
ERK5 Is a Major Determinant of Chemical Sarcomagenesis: Implications in Human Pathology

 , , , , , ,
, , , , , ,  , , ,
, , ,  , and add
Show full author list
, and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Antibodies and Chemicals
2.3. Animal Studies
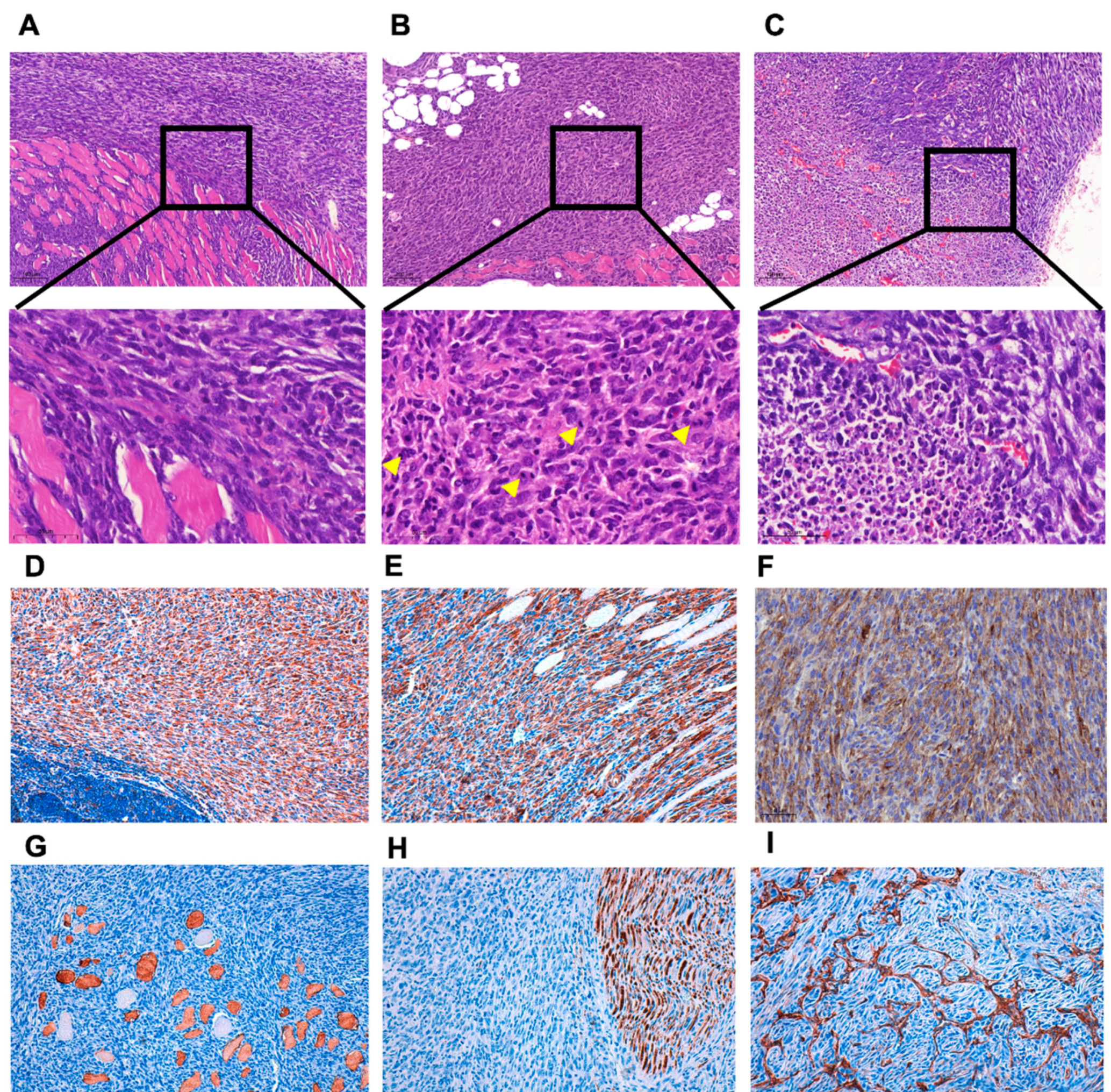
2.4. Induction of Murine Sarcoma and Cell Lines Derivation
2.5. Xenograft Assays
2.6. RNA Interference Assays, Lentiviral Production, and Infections
2.7. Western Blotting
2.8. Clonogenic Assay
2.9. Growth Curves
2.10. Adhesion Assay
2.11. RNA Isolation, Reverse Transcription, and Real-Time Quantitative PCR (RT-qPCR)
2.12. Histology and Immunohistochemistry
2.13. RNA Sequencing
2.14. Functional Enrichment Analysis
2.15. In Silico Analysis
2.16. Statistical Analysis
3. Results
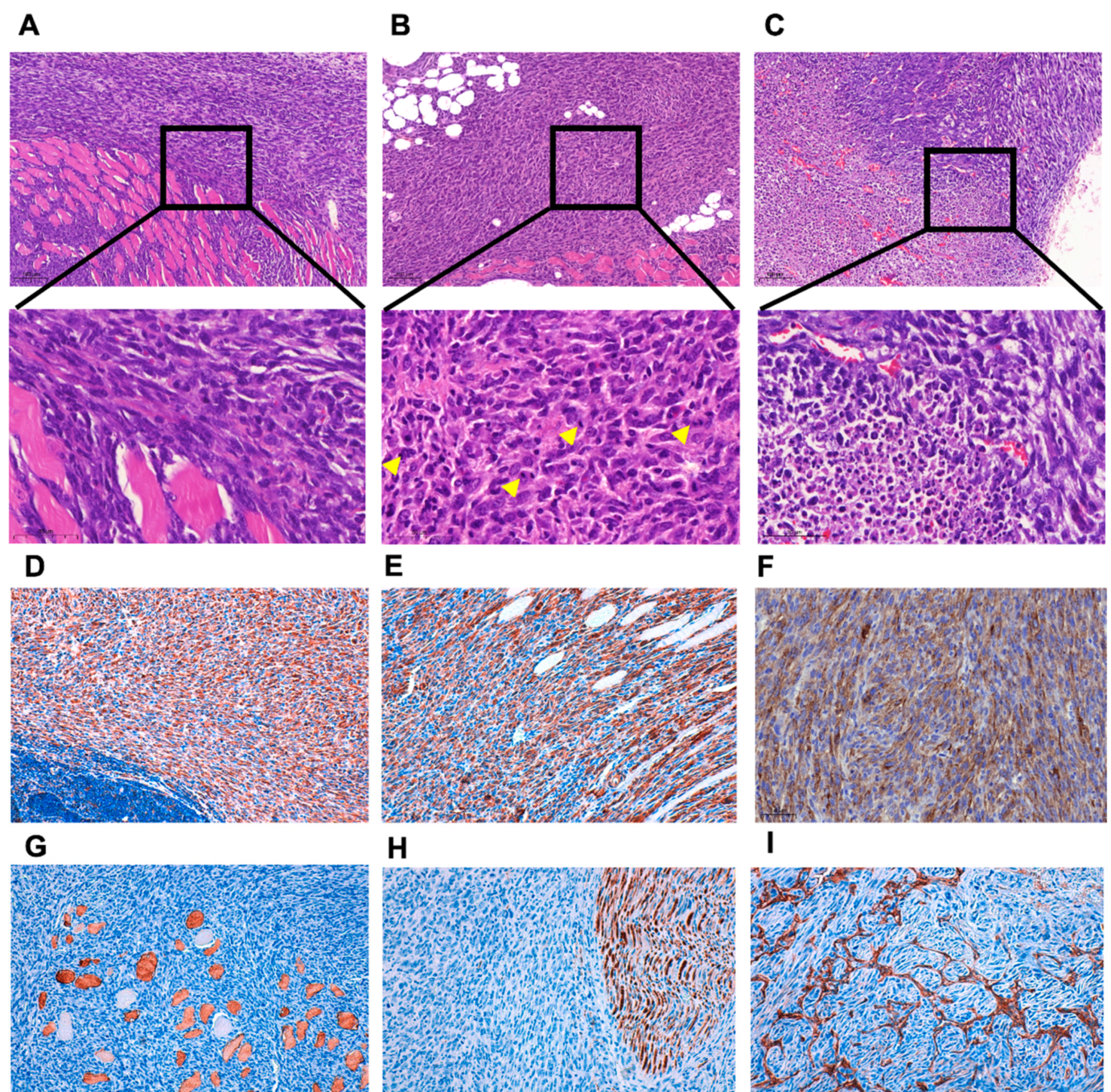
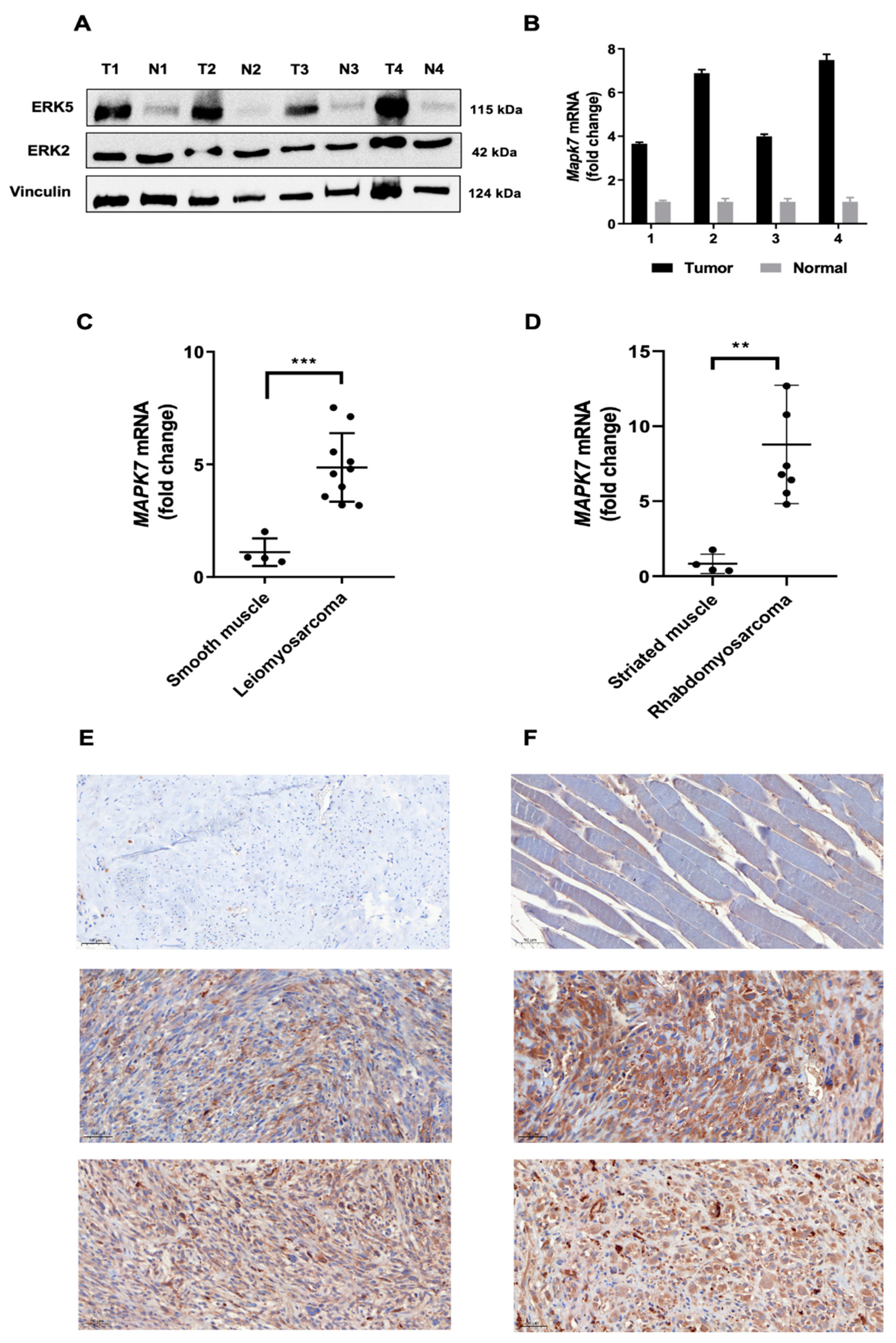
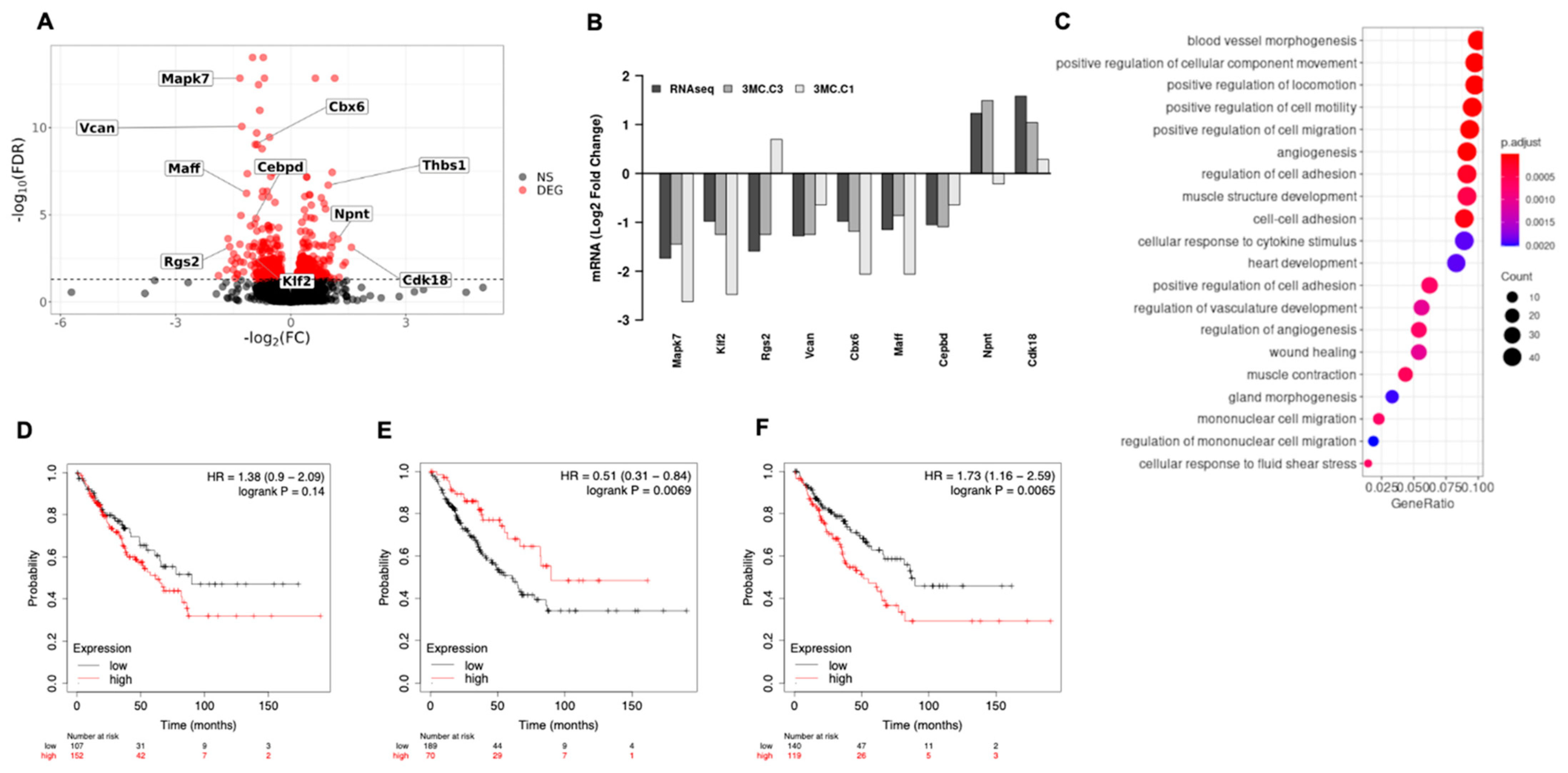
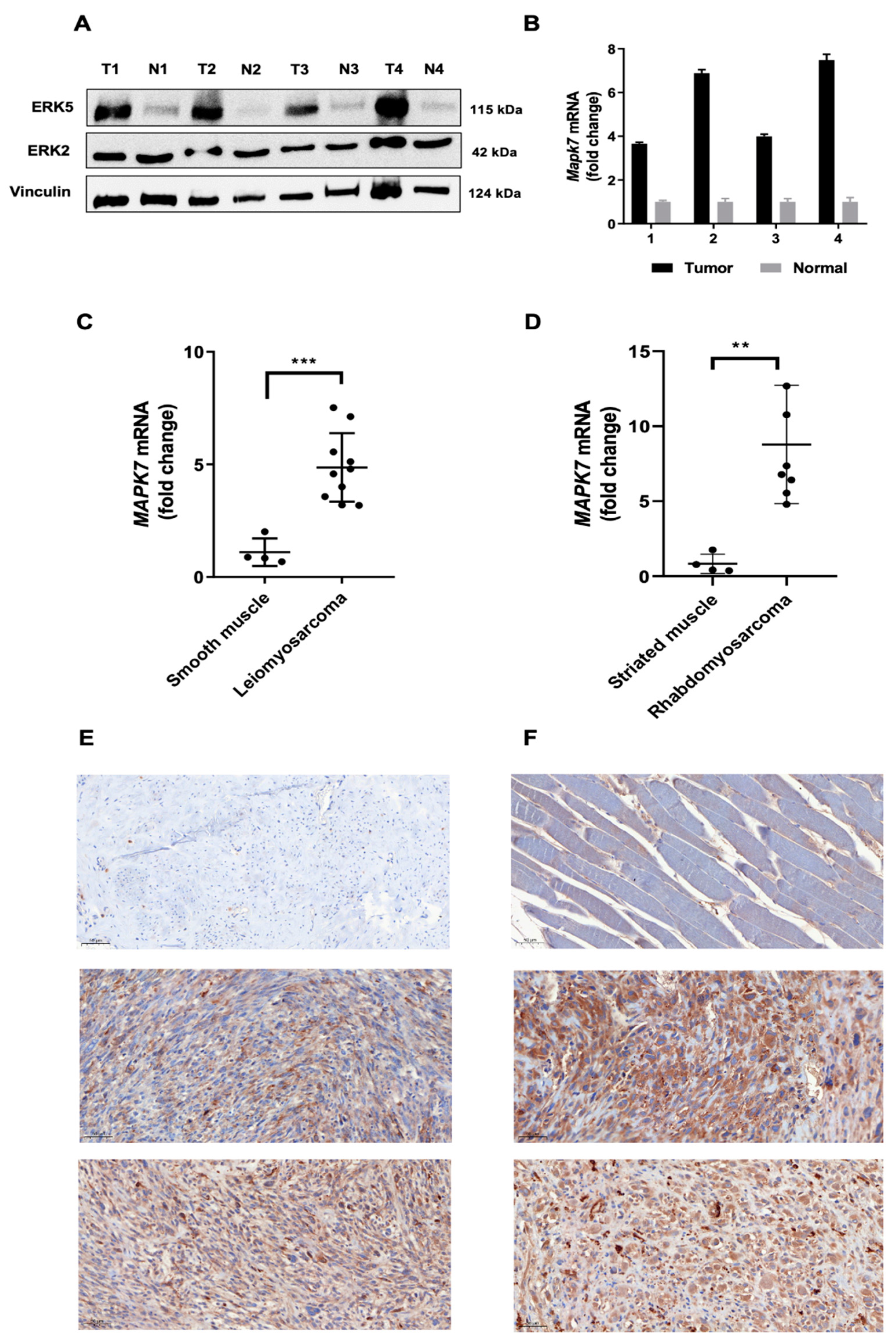
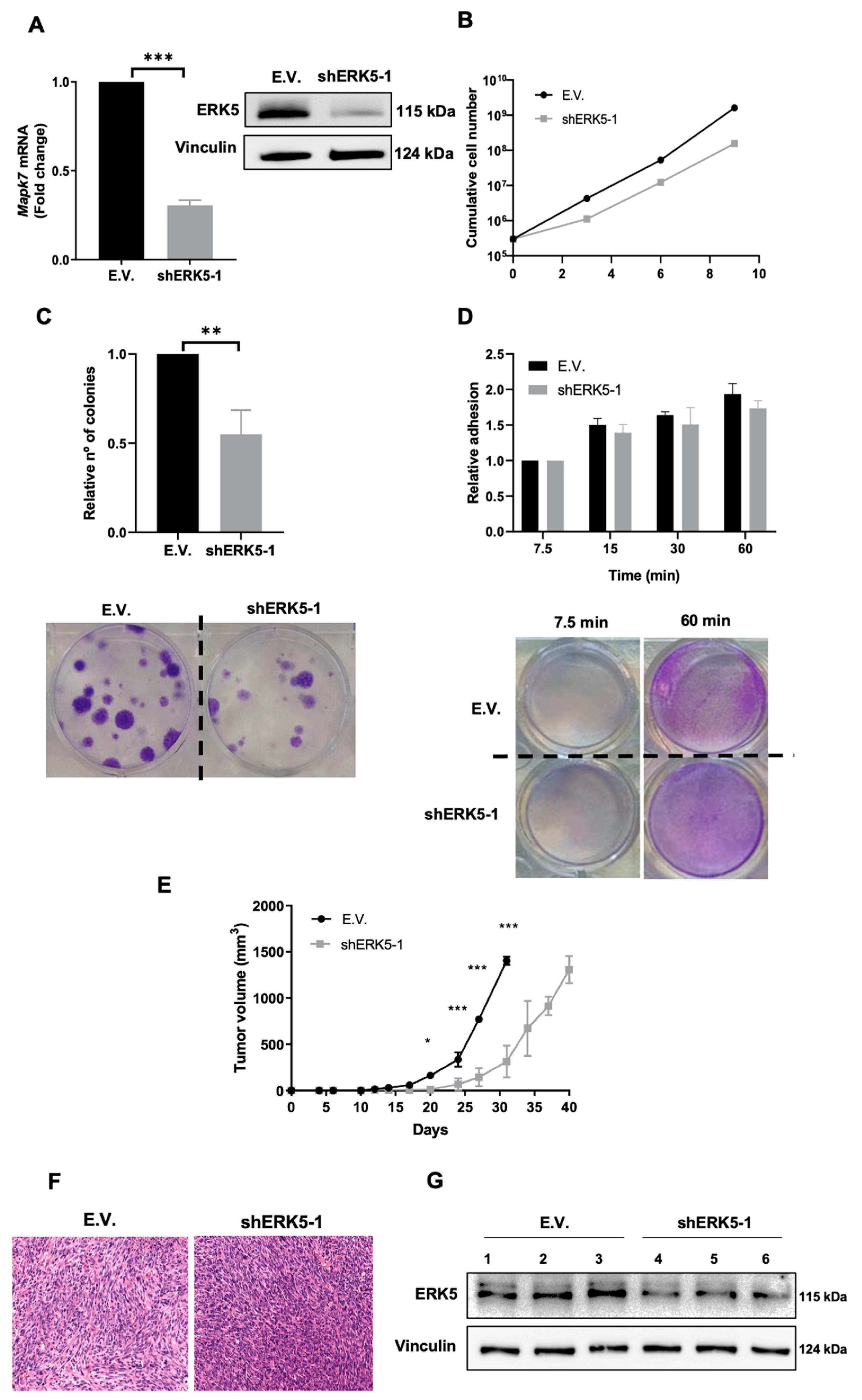
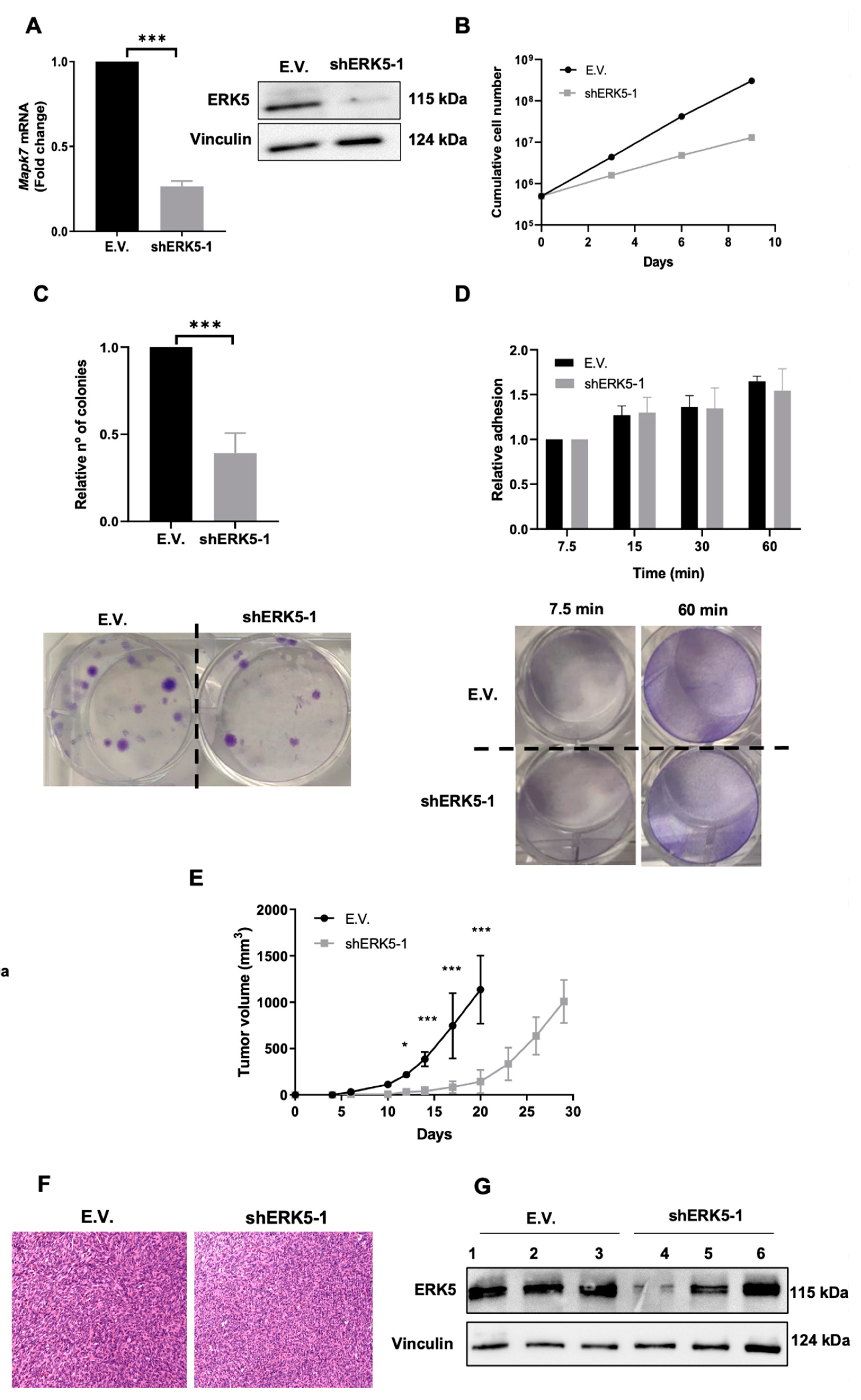
3.1. ERK5 Is Upregulated in 3MC-Induced Sarcoma and in Human Leiomyosarcoma
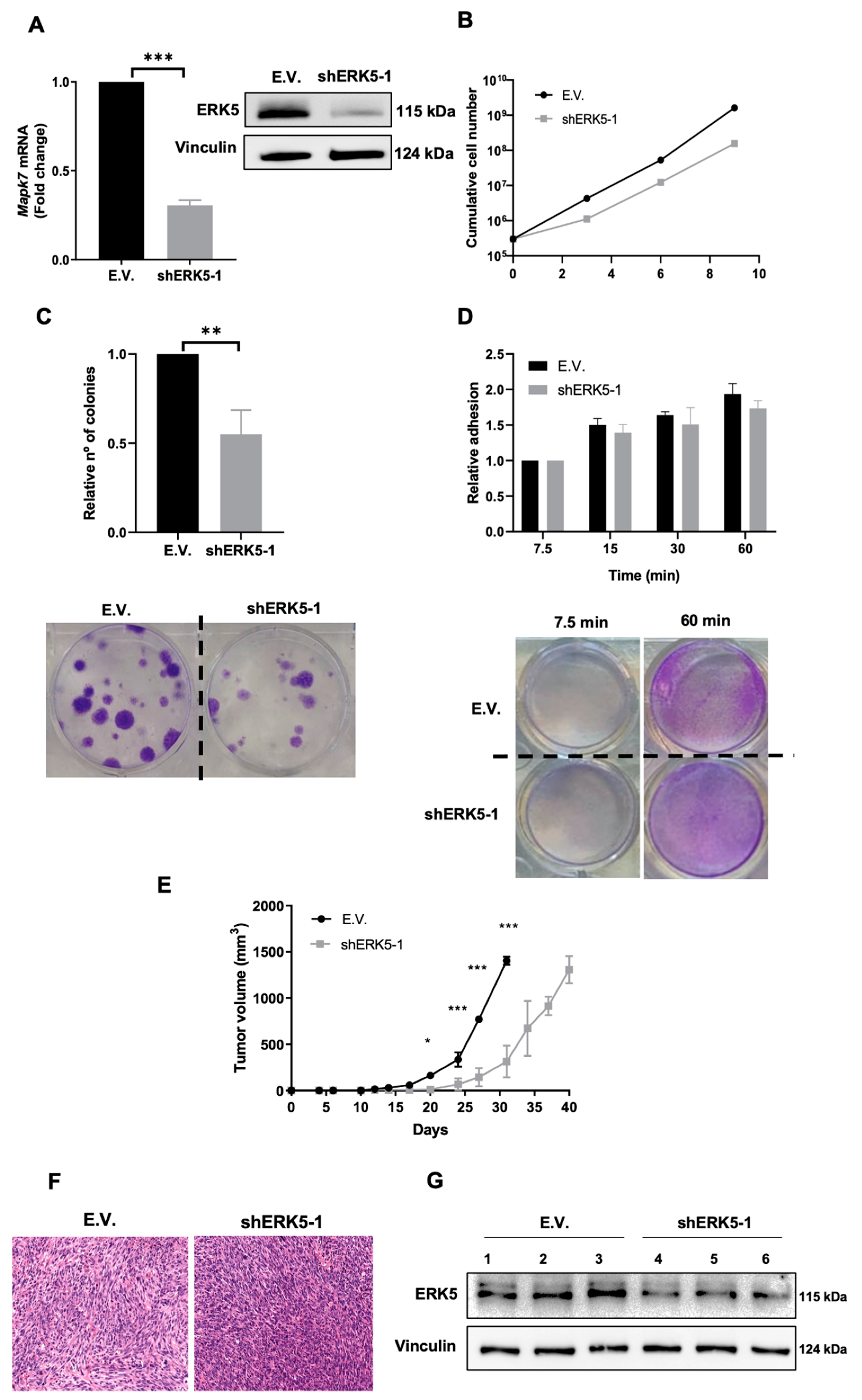
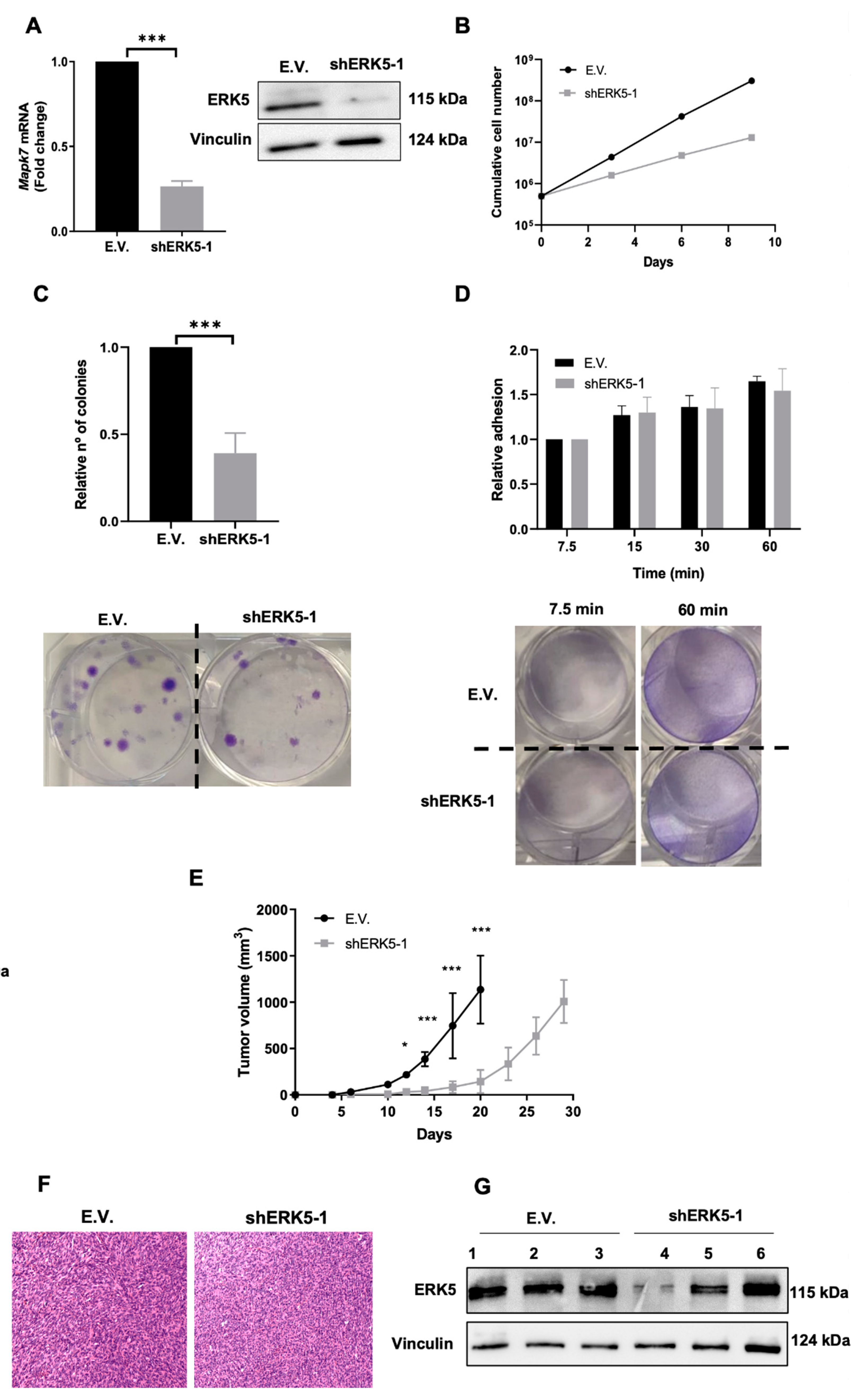
3.2. ERK5 Is a Major Determinant in the Biology of Sarcoma-Derived Cell Lines Induced by 3MC
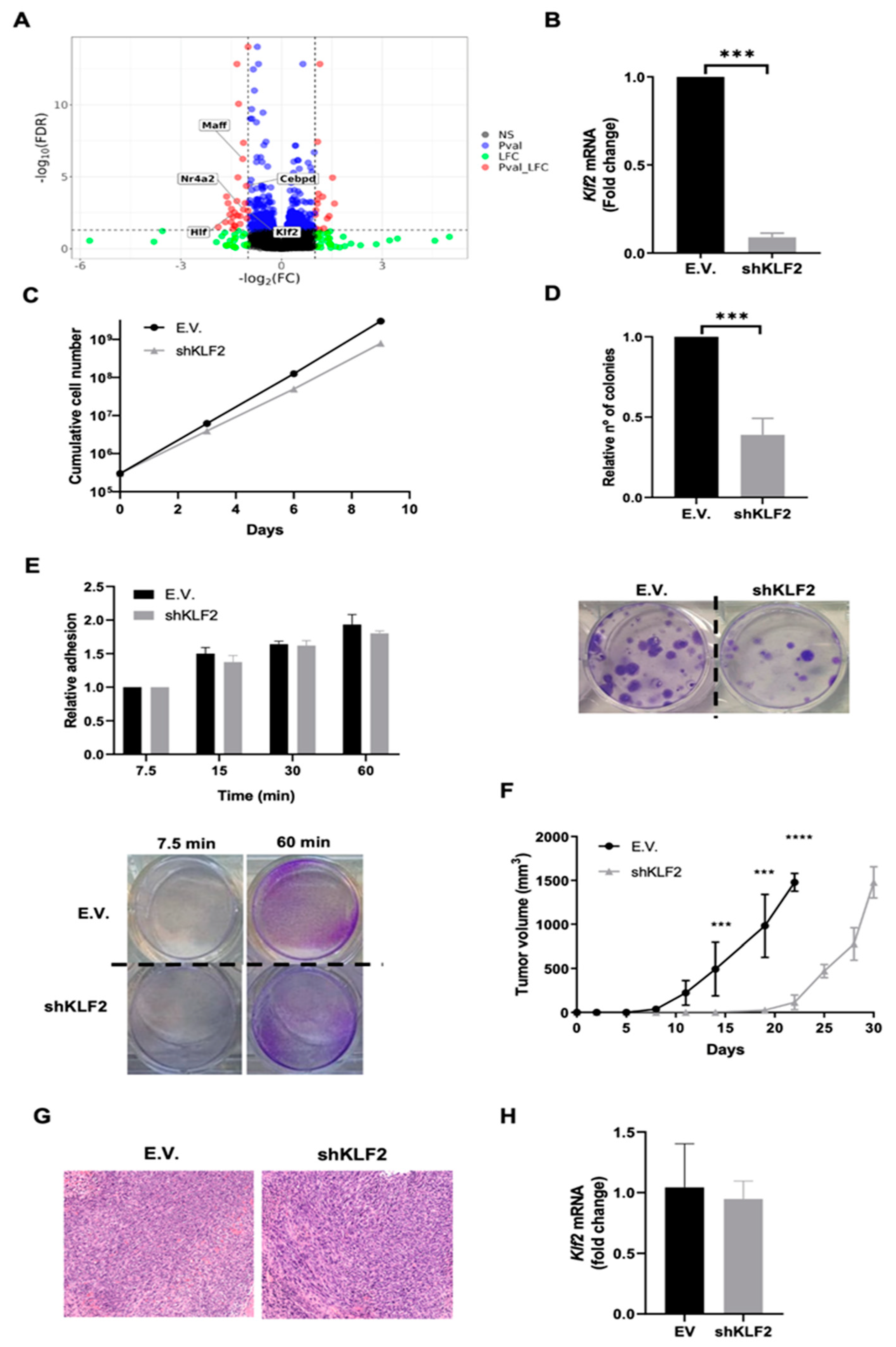
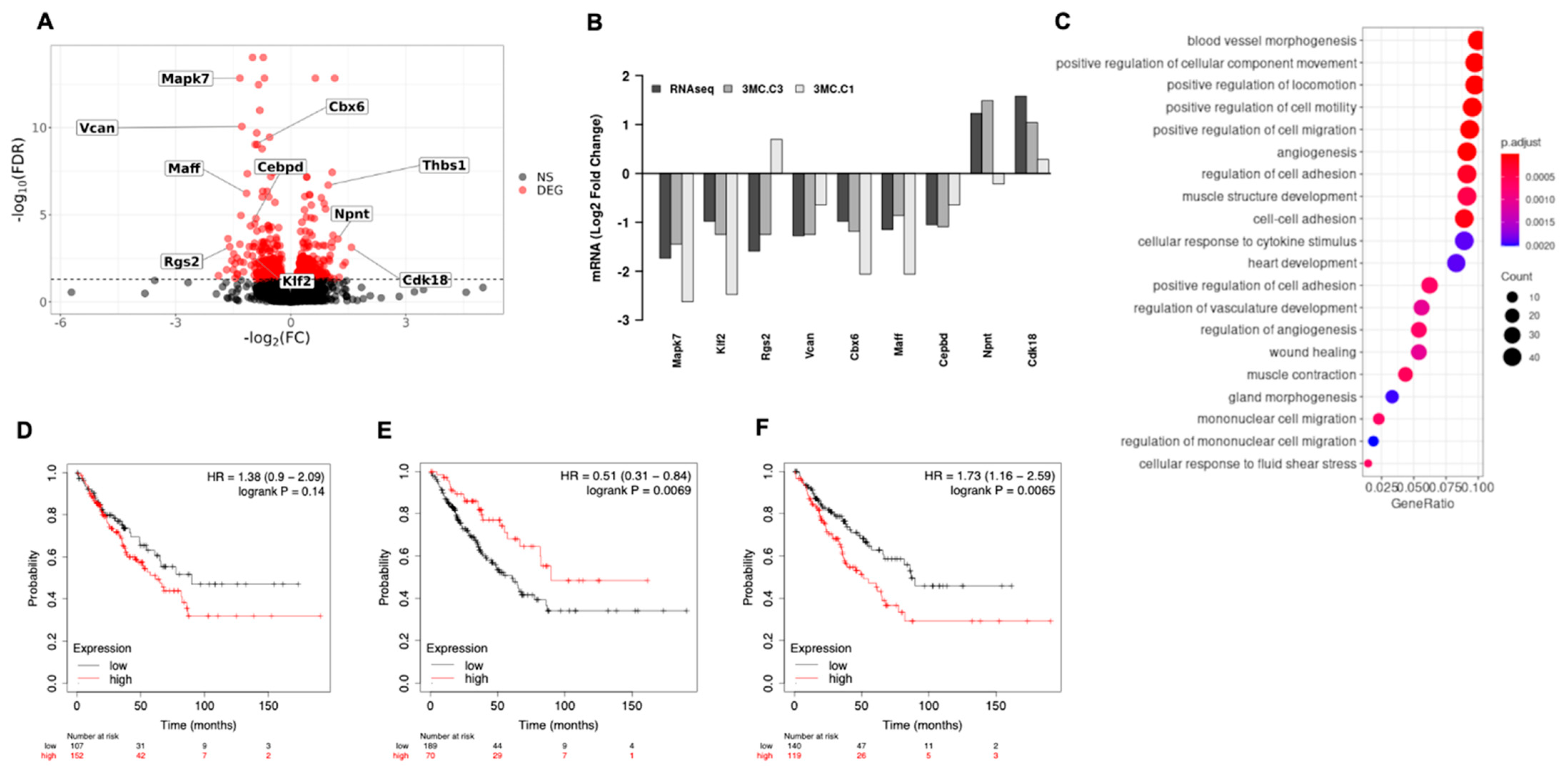
3.3. ERK5 Modulates Transcriptional Landscape in Sarcoma-Derived Cell Lines Induced by 3MC
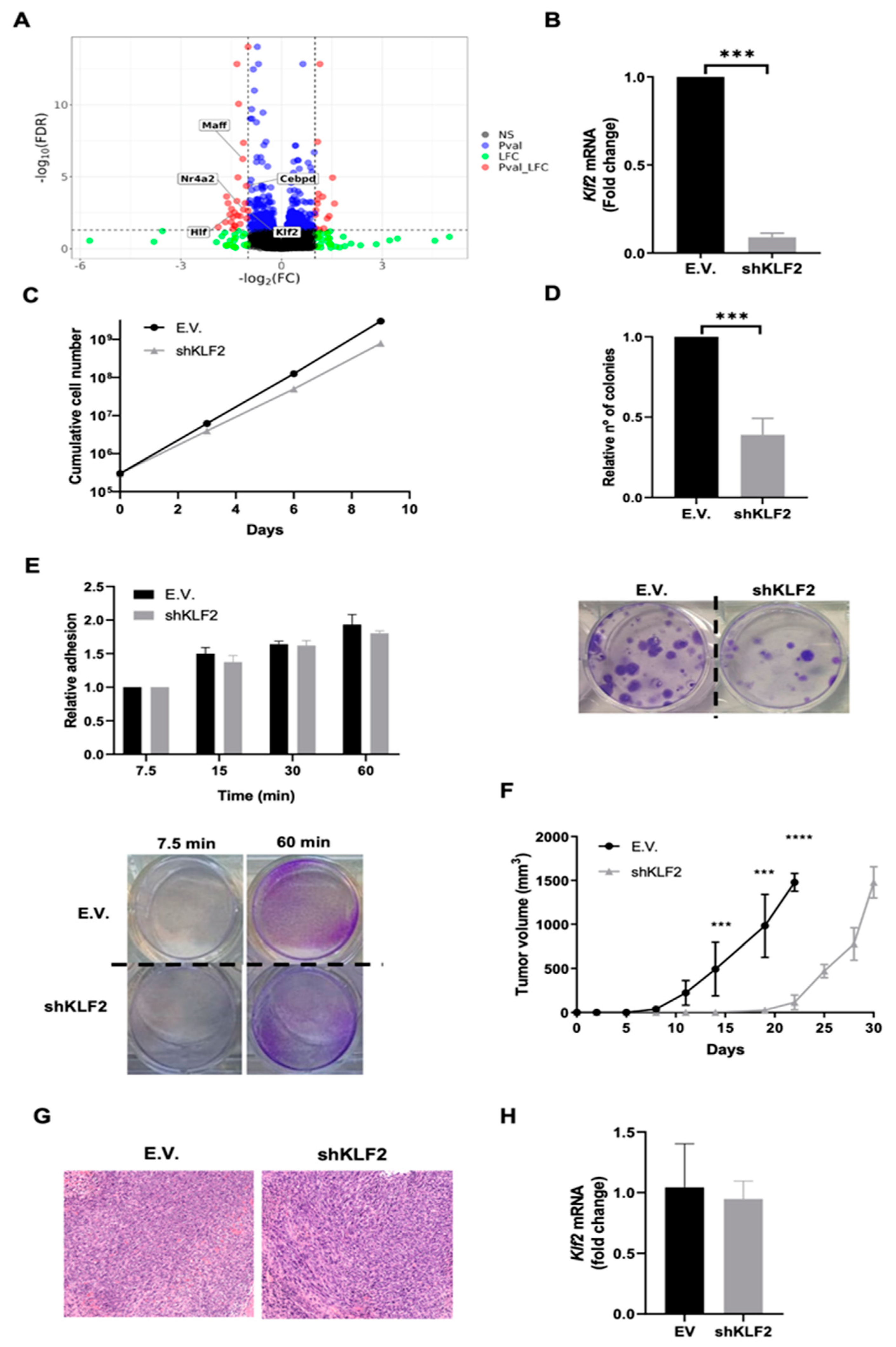
3.4. KLF2 Is a Critical Mediator of the Biological Effects of ERK5 in Sarcoma-Derived Cell Lines Induced by 3MC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blay, J.-Y.; Kang, Y.-K.; Nishida, T.; von Mehren, M. Gastrointestinal stromal tumours. Nat. Rev. Dis. Primers 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef] [PubMed]
- Dodd, R.D.; Mito, J.K.; Kirsch, D.G. Animal models of soft-tissue sarcoma. Dis. Model. Mech. 2010, 3, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Sampson, V.B.; Kamara, D.F.; Kolb, E.A. Xenograft and genetically engineered mouse model systems of osteosarcoma and Ewing’s sarcoma: Tumor models for cancer drug discovery. Expert Opin. Drug Discov. 