
Conjoined Genes as Common Events in Childhood Acute Lymphoblastic Leukemia

 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
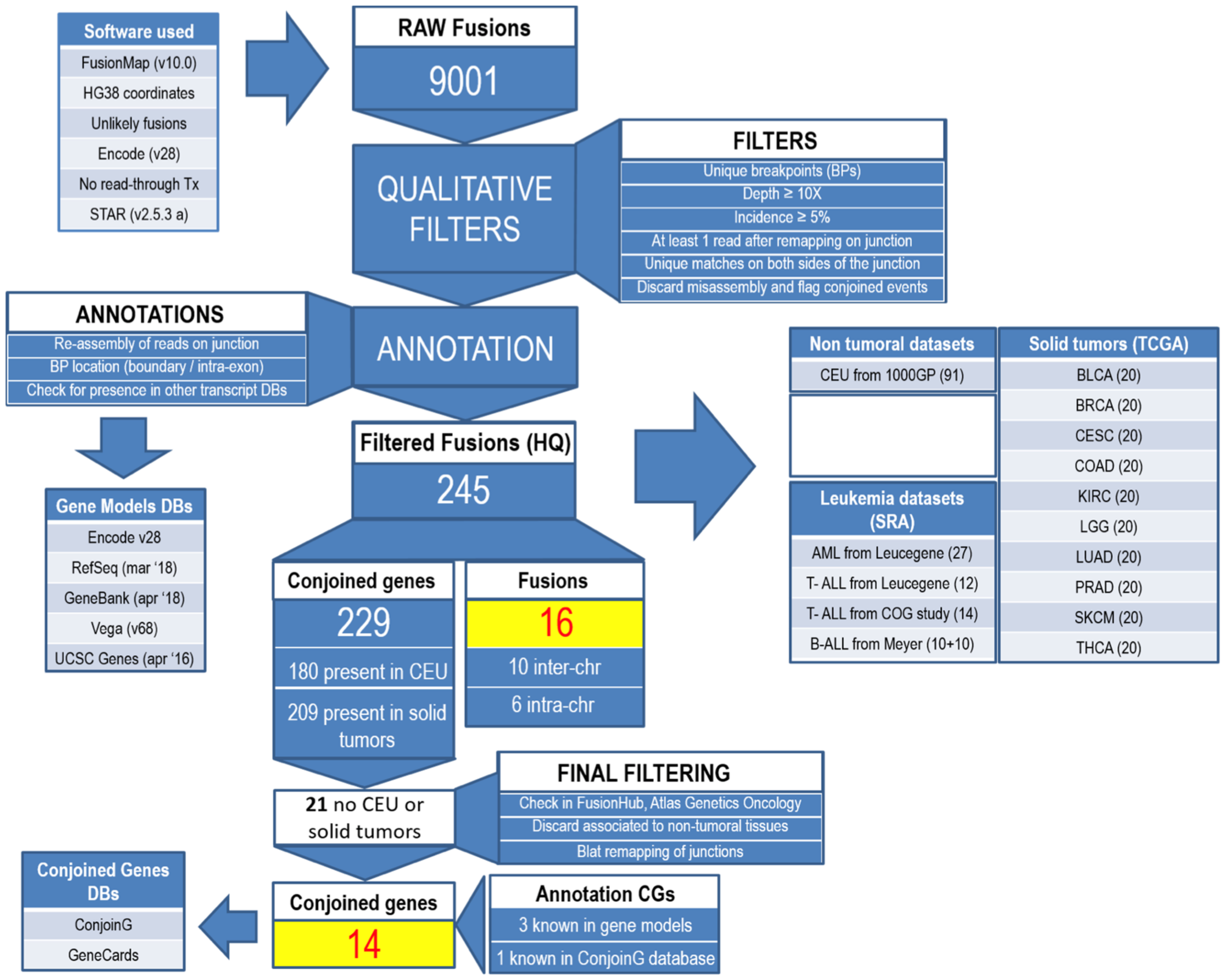
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inaba, H.; Mullighan, C.G. Pediatric Acute Lymphoblastic Leukemia. Hematologica 2020, 105, 2524–2539. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Mullighan, C.G.; Evans, W.E.; Relling, M.V. Pediatric acute lymphoblastic leukemia: Where are we going and how do we get there? Blood 2012, 120, 1165–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhojwani, D.; Yang, J.J.; Pui, C.H. Biology of Childhood Acute Lymphoblastic Leukemia. Pediatr. Clin. North Am. 2015, 62, 47–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, K.R.; Pullen, D.J.; Sather, H.N.; Shuster, J.J.; Devidas, M.; Borowitz, M.J.; Carroll, A.J.; Heerema, N.A.; Rubnitz, J.E.; Loh, M.L.; et al. Risk- and response-based classification of childhood B- precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG). Blood 2007, 109, 926–935. [Google Scholar] [CrossRef] [Green Version]
- Chouvarine, P.; Antic, Z.; Lentes, J.; Schröder, C.; Alten, J.; Brüggemann, M.; Carrillo-de Santa Pau, E.; Illig, T.; Laguna, T.; Schewe, D.; et al. Transcriptional and Mutational Profiling of B-Other Acute Lymphoblastic Leukemia for Improved Diagnostics. Cancers 2021, 13, 5653. [Google Scholar] [CrossRef]
- Li, J.F.; Dai, Y.T.; Lilljebjörn, H.; Shen, S.H.; Cui, B.W.; Bai, L.; Liu, Y.F.; Qian, M.X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1223 cases. Proc. Natl. Acad. Sci. USA 2018, 115, E11711–E11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.R.; Li, Z.; Tai, S.T.; Oh, B.L.Z.; Yeoh, A.E.J. Genetic Alterations in Childhood Acute Lymphoblastic Leukemia: Interactions with Clinical Features and Treatment Response. Cancers 2021, 13, 4068. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.N.; Kim, A.; Choi, S.H.; Kim, D.S.; Nam, S.H.; Kim, D.W.; Kim, D.W.; Kang, A.; Kim, M.Y.; Park, K.H.; et al. Novel mechanism of conjoined gene formation in the human genome. Funct. Integr. Genom. 2012, 12, 45–61. [Google Scholar] [CrossRef]
