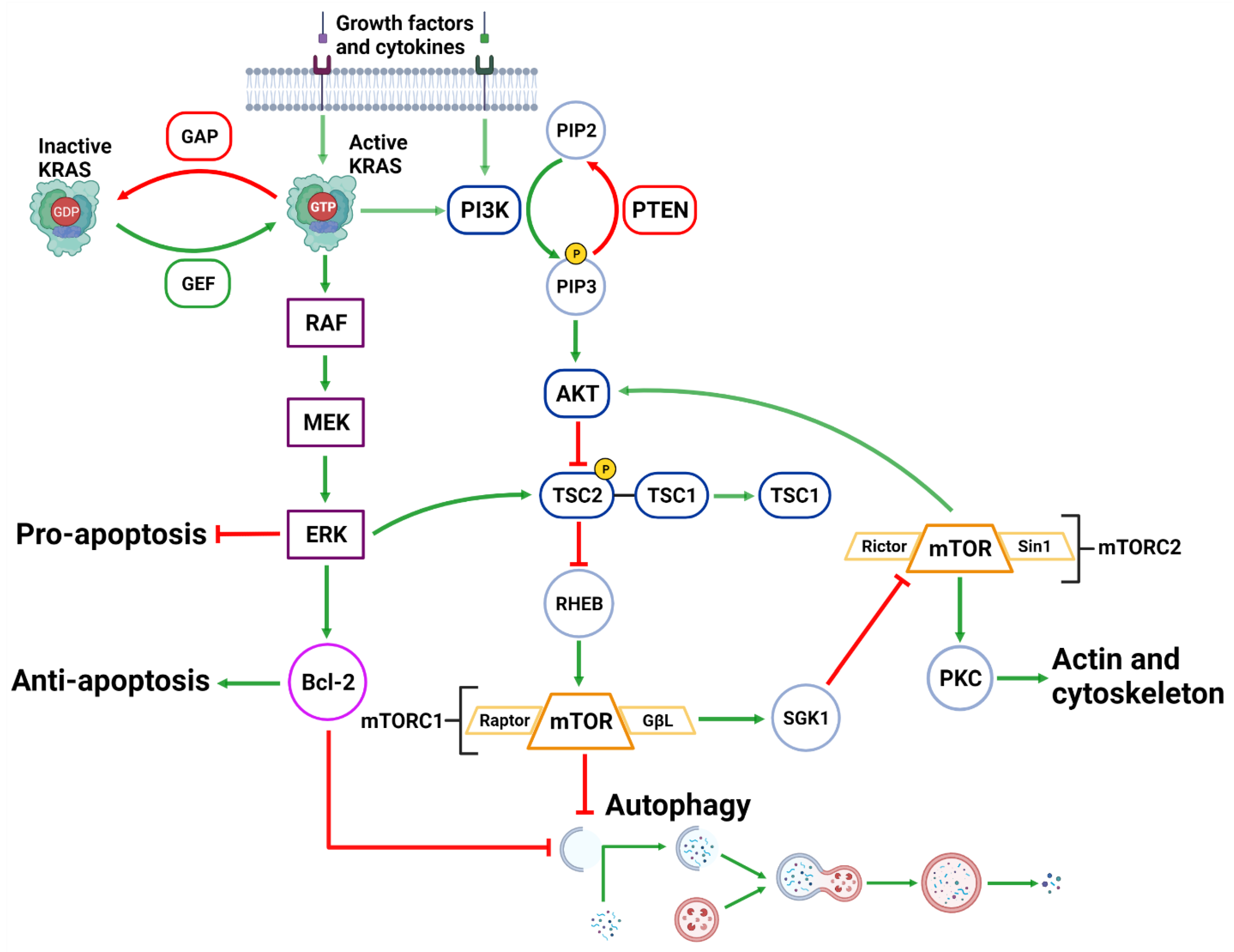

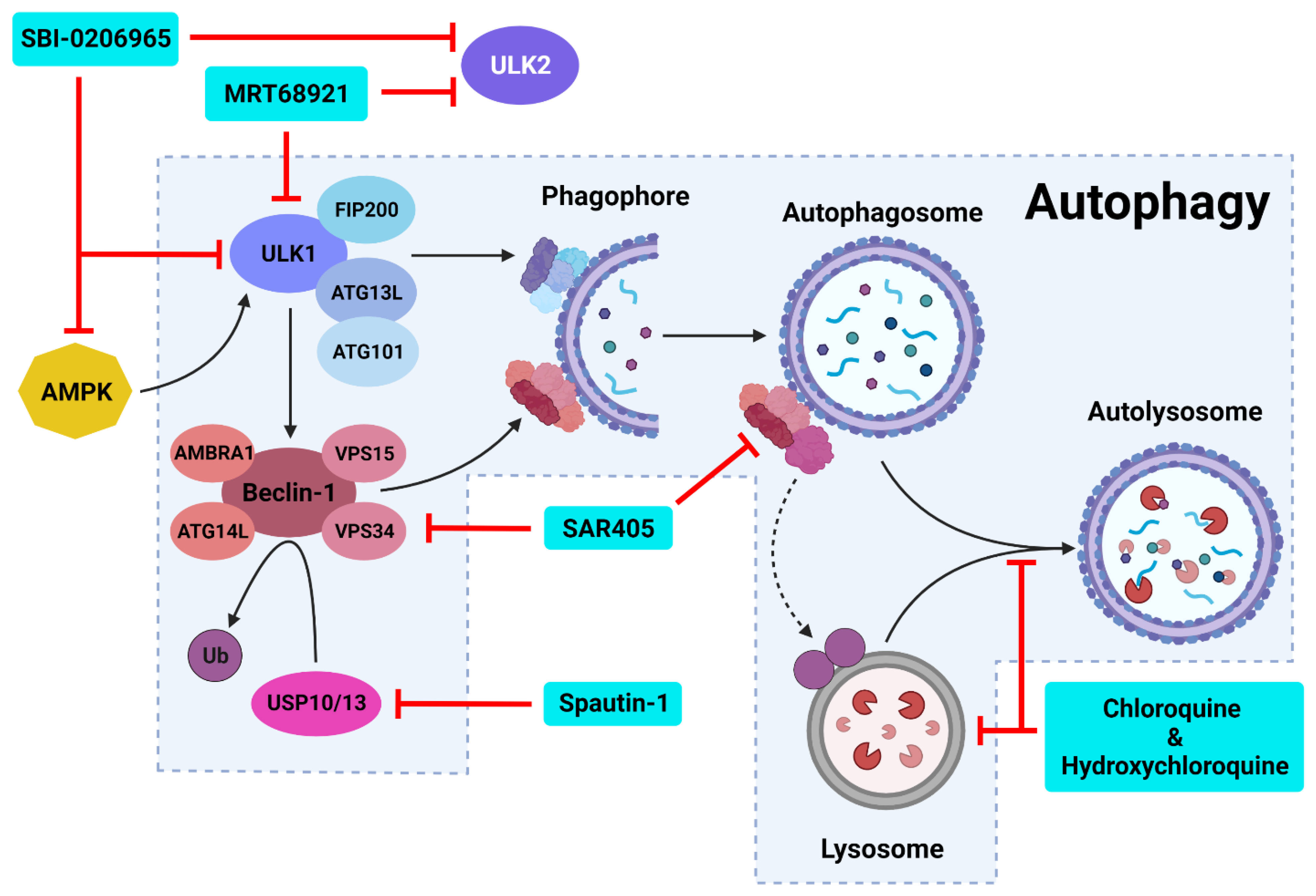

{kind=link}
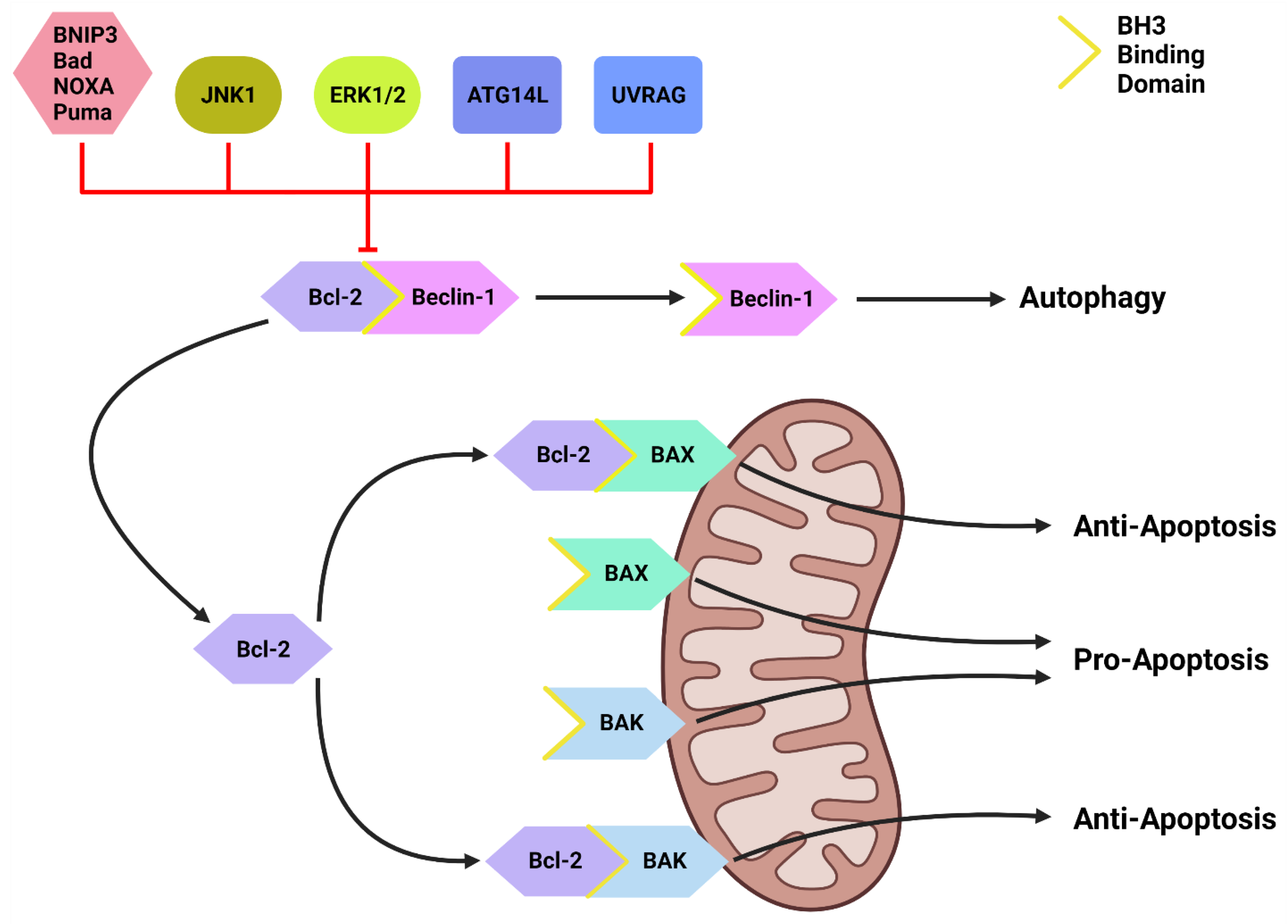{kind=link}
{kind=link}
{kind=link}
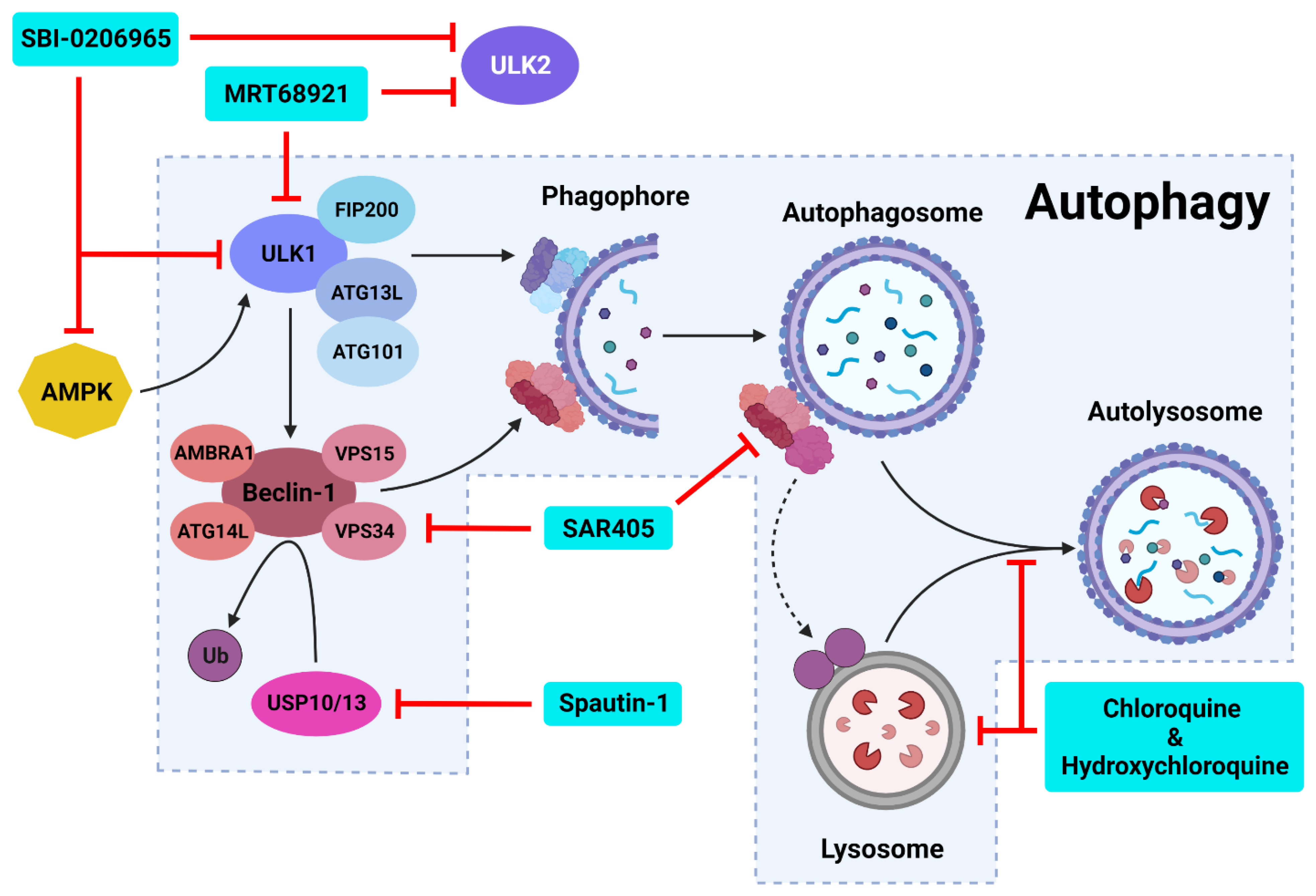{kind=link}
| Type of PC | Study Design | Drug and Dose | Status | Serial No. |
|---|---|---|---|---|
| Metastatic PDAC | Phase 2, Non-randomized, Open label | 400 mg HCQ OR 600 mg HCQ, BID for 4 weeks | Completed | NCT01273805 |
| Inoperable locally advanced and metastatic PC | Phase 1, Open label, | mFOLFIRINOX (backbone) + 250 mg chlorphenesin carbamate + 200 mg HCQ, BID for 48 weeks | Recruiting | NCT05083780 |
| Resectable PC | Phase 2, Open label | Photon/proton radiation during week 2 for 5 days + 825 mg/m2 capecitabine BID for 10 days + 400 mg HCQ BID from day 1 until surgery | Active not recruiting | NCT01494155 |