2013, 8, 1181–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, R.D.; Añó, L.; Blum, J.M.; Li, Z.; Van Mater, D.; Kirsch, D.G. Methods to generate genetically engineered mouse models of soft tissue sarcoma. Methods Mol. Biol. 2015, 1267, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Hernando, E.; Charytonowicz, E.; Dudas, M.E.; Menendez, S.; Matushansky, I.; Mills, J.; Socci, N.D.; Behrendt, N.; Ma, L.; Maki, R.G.; et al. The AKT-mTOR pathway plays a critical role in the development of leiomyosarcomas. Nat. Med. 2007, 13, 748–753. [Google Scholar] [CrossRef]
- Langdon, C.G.; Gadek, K.E.; Garcia, M.R.; Evans, M.K.; Reed, K.B.; Bush, M.; Hanna, J.A.; Drummond, C.J.; Maguire, M.C.; Leavey, P.J.; et al. Synthetic essentiality between PTEN and core dependency factor PAX7 dictates rhabdomyosarcoma identity. Nat. Commun. 2021, 12, 5520. [Google Scholar] [CrossRef]
- Syverton, J.T.; Berry, G.P. Sarcomata and carcinomata induced in cottontail rabbits by methylcholanthrene. Science 1940, 91, 298. [Google Scholar] [CrossRef]
- Efeyan, A.; Murga, M.; Martinez-Pastor, B.; Ortega-Molina, A.; Soria, R.; Collado, M.; Fernandez-Capetillo, O.; Serrano, M. Limited role of murine ATM in oncogene-induced senescence and p53-dependent tumor suppression. PLoS ONE 2009, 4, e5475. [Google Scholar] [CrossRef]
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Cecilia, Y.; Peregrina, S.; Garcia-Serelde, B.; Muñoz-Galvan, S.; Cañamero, M.; Carnero, A. The essential role of PIM kinases in sarcoma growth and bone invasion. Carcinogenesis 2012, 33, 1479–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remeseiro, S.; Cuadrado, A.; Gómez-López, G.; Pisano, D.G.; Losada, A. A unique role of cohesin-SA1 in gene regulation and development. EMBO J. 2012, 31, 2090–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-L.; Mowery, Y.M.; Daniel, A.R.; Zhang, D.; Sibley, A.B.; Delaney, J.R.; Wisdom, A.J.; Qin, X.; Wang, X.; Caraballo, I.; et al. Mutational landscape in genetically engineered, carcinogen-induced, and radiation-induced mouse sarcoma. JCI Insight 2019, 4, 128698. [Google Scholar] [CrossRef] [Green Version]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano, C.; Romagosa, C.; Hernández-Losa, J.; Simonetti, S.; Valverde, C.; Moliné, T.; Somoza, R.; Pérez, M.; Vélez, R.; Vergés, R.; et al. RAS/MAPK pathway hyperactivation determines poor prognosis in undifferentiated pleomorphic sarcomas. Cancer 2016, 122, 99–107. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular Biology of Osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef]