- Kim, D.S.; Kim, D.W.; Kim, M.Y.; Nam, S.H.; Choi, S.H.; Kim, R.N.; Kang, A.; Kim, A.; Park, H.S. CACG: A database for comparative analysis of conjoined genes. Genomics 2012, 100, 14–17. [Google Scholar] [CrossRef] [Green Version]
- Prakash, T.; Sharma, V.K.; Adati, N.; Ozawa, R.; Kumar, N.; Nishida, Y.; Fujikake, T.; Takeda, T.; Taylor, T.D. Expression of conjoined genes: Another mechanism for gene regulation in eukaryotes. PLoS ONE 2010, 5, e13284. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Xie, Z.; Li, H. Intergenically Spliced Chimeric RNAs in Cancer. Trends Cancer 2016, 2, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufflé, F.; Audoux, J.; Boureux, A.; Beaumeunier, S.; Gaillard, J.B.; Samra, E.B.; Megarbane, A.; Cassinat, B.; Chomienne, C.; Alves, R.; et al. New chimeric RNAs in acute myeloid leukemia. F1000Research 2017, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, Y.; Tsukamoto, T.; Chinen, Y.; Shimura, Y.; Sasaki, N.; Nagoshi, H.; Sato, R.; Adachi, H.; Nakano, M.; Horiike, S.; et al. Detection of novel and recurrent conjoined genes in non-Hodgkin B-cell lymphoma. J. Clin. Exp. Hematop. 2021, 61, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Liu, K.; Juan, T.; Fang, F.; Newman, M.; Hoeck, W. FusionMap: Detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics 2011, 27, 1922–1928. [Google Scholar] [CrossRef] [Green Version]
- Panigrahi, P.; Jere, A.; Anamika, K. FusionHub: A unified web platform for annotation and visualization of gene fusion events in human cancer. PLoS ONE 2018, 13, e0196588. [Google Scholar] [CrossRef] [PubMed]
- Huret, J.L.; Ahmad, M.; Arsaban, M.; Bernheim, A.; Cigna, J.; Desangles, F.; Guignard, J.C.; Jacquemot-Perbal, M.C.; Labarussias, M.; Leberre, V.; et al. Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res. 2013, 41, D920–D924. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Dupain, C.; Harttrampf, A.C.; Urbinati, G.; Geoerger, B.; Massaad-Massade, L. Relevance of Fusion Genes in Pediatric Cancers: Toward Precision Medicine. Mol. Ther. Nucleic Acids 2017, 6, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Fazio, G.; Daniele, G.; Cazzaniga, V.; Impera, L.; Severgnini, M.; Iacobucci, I.; Galbiati, M.; Leszl, A.; Cifola, I.; De Bellis, G.; et al. Three novel fusion transcripts of the paired box 5 gene in B-cell precursor acute lymphoblastic leukemia. Haematologica 2015, 100, e14–e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltri, G.; Sandei, M.; Silvestri, D.; Bresolin, S.; Pession, A.; Santoro, N.; Ziino, O.; Veltroni, M.; Rizzari, C.; Biffi, A.; et al. NUP214-ABL1 fusion in childhood T-ALL. Pediatr. Blood Cancer 2022, e29643. