
The Epigenetic Regulatory Protein CBX2 Promotes mTORC1 Signalling and Inhibits DREAM Complex Activity to Drive Breast Cancer Cell Growth

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Line Transfection and CBX2 Inhibitor Treatments
2.3. Protein and Gene Expression Analysis
2.4. Cell Growth and Cell Cycle Analysis
2.5. RNA Sequencing and Gene Set Enrichment Analysis
2.6. Chromatin Immunoprecipitation-qPCR (ChIP-qPCR) and Cleavage under Targets and Release Using Nuclease (CUTandRUN)-qPCR
2.7. CBX2 Correlation Analysis of Patient Data Sets
3. Results
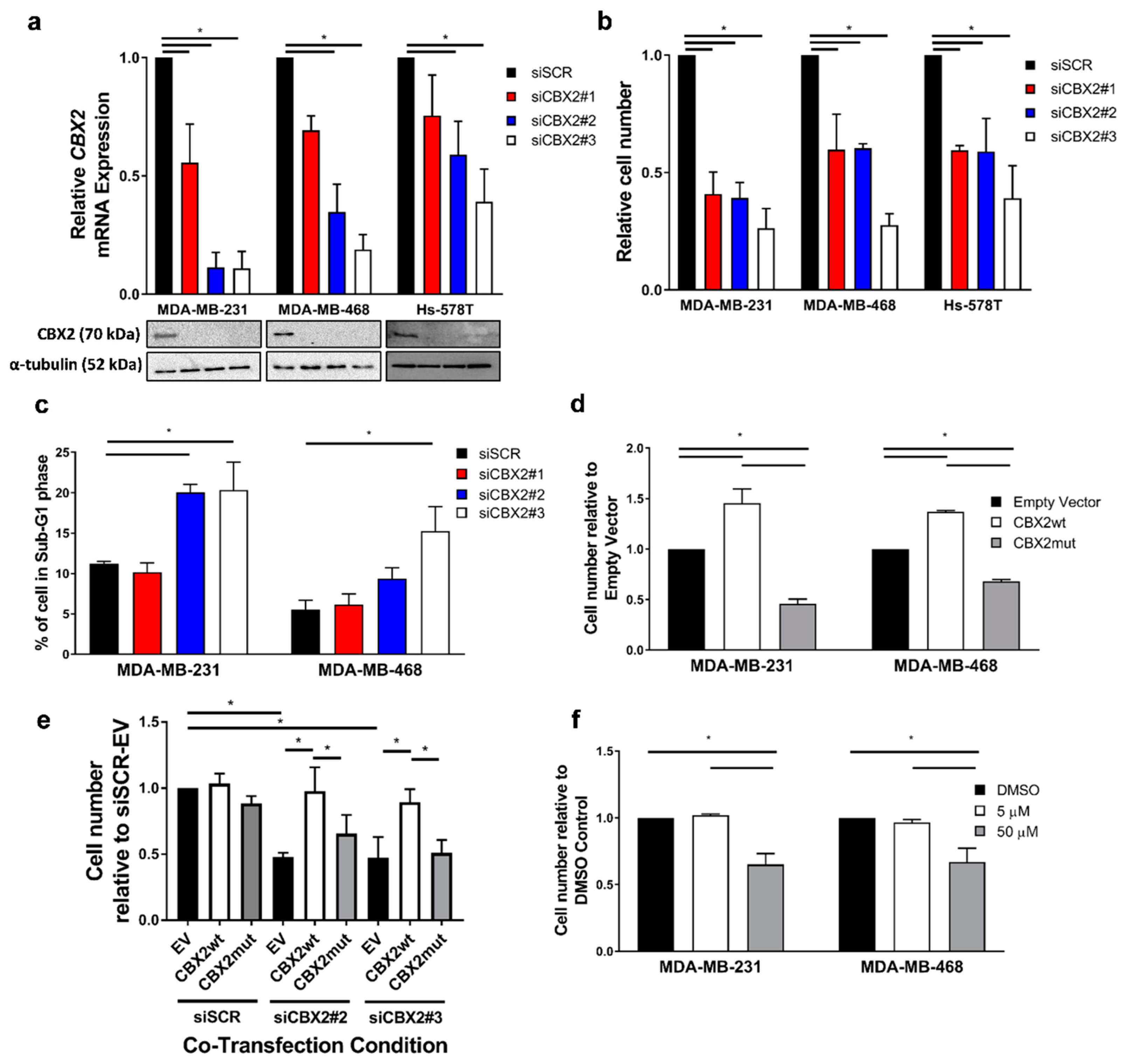
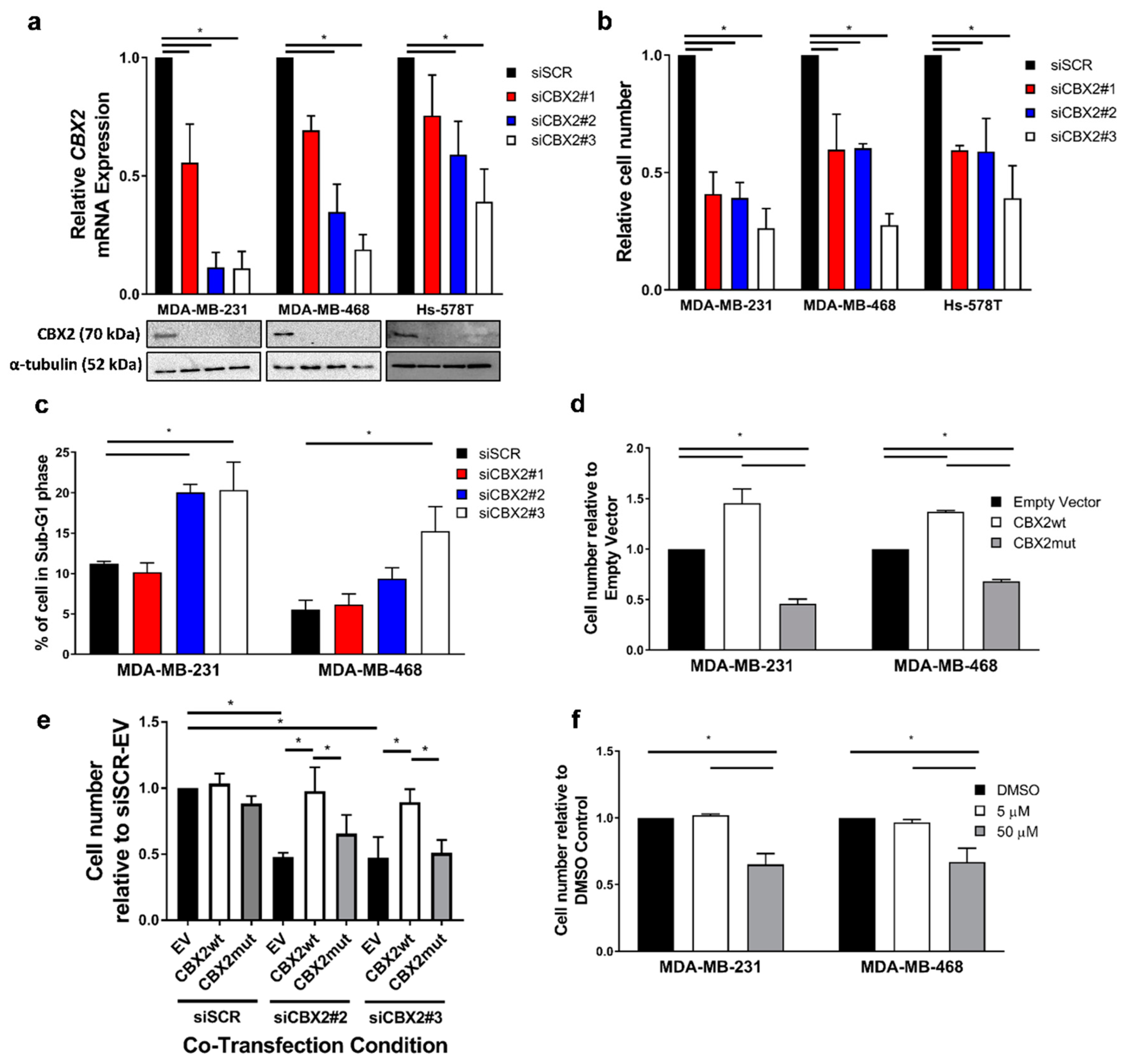
3.1. CBX2 Chromatin Interactions Are Required for TNBC Cell Growth and Viability
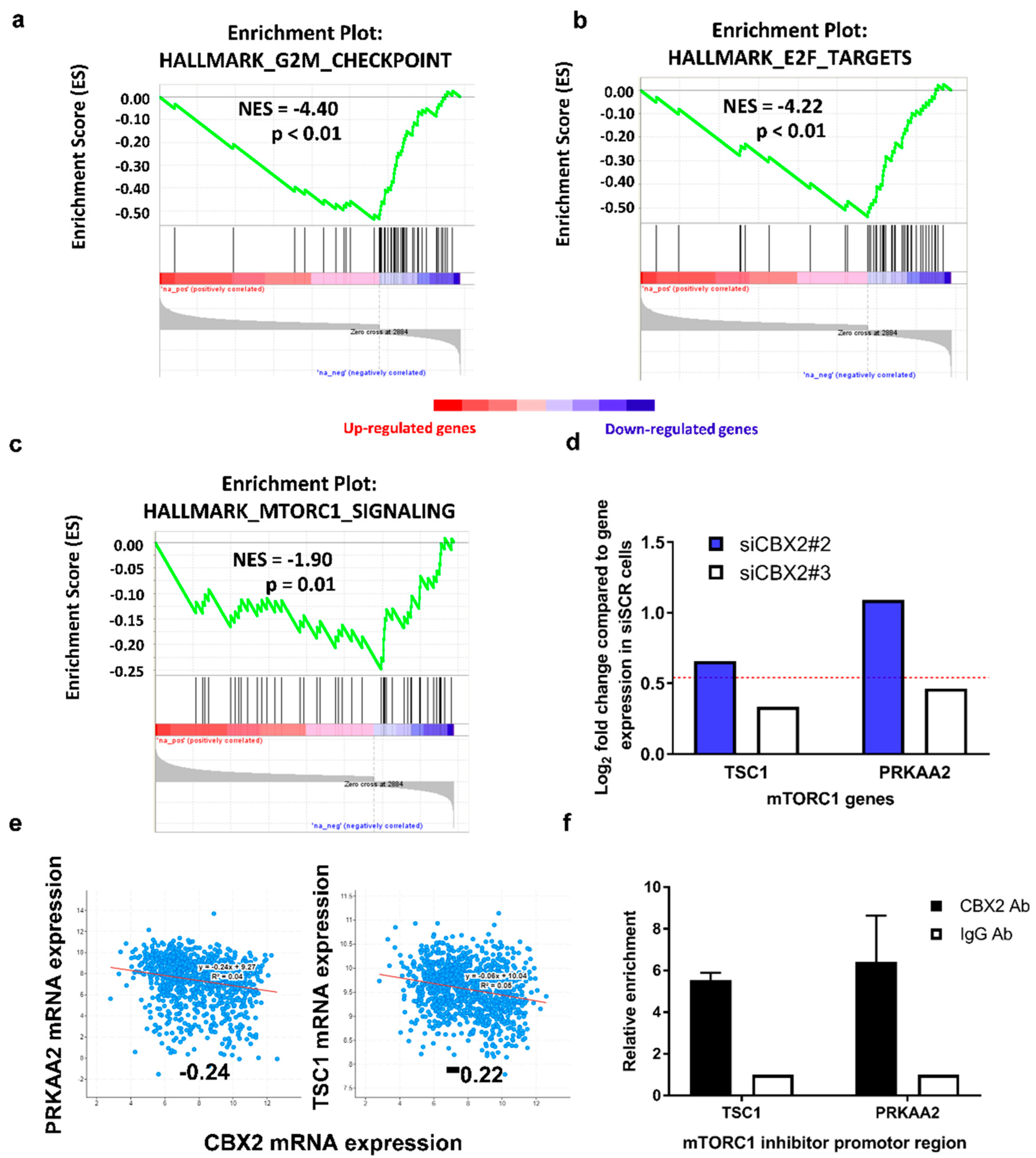
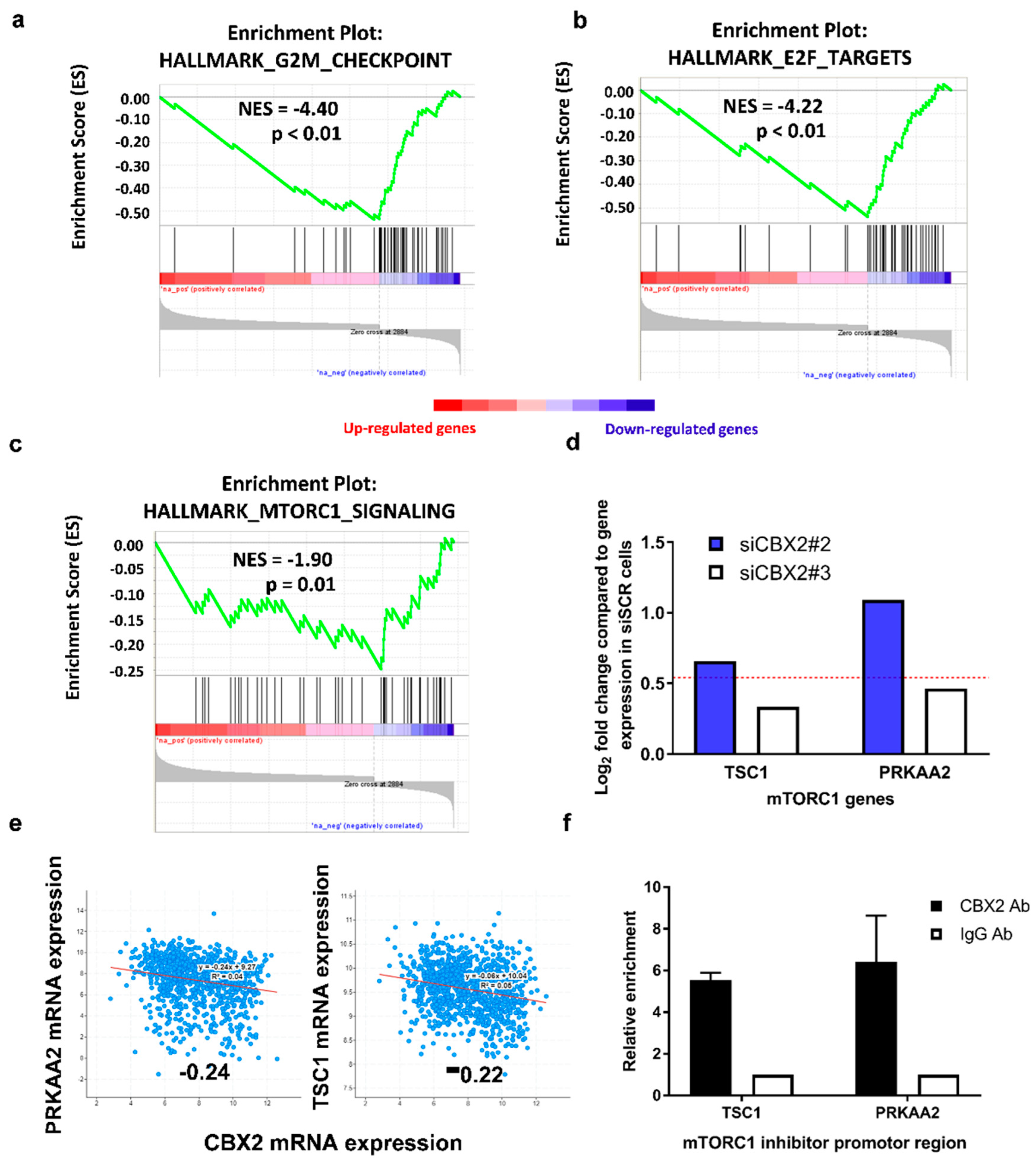