- Lin, Y.K.; Wu, W.; Ponce, R.K.; Kim, J.W.; Okimoto, R.A. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc. Natl. Acad. Sci. USA 2020, 117, 20776–20784. [Google Scholar] [CrossRef]
- Liu, H.; Nazmun, N.; Hassan, S.; Liu, X.; Yang, J. BRAF mutation and its inhibitors in sarcoma treatment. Cancer Med. 2020, 9, 4881–4896. [Google Scholar] [CrossRef]
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK inhibition in a recurrent malignant peripheral nerve sheath tumor. NPJ Precis. Oncol. 2021, 5, 9. [Google Scholar] [CrossRef]
- Zhou, G.; Bao, Z.Q.; Dixon, J.E. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 1995, 270, 12665–12669. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.D.; Ulevitch, R.J.; Han, J. Primary structure of BMK1: A new mammalian map kinase. Biochem. Biophys. Res. Commun. 1995, 213, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.T.; Yan, T.J.; Cavanaugh, J.E.; Flaherty, P.T.; Beckman, B.S.; Burow, M.E. Oncogenic signaling of MEK5-ERK5. Cancer Lett. 2017, 392, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Rovida, E. Impact of ERK5 on the Hallmarks of Cancer. Int. J. Mol. Sci. 2019, 20, 1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, M.; Celli, J.; Missale, F.; Cersosimo, F.; Russo, M.; Belloni, E.; Di Matteo, A.; Lonardi, S.; Vermi, W.; Ghigna, C.; et al. Clinical Significance and Regulation of ERK5 Expression and Function in Cancer. Cancers 2022, 14, 348. [Google Scholar] [CrossRef]
- Pavan, S.; Meyer-Schaller, N.; Diepenbruck, M.; Kalathur, R.K.R.; Saxena, M.; Christofori, G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene 2018, 37, 4197–4213. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Vergara, X.; Tabera, S.; Montero, J.C.; Esparís-Ogando, A.; López-Pérez, R.; Mateo, G.; Gutiérrez, N.; Parmo-Cabañas, M.; Teixidó, J.; San Miguel, J.F.; et al. Multifunctional role of Erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. [Google Scholar] [CrossRef] [Green Version]
- Gentilini, A.; Lori, G.; Caligiuri, A.; Raggi, C.; Di Maira, G.; Pastore, M.; Piombanti, B.; Lottini, T.; Arcangeli, A.; Madiai, S.; et al. Extracellular Signal-Regulated Kinase 5 Regulates the Malignant Phenotype of Cholangiocarcinoma Cells. Hepatology 2021, 74, 2007–2020. [Google Scholar] [CrossRef]