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; Saint-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022, 6, 1329–1341. [Google Scholar] [CrossRef]
- Eyre, T.; Schwab, C.J.; Kinstrie, R.; McGuire, A.K.; Strefford, J.; Peniket, A.; Mead, A.; Littlewood, T.; Holyoake, T.L.; Copland, M.; et al. Episomal amplification of NUP214-ABL1 fusion gene in B-cell acute lymphoblastic leukemia. Blood 2012, 120, 4441–4443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duployez, N.; Grzych, G.; Ducourneau, B.; Fuentes, M.A.; Grardel, N.; Boyer, T.; Chahla, W.A.; Bruno, B.; Nelken, B.; Clappier, E.; et al. NUP214-ABL1 fusion defines a rare subtype of B-cellprecursor acute lymphoblasticleukemiathatcould benefit from tyrosinekinaseinhibitors. Haematologica 2016, 101, e133–e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Wang, Q.; Torres-Garcia, W.; Zheng, S.; Vegesna, R.; Kim, H.; Verhaak, R.G. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 2015, 34, 4845–4854. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar] [CrossRef]
- Chase, A.; Ernst, T.; Fiebig, A.; Collins, A.; Grand, F.; Erben, P.; Reiter, A.; Schreiber, S.; Cross, N.C. TFG, a target of chromosome translocations in lymphoma and soft tissue tumors, fuses to GPR128 in healthy individuals. Haematologica 2010, 9, 20–26. [Google Scholar] [CrossRef]
- Yuan, H.; Qin, F.; Movassagh, M.; Park, H.; Golden, W.; Xie, Z.; Zhang, P.; Sklar, J.; Li, H. A chimeric RNA characteristic of rhabdomyosarcoma in normal myogenesis Process. Cancer Discov. 2013, 3, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.S.; Yu, C.Y.; Chuang, C.Y.; Hsiao, M.; Kao, C.F.; Kuo, H.C.; Chuang, T.J. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014, 24, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Barresi, V.; Cosentini, I.; Scuderi, C.; Napoli, S.; Di Bella, V.; Spampinato, G.; Condorelli, D.F. Fusion Transcripts of Adjacent Genes: New Insights into the World of Human Complex Transcripts in Cancer. Int. J. Mol. Sci. 2019, 20, 5252. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.D.; Paculova, H.; Kogut, S.; Heath, J.; Schjerven, H.; Frietze, S. Non-Coding RNA Signatures of B-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2021, 22, 2683. [Google Scholar] [CrossRef]