3.2. CBX2 Promotes Proliferative and Oncogenic Gene Signatures
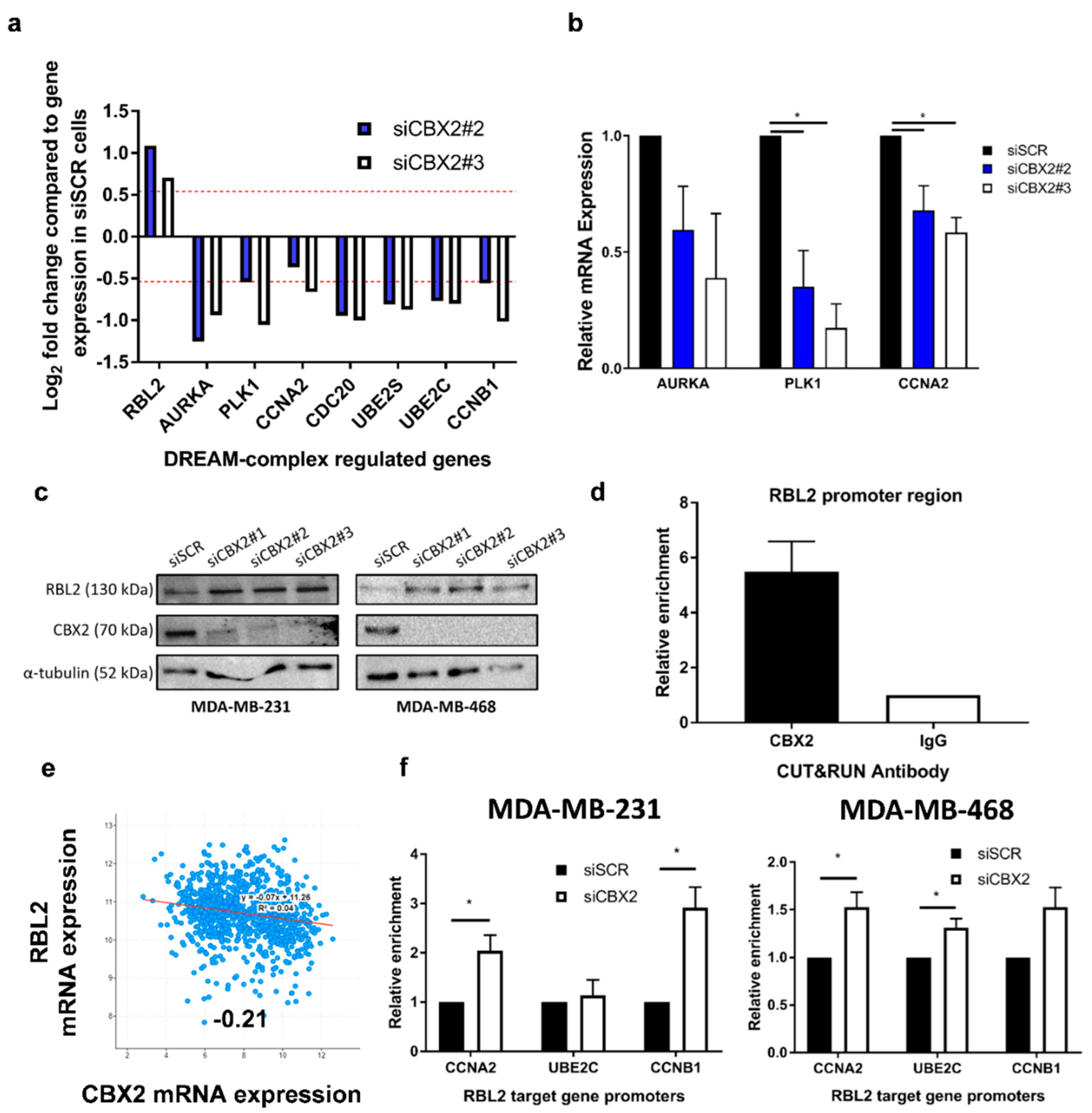
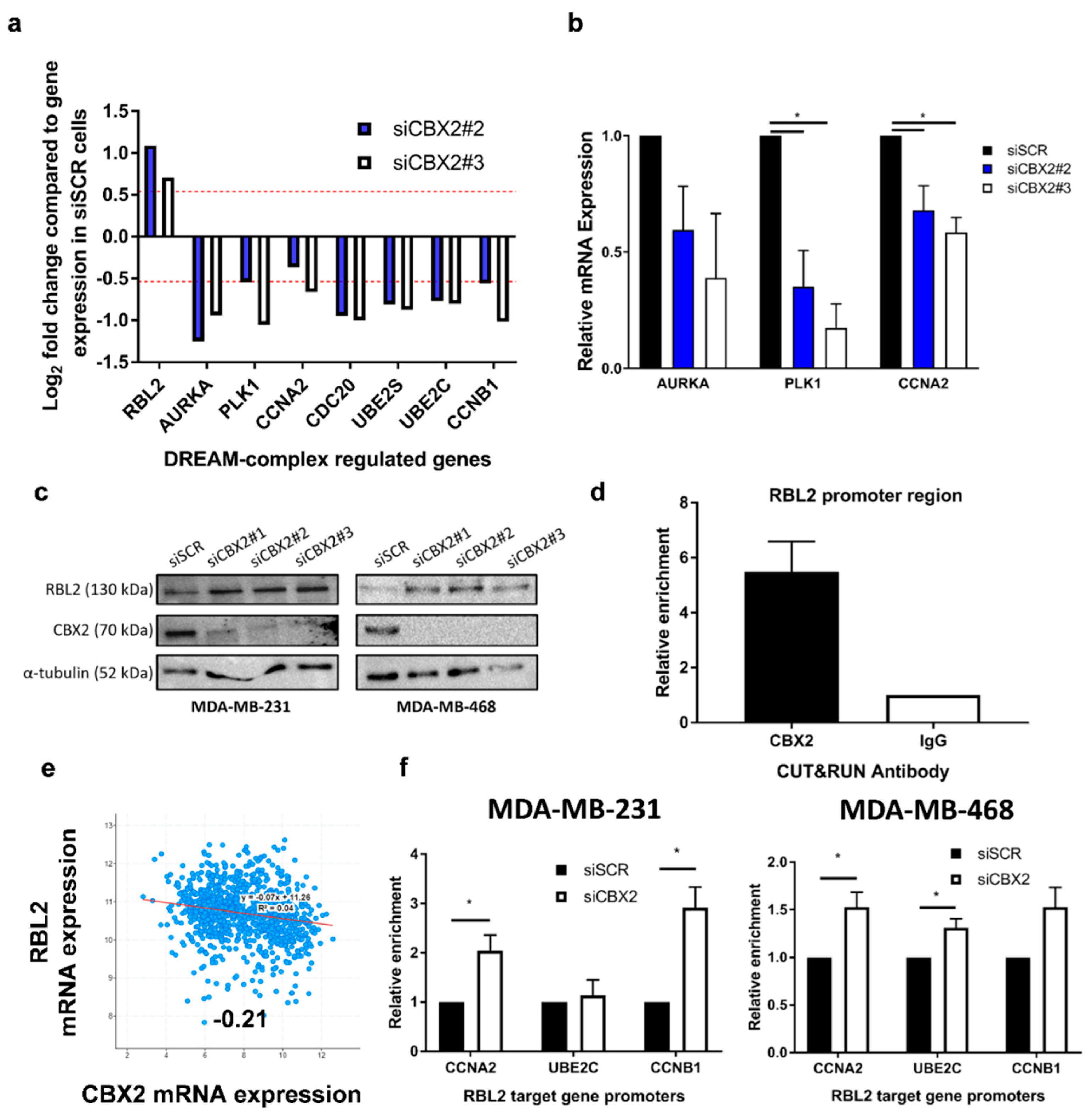
3.3. CBX2 Inhibits RBL2 Expression and DREAM Complex Activity to Promote TNBC Cell Growth