- Pascual-Serra, R.; Fernández-Aroca, D.M.; Sabater, S.; Roche, O.; Andrés, I.; Ortega-Muelas, M.; Arconada-Luque, E.; Garcia-Flores, N.; Bossi, G.; Belandia, B.; et al. p38β (MAPK11) mediates gemcitabine-associated radiosensitivity in sarcoma experimental models. Radiother. Oncol. 2021, 156, 136–144. [Google Scholar] [CrossRef]
- Ortega-Muelas, M.; Roche, O.; Fernández-Aroca, D.M.; Encinar, J.A.; Albandea-Rodríguez, D.; Arconada-Luque, E.; Pascual-Serra, R.; Muñoz, I.; Sánchez-Pérez, I.; Belandia, B.; et al. ERK5 signalling pathway is a novel target of sorafenib: Implication in EGF biology. J. Cell Mol. Med. 2021, 25, 10591–10603. [Google Scholar] [CrossRef]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjö, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Choi, J.; Manzano, A.; Dong, W.; Bellone, S.; Bonazzoli, E.; Zammataro, L.; Yao, X.; Deshpande, A.; Zaidi, S.; Guglielmi, A.; et al. Integrated mutational landscape analysis of uterine leiomyosarcomas. Proc. Natl. Acad. Sci. USA 2021, 118, e2025182118. [Google Scholar] [CrossRef]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.-H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Cai, F.; Xu, H.; Lu, Y.; Chen, J.; Liu, J.; Cao, N.; Zhang, X.; Chen, X.; Huang, Q.; et al. Extracellular signal regulated kinase 5 promotes cell migration, invasion and lung metastasis in a FAK-dependent manner. Protein Cell 2020, 11, 825–845. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Zhang, J.; Telfer, B.A.; Zhang, H.; Ali, N.; Chen, F.; Risa, B.; Pearson, A.J.; Zhang, W.; Finegan, K.G.; et al. The extracellular-regulated protein kinase 5 (ERK5) enhances metastatic burden in triple-negative breast cancer through focal adhesion protein kinase (FAK)-mediated regulation of cell adhesion. Oncogene 2021, 40, 3929–3941. [Google Scholar] [CrossRef]
- Zhou, Q.; Liu, M.; Xia, X.; Gong, T.; Feng, J.; Liu, W.; Liu, Y.; Zhen, B.; Wang, Y.; Ding, C.; et al. A mouse tissue transcription factor atlas. Nat. Commun. 2017, 8, 15089. [Google Scholar] [CrossRef]
- Ananieva, O.; Macdonald, A.; Wang, X.; McCoy, C.E.; McIlrath, J.; Tournier, C.; Arthur, J.S.C. ERK5 regulation in naïve T-cell activation and survival. Eur. J. Immunol. 2008, 38, 2534–2547. [Google Scholar] [CrossRef]
- Paez-Mayorga, J.; Chen, A.L.; Kotla, S.; Tao, Y.; Abe, R.J.; He, E.D.; Danysh, B.P.; Hofmann, M.-C.C.; Le, N.-T. Ponatinib Activates an Inflammatory Response in Endothelial Cells via ERK5 SUMOylation. Front. Cardiovasc. Med. 2018, 5, 125. [Google Scholar] [CrossRef]