- Nagoshi, H.; Taki, T.; Hanamura, I.; Nitta, M.; Otsuki, T.; Nishida, K.; Okuda, K.; Sakamoto, N.; Kobayashi, S.; Yamamoto-Sugitani, M.; et al. Frequent PVT1 rearrangement and novel chimeric genes PVT1-NBEA and PVT1- WWOX occur in multiple myeloma with 8q24 Abnormality. Cancer Res. 2012, 72, 4954–4962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinen, Y.; Sakamoto, N.; Nagoshi, H.; Taki, T.; Maegawa, S.; Tatekawa, S.; Tsukamoto, T.; Mizutani, S.; Shimura, Y.; Yamamoto-Sugitani, M.; et al. 8q24 amplified segments involve novel fusion genes between NSMCE2 and long noncoding RNAs in acute myelogenous leukemia. J. Hematol. Oncol. 2014, 7, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Code | Age | Sex | Phenotype | BM Blasts % | Translocations | DNA Index | Steroid Response | MRD Risk Stratification |
|---|---|---|---|---|---|---|---|---|
| #8 | 16 | M | BII | 86 | NEG | 1 | PGR | HR |
| #11 | 15 | M | BII | 80 | NEG | 1 | PGR | HR |
| #12 | 3 | F | BI | 73 | NEG | 1 | PGR | HR |
| #13 | 4 | F | BIII | 78 | NEG | 1 | PGR | HR |
| #15 | 9 | F | BII | 74 | NEG | 1 | PGR | HR |
| #16 | 11 | M | BIII | 90 | NEG | 1 | PGR | HR |
| #6 | 1 | M | BII | 94 | NEG | 1 | PGR | SR |
| #7 | 5 | M | BII | 70 | NEG | 1 | PGR | SR |
| #9 | 15 | M | BII | 94 | NEG | 1 | PGR | SR |
| #14 | 8 | M | BII/BIII | 90 | NEG | 1 | PGR | SR |
| Conjoined Genes | Gene Orientation * | 5′ Gene | 3′ Gene | 5′ Breakpoint | 3′ Breakpoint | Annotation | Our ALL Cohort (Case ID#) | AML ** | T-ALL (Leucegene) ** | T-ALL (COG) ** | B-ALL** | TCGA Solid Tumors *** |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| LRRC61::ZBED6CL | +/+ | LRRC61 | ZBED6CL | 7:150328742 | 7:150330107 | - | 1/10 (#14) | 17/27 | 8/12 | 6/14 | 0/20 | THCA (1/20) |
| ZNF250::COMMD5 | +/+ | ZNF250 | COMMD5 | 8:144880365 | 8:144851395 | GenBank | 1/10 (#14) | 15/27 | 9/12 | 4/14 | 4/20 | LGG (1/20), PRAD (1/20) |