| Metastatic PDAC, stage IV PC | Phase 1 pilot, Open label, | Binimetinib + HCQ, BID for 2 weeks | Recruiting | NCT04132505 |
| Advanced PDAC, metastatic PDAC, stage IV PC | Phase 2, Open label | Paricalcitol three times weekly + HCQ BID + gemcitabine weekly + nab-paclitaxel 30 min weekly | Recruiting | NCT04524702 |
| Metastatic PDAC, Stage II, Stage IIA, Stage IIB, Stage III, Stage IV PC, Unresectable PDAC | Phase 1, Open label | Trametinib QD + HCQ QD or BID for 4 weeks | Recruiting | NCT03825289 |
| Advanced PDAC Metastatic PDAC | Phase 1/2, Randomized, Open label | Gemcitabine 1000 mg/m2 weekly + nab-paclitaxel 125 mg/m2 weekly OR gemcitabine 1000 mg/m2 weekly + nab-paclitaxel 125 mg/m2 weekly + HCQ 600 mg/m2 BID, for 15 days | Completed | NCT01506973 |
| Pancreatic cancer | Phase 2, Randomized, Open label | Gemcitabine 1000 mg/m2 + nab-paclitaxel 125 mg/m2, weekly for 45 days OR gemcitabine 1000 mg/m2 + nab-paclitaxel 125 mg/m, weekly for 45 days + HCQ 600 mg/m2 QD or BID until surgery | Completed | NCT01978184 |
| Pancreatic cancer | Phase 1/2, Open label | Gemcitabine 10 mg/m2/min (dependent on dose) on day 1 and 15 + HCQ 200, 400, 600, 800, 1000, or 1200 mg BID for 31 days | Completed | NCT01128296 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gillson, J.; Abd El-Aziz, Y.S.; Leck, L.Y.W.; Jansson, P.J.; Pavlakis, N.; Samra, J.S.; Mittal, A.; Sahni, S. Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers 2022, 14, 3528. https://doi.org/10.3390/cancers14143528
Gillson J, Abd El-Aziz YS, Leck LYW, Jansson PJ, Pavlakis N, Samra JS, Mittal A, Sahni S. Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers. 2022; 14(14):3528. https://doi.org/10.3390/cancers14143528
Chicago/Turabian StyleGillson, Josef, Yomna S. Abd El-Aziz, Lionel Y. W. Leck, Patric J. Jansson, Nick Pavlakis, Jaswinder S. Samra, Anubhav Mittal, and Sumit Sahni. 2022. "Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target" Cancers 14, no. 14: 3528. https://doi.org/10.3390/cancers14143528
APA StyleGillson, J., Abd El-Aziz, Y. S., Leck, L. Y. W., Jansson, P. J., Pavlakis, N., Samra, J. S., Mittal, A., & Sahni, S. (2022). Autophagy: A Key Player in Pancreatic Cancer Progression and a Potential Drug Target. Cancers, 14(14), 3528. https://doi.org/10.3390/cancers14143528