- Paolini, A.; Fontana, F.; Pham, V.-C.; Rödel, C.J.; Abdelilah-Seyfried, S. Mechanosensitive Notch-Dll4 and Klf2-Wnt9 signaling pathways intersect in guiding valvulogenesis in zebrafish. Cell Rep. 2021, 37, 109782. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.A.; Williams, C.A.C.; Zhou, H.; Rios-Szwed, D.; Fernandez-Alonso, R.; Mansoor, S.; McMulkin, L.; Toth, R.; Gourlay, R.; Peltier, J.; et al. An ERK5-KLF2 signalling module regulates early embryonic gene expression and telomere rejuvenation in stem cells. Biochem. J. 2021, 478, 4119–4136. [Google Scholar] [CrossRef] [PubMed]
- Astolfi, A.; Nannini, M.; Indio, V.; Schipani, A.; Rizzo, A.; Perrone, A.M.; De Iaco, P.; Pirini, M.G.; De Leo, A.; Urbini, M.; et al. Genomic Database Analysis of Uterine Leiomyosarcoma Mutational Profile. Cancers 2020, 12, 2126. [Google Scholar] [CrossRef] [PubMed]
- Cuppens, T.; Moisse, M.; Depreeuw, J.; Annibali, D.; Colas, E.; Gil-Moreno, A.; Huvila, J.; Carpén, O.; Zikán, M.; Matias-Guiu, X.; et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int. J. Cancer 2018, 142, 1230–1243. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Inoue, M. Immunohistochemical analysis for Mdm2 and p53 proteins in methylcholanthrene-induced mouse rhabdomyosarcomas. J. Vet. Med. Sci. 2006, 68, 427–431. [Google Scholar] [CrossRef] [Green Version]
- Rovida, E.; Di Maira, G.; Tusa, I.; Cannito, S.; Paternostro, C.; Navari, N.; Vivoli, E.; Deng, X.; Gray, N.S.; Esparís-Ogando, A.; et al. The mitogen-activated protein kinase ERK5 regulates the development and growth of hepatocellular carcinoma. Gut 2015, 64, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- Esparís-Ogando, A.; Díaz-Rodríguez, E.; Montero, J.C.; Yuste, L.; Crespo, P.; Pandiella, A. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol. Cell Biol. 2002, 22, 270–285. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Fdez, A.; Re-Louhau, M.F.; Rodríguez-Núñez, P.; Ludeña, D.; Matilla-Almazán, S.; Pandiella, A.; Esparís-Ogando, A. Clinical, genetic and pharmacological data support targeting the MEK5/ERK5 module in lung cancer. NPJ Precis. Oncol. 2021, 5, 78. [Google Scholar] [CrossRef]
- Ali, M.; Mutahir, Z.; Riaz, A. CRISPR/Cas9 engineering of ERK5 identifies its FAK/PYK2 dependent role in adhesion-mediated cell survival. Biochem. Biophys. Res. Commun. 2019, 513, 179–185. [Google Scholar] [CrossRef]