| RP11-305L7.3::RP11-305L7.1 | +/+ | RP11-305L7.3 | RP11-305L7.1 | 9:91163005 | 9:91106671 | RefSeq | 1/10 (#13) | 10/27 | 8/12 | 3/14 | 1/20 | BLCA (1/20), BRCA (1/20), SKCM (1/20) |
| RP11-556E13.1::RP11-346D6.6 | +/+ | RP11-556E13.1 | RP11-346D6.6 | 10:52561006 | 10:52463332 | 1/10 (#11) | 9/27 | 2/12 | 0/14 | 0/20 | LUAD (1/20), SKCM (1/20) | |
| OPN4::LDB3 | +/+ | OPN4 | LDB3 | 10:86666848 | 10:86668669 | GenBank, RefSeq | 1/10 (#16) | 0/27 | 0/12 | 0/14 | 2/20 | BRCA (1/20), CESC (1/20), KIRC (6/20), LGG (9/20), SKCM (1/20) |
| C14orf37::RP11-999E24.3 | +/+ | C14orf37 | RP11-999E24.3 | 14:58003907 | 14:57994417 | RefSeq | 1/10 (#13) | 2/27 | 0/12 | 0/14 | 0/20 | KIRC (1/20), LGG (1/20) |
| RP11-1360M22.11::RP11-810K23.8 | +/+ | RP11-1360M22.11 | RP11-810K23.8 | 15:20012876 | 15:21019524 | - | 1/10 (#13) | 5/27 | 3/12 | 4/14 | 0/20 | KIRC (1/20), LGG (1/20) |
| IGF1R::RP11-35O15.2 | +/+ | IGF1R | RP11-35O15.2 | 15:98649675 | 15:98660230 | GenBank | 2/10 (#6,#7) | 8/27 | 3/12 | 0/14 | 3/20 | BRCA (1/20), CESC (2/20), LGG (1/20) |
| NHLRC4::PIGQ | +/+ | NHLRC4 | PIGQ | 16:568989 | 16:574066 | Ensembl, UCSC, Vega, ConjoinG, FusionHub | 2/10 (#7,#8) | 12/27 | 7/12 | 6/14 | 10/20 | BRCA (2/20), COAD (2/20), KIRC (1/20) |
| MAPK3::GDPD3 | +/+ | MAPK3 | GDPD3 | 16:30114203 | 16:30113437 | - | 1/10 (#11) | 10/27 | 5/12 | 4/14 | 4/20 | BRCA (1/20), LGG (1/20) |
| TRAPPC1::KCNAB3 | +/+ | TRAPPC1 | KCNAB3 | 17:7930527 | 17:7927826 | ConjoinG, FusionHub | 1/10 (#11) | 0/27 | 1/12 | 2/14 | 0/20 | CESC (2/20) |
| Conjoined Genes | Gene Orientation * | 5′ Gene | 3′ Gene | 5′ Breakpoint | 3′ Breakpoint | Annotation | Our ALL Cohort (Case Id#) | AML ** | T-ALL (Leucegene) ** | T-ALL (COG) ** | B-ALL ** |
|---|---|---|---|---|---|---|---|---|---|---|---|
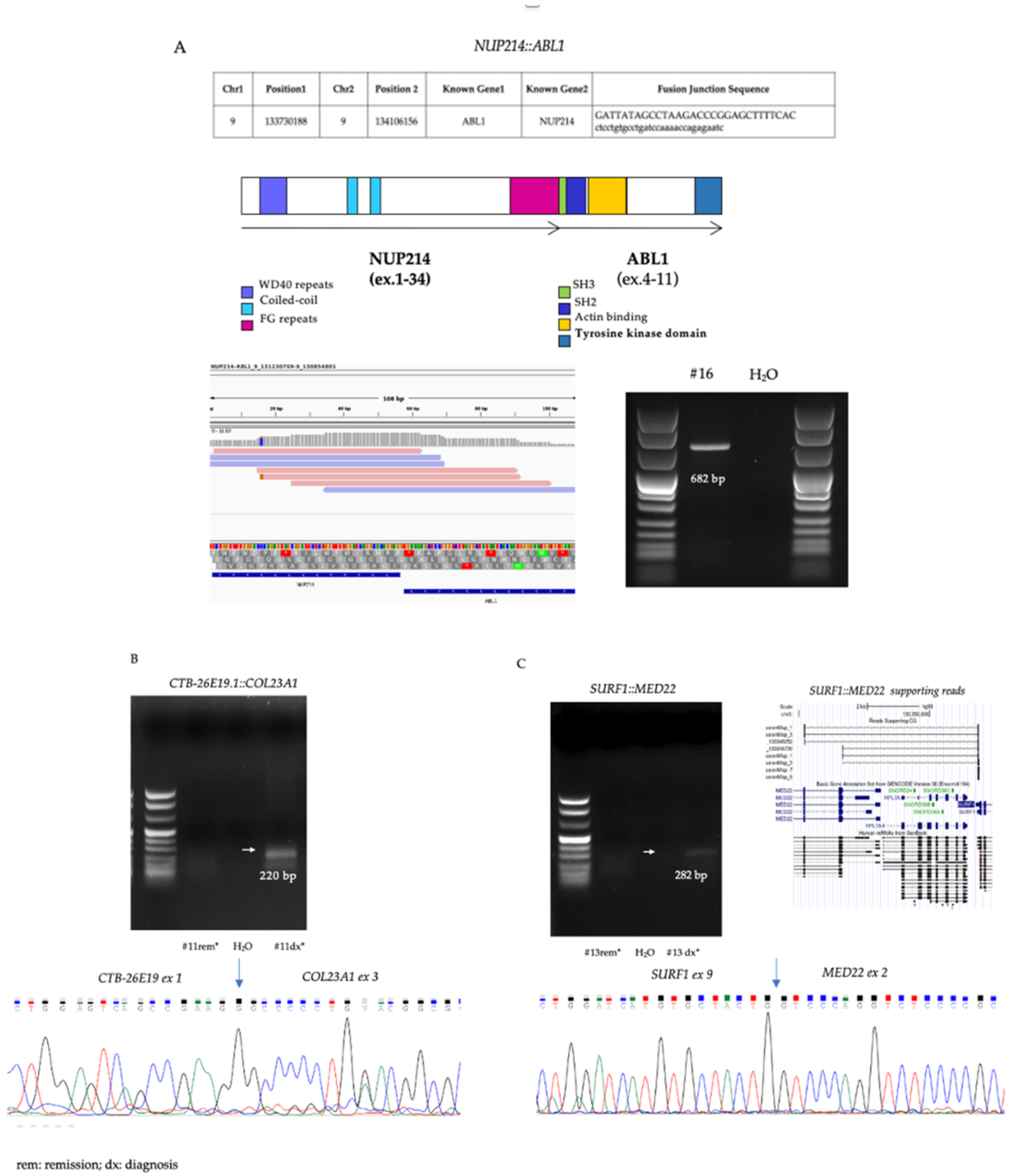
| CTB-26E19.1::COL23A1 | +/+ | CTB-26E19.1 | COL23A1 | 5:178442931 | 5:178306919 | - | 1/10 (#11) | 2/27 | 7/12 | 7/14 | 3/20 |
| SURF1::MED22 | +/+ | SURF1 | MED22 | 9:133351836 | 9:133345252 | - | 2/10 (#6,#7) | 1/27 | 2/12 | 0/14 | 0/20 |
| SURF1::MED22 | +/+ | SURF1 | MED22 | 9:133351836 | 9:133346700 | - | 1/10 (#13) | 6/27 | 4/12 | 0/14 | 1/20 |
| CACUL1::RP11-427L15.2 | +/+ | CACUL1 | RP11-427L15.2 | 10:118693702 | 10:118692438 | - | 2/10 (#7,#11) | 3/27 | 1/12 | 0/14 | 1/20 |