- Matossian, M.D.; Hoang, V.T.; Burks, H.E.; La, J.; Elliott, S.; Brock, C.; Rusch, D.B.; Buechlein, A.; Nephew, K.P.; Bhatt, A.; et al. Constitutive activation of MEK5 promotes a mesenchymal and migratory cell phenotype in triple negative breast cancer. Oncoscience 2021, 8, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Lemos, C.; Wortmann, L.; Eis, K.; Holton, S.J.; Boemer, U.; Moosmayer, D.; Eberspaecher, U.; Weiske, J.; Lechner, C.; et al. Discovery and Characterization of the Potent and Highly Selective (Piperidin-4-yl)pyrido[3,2-d]pyrimidine Based in Vitro Probe BAY-885 for the Kinase ERK5. J. Med. Chem. 2019, 62, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Tusa, I.; Gagliardi, S.; Tubita, A.; Pandolfi, S.; Menconi, A.; Lulli, M.; Dello Sbarba, P.; Stecca, B.; Rovida, E. The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells. Int. J. Mol. Sci. 2021, 22, 11259. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Schulze-Hoepfner, F.T.; Hong, J.; Chlenski, A.; Zeitlin, B.D.; Goel, K.; Gomes, S.; Liu, Y.; Abe, M.K.; Nor, J.E.; et al. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood 2009, 114, 4592–4600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultgren, N.W.; Fang, J.S.; Ziegler, M.E.; Ramirez, R.N.; Phan, D.T.T.; Hatch, M.M.S.; Welch-Reardon, K.M.; Paniagua, A.E.; Kim, L.S.; Shon, N.N.; et al. Slug regulates the Dll4-Notch-VEGFR2 axis to control endothelial cell activation and angiogenesis. Nat. Commun. 2020, 11, 5400. [Google Scholar] [CrossRef]
- Radu, M.; Lyle, K.; Hoeflich, K.P.; Villamar-Cruz, O.; Koeppen, H.; Chernoff, J. p21-Activated Kinase 2 Regulates Endothelial Development and Function through the Bmk1/Erk5 Pathway. Mol. Cell Biol. 2015, 35, 3990–4005. [Google Scholar] [CrossRef] [Green Version]
- Kedika, S.R.; Shukla, S.P.; Udugamasooriya, D.G. Design of a dual ERK5 kinase activation and autophosphorylation inhibitor to block cancer stem cell activity. Bioorg. Med. Chem. Lett. 2020, 30, 127552. [Google Scholar] [CrossRef]
- Pereira, D.M.; Gomes, S.E.; Borralho, P.M.; Rodrigues, C.M.P. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov. 2019, 5, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xiong, X.; Yao, Z.; Zhu, J.; Lin, Y.; Lin, W.; Li, K.; Xu, X.; Guo, Y.; Chen, Y.; et al. Chimeric RNA ASTN2-PAPPAas aggravates tumor progression and metastasis in human esophageal cancer. Cancer Lett. 2021, 501, 1–11. [Google Scholar] [CrossRef]
- Salama, Y.; Heida, A.H.; Yokoyama, K.; Takahashi, S.; Hattori, K.; Heissig, B. The EGFL7-ITGB3-KLF2 axis enhances survival of multiple myeloma in preclinical models. Blood Adv. 2020, 4, 1021–1037. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Kong, L.; Xiao, Y.; Yuan, H.; Song, Y.; Wang, J.; Yu, H.; Mao, S.; Xu, W. CDK8 regulates the angiogenesis of pancreatic cancer cells in part via the CDK8-β-catenin-KLF2 signal axis. Exp. Cell Res. 2018, 369, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-D.; Chen, W.-M.; Qi, F.-Z.; Sun, M.; Xu, T.-P.; Ma, P.; Shu, Y.-Q. Long non-coding RNA TUG1 is up-regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol. Cancer 2015, 14, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Tu, S.; Zeng, Y.; Zhang, C.; Deng, T.; Luo, W.; Lian, L.; Chen, L.; Xiong, X.; Yan, X. KLF2 inhibits TGF-β-mediated cancer cell motility in hepatocellular carcinoma. Acta Biochim. Biophys. Sin. 2020, 52, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Qin, H.; Jiang, B.; Lu, W.; Hao, J.; Cao, W.; Du, L.; Chen, W.; Zhao, X.; Guo, H. KLF2 inhibits cancer cell migration and invasion by regulating ferroptosis through GPX4 in clear cell renal cell carcinoma. Cancer Lett. 2021, 522, 1–13. [Google Scholar] [CrossRef]
- Morikawa, M.; Koinuma, D.; Mizutani, A.; Kawasaki, N.; Holmborn, K.; Sundqvist, A.; Tsutsumi, S.; Watabe, T.; Aburatani, H.; Heldin, C.-H.; et al. BMP Sustains Embryonic Stem Cell Self-Renewal through Distinct Functions of Different Krüppel-like Factors. Stem Cell Rep. 2016, 6, 64–73. [Google Scholar] [CrossRef]
- Wu, N.; Sun, H.; Zhao, X.; Zhang, Y.; Tan, J.; Qi, Y.; Wang, Q.; Ng, M.; Liu, Z.; He, L.; et al. MAP3K2-regulated intestinal stromal cells define a distinct stem cell niche. Nature 2021, 592, 606–610. [Google Scholar] [CrossRef]
- Turpaev, K.T. Transcription Factor KLF2 and Its Role in the Regulation of Inflammatory Processes. Biochemistry 2020, 85, 54–67. [Google Scholar] [CrossRef]
- Wittner, J.; Schuh, W. Krüppel-like Factor 2 (KLF2) in Immune Cell Migration. Vaccines 2021, 9, 1171. [Google Scholar] [CrossRef]
- Chen, G.-H.; Li, X.-L.; Deng, Y.-Q.; Zhou, F.-M.; Zou, W.-Q.; Jiang, W.-X.; Shangguan, S.-Q.; Lu, Z.-N. The Molecular Mechanism of EPO Regulates the Angiogenesis after Cerebral Ischemia through AMPK-KLF2 Signaling Pathway. Crit. Rev. Eukaryot. Gene Expr. 2019, 29, 105–112. [Google Scholar] [CrossRef]