| TMEM86A::RP11-1081L13.4 | +/+ | TMEM86A | RP11-1081L13.4 | 11:18702116 | 11:18706818 | - | 1/10 (#7) | 1/27 | 1/12 | 1/14 | 0/20 |
| RP11-20D14.3::RIMKLB | +/+ | RP11-20D14.3 | RIMKLB | 12:8668831 | 12:8713811 | - | 9/10 (#6,#7,#9,#8,#11,#12,#13,#15,#16) | 17/27 | 1/12 | 1/14 | 16/20 |
| RP11-397H6.1::RP11-541G9.1 | +/+ | RP11-397H6.1 | RP11-541G9.1 | 12:97033423 | 12:97185076 | - | 1/10 (#13) | 0/27 | 0/12 | 0/14 | 0/20 |
| CCPG1::PIGBOS1 | +/+ | CCPG1 | PIGBOS1 | 15:55355634 | 15:55317867 | - | 2/10 (#6,#13) | 14/27 | 2/12 | 6/14 | 0/20 |
| AC019118.3::AC019118.2 | +/+ | AC019118.3 | AC019118.2 | 2:3145547 | 2:2966675 | - | 2/10 (#14,#16) | 0/27 | 1/12 | 0/14 | 4/20 |
| RP11-87G24.3::RP11-87G24.6 | +/+ | RP11-87G24.3 | RP11-87G24.6 | 17:76963893 | 17:76957657 | - | 6/10 (#7,#8,#12,#13,#14,#16) | 3/27 | 9/12 | 6/14 | 6/20 |
| KLHL22::SCARF2 | +/+ | KLHL22 | SCARF2 | 22:20446443 | 22:20430557 | Ensembl, UCSC, Vega, ConjoinG, FusionHub | 4/10 (#9,#12,#13,#15) | 14/27 | 8/12 | 5/14 | 4/20 |
| PPP1R3F::LL0XNC01-7P3.1 | +/+ | PPP1R3F | LL0XNC01-7P3.1 | X:49270873 | X:49273514 | GenBank | 1/10 (#7) | 2/27 | 0/12 | 0/14 | 0/20 |
| MYNN::RP11-362K14.7 | +/+ | MYNN | RP11-362K14.7 | 3:169786730 | 3:169793627 | - | 2/10 (#8,#14) | 8/27 | 4/12 | 4/14 | 0/20 |
| FAM200B::BST1 | +/+ | FAM200B | BST1 | 4:15687148 | 4:15711807 | RefSeq | 2/10 (#13,#14) | 3/27 | 2/12 | 0/14 | 0/20 |
| Fusions | Gene Orientation * | 5′ Gene | 3′ Gene | 5′ Breakpoint | 3′ Breakpoint | Annotation | Our ALL Cohort (Case ID#) | AML ** | T-ALL (Leucegene) ** | T-ALL (COG) ** | B-ALL ** |
|---|---|---|---|---|---|---|---|---|---|---|---|
| IK::FBXW2 | +/− | IK | FBXW2 | 5:140659164 | 9:120792947 | - | 1/10 (#13) | 0/27 | 0/12 | 0/14 | 0/20 |
| ZNF444::HLA-B | +/− | ZNF444 | HLA-B | 19:56160570 | 6:31355372 | - | 1/10 (#9) | 1/27 | 3/12 | 0/14 | 0/20 |
| PAX5::POM121C | +/+ | PAX5 | POM121C | 9:36966549 | 7:75441583 | - | 1/10 (#6) | 0/27 | 0/12 | 0/14 | 0/20 |
| NFX1::DICER1 | +/− | NFX1 | DICER1 | 9:33290597 | 14:95141748 | - | 1/10 (#9) | 0/27 | 0/12 | 0/14 | 0/20 |
| DCAF8::ZNF836 | −/− | DCAF8 | ZNF836 | 1:160220067 | 19:52156511 | - | 1/10 (#14) | 0/27 | 0/12 | 0/14 | 0/20 |
| DMD::STAMBPL1 | −/+ | DMD | STAMBPL1 | X:32216916 | 10:88893871 | - | 1/10 (#13) | 0/27 | 0/12 | 0/14 | 0/20 |