- Myers, S.M.; Miller, D.C.; Molyneux, L.; Arasta, M.; Bawn, R.H.; Blackburn, T.J.; Cook, S.J.; Edwards, N.; Endicott, J.A.; Golding, B.T.; et al. Identification of a novel orally bioavailable ERK5 inhibitor with selectivity over p38α and BRD4. Eur. J. Med. Chem. 2019, 178, 530–543. [Google Scholar] [CrossRef]
- Yang, Q.; Deng, X.; Lu, B.; Cameron, M.; Fearns, C.; Patricelli, M.P.; Yates, J.R.; Gray, N.S.; Lee, J.-D. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochhead, P.A.; Tucker, J.A.; Tatum, N.J.; Wang, J.; Oxley, D.; Kidger, A.M.; Johnson, V.P.; Cassidy, M.A.; Gray, N.S.; Noble, M.E.M.; et al. Paradoxical activation of the protein kinase-transcription factor ERK5 by ERK5 kinase inhibitors. Nat. Commun. 2020, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arconada-Luque, E.; Jiménez-Suarez, J.; Pascual-Serra, R.; Nam-Cha, S.H.; Moline, T.; Cimas, F.J.; Fliquete, G.; Ortega-Muelas, M.; Roche, O.; Fernández-Aroca, D.M.; et al. ERK5 Is a Major Determinant of Chemical Sarcomagenesis: Implications in Human Pathology. Cancers 2022, 14, 3509. https://doi.org/10.3390/cancers14143509
Arconada-Luque E, Jiménez-Suarez J, Pascual-Serra R, Nam-Cha SH, Moline T, Cimas FJ, Fliquete G, Ortega-Muelas M, Roche O, Fernández-Aroca DM, et al. ERK5 Is a Major Determinant of Chemical Sarcomagenesis: Implications in Human Pathology. Cancers. 2022; 14(14):3509. https://doi.org/10.3390/cancers14143509
Chicago/Turabian StyleArconada-Luque, Elena, Jaime Jiménez-Suarez, Raquel Pascual-Serra, Syong Hyun Nam-Cha, Teresa Moline, Francisco J. Cimas, Germán Fliquete, Marta Ortega-Muelas, Olga Roche, Diego M. Fernández-Aroca, and et al. 2022. "ERK5 Is a Major Determinant of Chemical Sarcomagenesis: Implications in Human Pathology" Cancers 14, no. 14: 3509. https://doi.org/10.3390/cancers14143509
APA StyleArconada-Luque, E., Jiménez-Suarez, J., Pascual-Serra, R., Nam-Cha, S. H., Moline, T., Cimas, F. J., Fliquete, G., Ortega-Muelas, M., Roche, O., Fernández-Aroca, D. M., Muñoz Velasco, R., García-Flores, N., Garnés-García, C., Sánchez-Fdez, A., Matilla-Almazán, S., Sánchez-Arévalo Lobo, V. J., Hernández-Losa, J., Belandia, B., Pandiella, A., ... Ruiz-Hidalgo, M. J. (2022). ERK5 Is a Major Determinant of Chemical Sarcomagenesis: Implications in Human Pathology. Cancers, 14(14), 3509. https://doi.org/10.3390/cancers14143509