| SLFNL1::SMPD2 | −/+ | SLFNL1 | SMPD2 | 1:41022123 | 6:109441113 | - | 1/10 (#16) | 0/27 | 0/12 | 0/14 | 0/20 |
| RP11-148O21.2::ATG4B | −/+ | RP11-148O21.2 | ATG4B | 8:11558702 | 2:241672559 | - | 1/10 (#7) | 0/27 | 0/12 | 0/14 | 0/20 |
| TMEM263::CD47 | +/− | TMEM263 | CD47 | 12:106971137 | 3:108055566 | - | 1/10 (#11) | 0/27 | 0/12 | 0/14 | 0/20 |
| INPP5A::SETD7 | +/− | INPP5A | SETD7 | 10:132607956 | 4:139548119 | - | 1/10 (#7) | 0/27 | 0/12 | 0/14 | 0/20 |
| ZC3H12D::RP11-445F6.2 | −/+ | ZC3H12D | RP11-445F6.2 | 6:149456666 | 6:139271659 | - | 1/10 (#14) | 0/27 | 0/12 | 0/14 | 0/20 |
| NUP214::ABL1 | −/− | NUP214 | ABL1 | 9:131230769 | 9:130854801 | FusionHub, Atlas Genetics Oncology | 1/10 (#16) | 0/27 | 0/12 | 0/14 | 0/20 |
| MAML2::FAT3 | −/+ | MAML2 | FAT3 | 11:96091892 | 11:92352096 | - | 1/10 (#15) | 0/27 | 0/12 | 0/14 | 0/20 |
| MNT::CLUH | −/− | MNT | CLUH | 17:2400640 | 17:2704564 | - | 2/10 (#9,#11) | 3/27 | 2/12 | 0/14 | 0/20 |
| TSKS::ARRDC2 | −/+ | TSKS | ARRDC2 | 19:49746470 | 19:18007338 | - | 1/10 (#11) | 0/27 | 0/12 | 0/14 | 0/20 |
| MAEA::CTBP1 | +/− | MAEA | CTBP1 | 4:1289982 | 4:1238337 | FusionHub, Atlas Genetics Oncology | 2/10 (#7,#12) | 9/27 | 3/12 | 6/14 | 2/20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Severgnini, M.; D’Angiò, M.; Bungaro, S.; Cazzaniga, G.; Cifola, I.; Fazio, G. Conjoined Genes as Common Events in Childhood Acute Lymphoblastic Leukemia. Cancers 2022, 14, 3523. https://doi.org/10.3390/cancers14143523
Severgnini M, D’Angiò M, Bungaro S, Cazzaniga G, Cifola I, Fazio G. Conjoined Genes as Common Events in Childhood Acute Lymphoblastic Leukemia. Cancers. 2022; 14(14):3523. https://doi.org/10.3390/cancers14143523
Chicago/Turabian StyleSevergnini, Marco, Mariella D’Angiò, Silvia Bungaro, Giovanni Cazzaniga, Ingrid Cifola, and Grazia Fazio. 2022. "Conjoined Genes as Common Events in Childhood Acute Lymphoblastic Leukemia" Cancers 14, no. 14: 3523. https://doi.org/10.3390/cancers14143523
APA StyleSevergnini, M., D’Angiò, M., Bungaro, S., Cazzaniga, G., Cifola, I., & Fazio, G. (2022). Conjoined Genes as Common Events in Childhood Acute Lymphoblastic Leukemia. Cancers, 14(14), 3523. https://doi.org/10.3390/cancers14143523