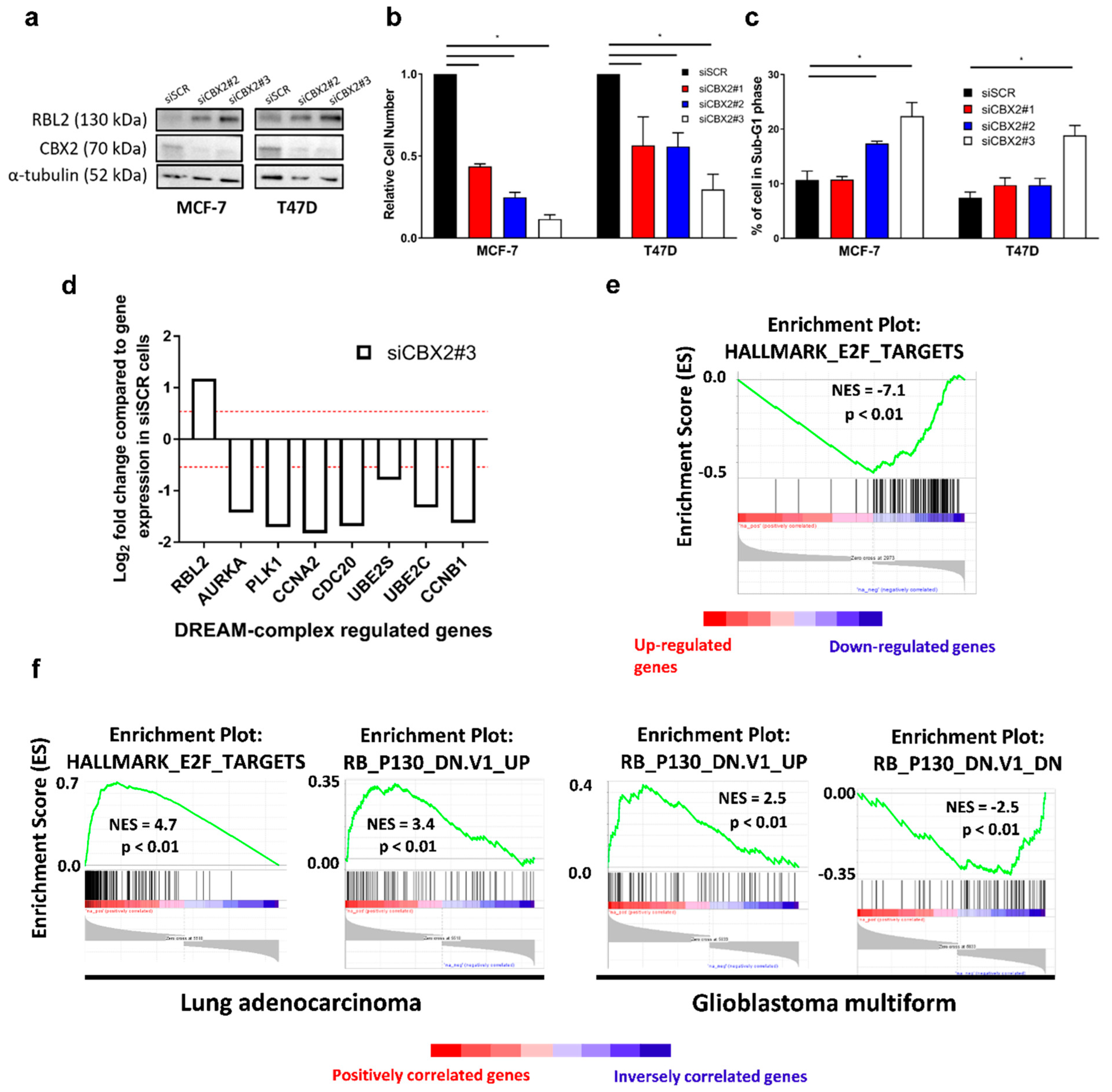
3.4. The CBX2-RBL2 Regulatory Axis May Be Common across Multiple Cancer Types
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernas, S.; Tolaney, S.M. HER2-positive breast cancer: New therapeutic frontiers and overcoming resistance. Ther. Adv. Med. Oncol. 2019, 11, 1758835919833519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Santanach, A.; Blanco, E.; Aloia, L.; Nora, E.P.; Bruneau, B.G.; Di Croce, L. Polycomb Regulates Mesoderm Cell Fate-Specification in Embryonic Stem Cells through Activation and Repression Mechanisms. Cell Stem Cell 2015, 17, 300–315. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.L.; Beckedorff, F.; Zhang, Y.; Garcia-Huidobro, J.; Jiang, H.; Colaprico, A.; Bilbao, D.; Figueroa, M.E.; LaCava, J.; Shiekhattar, R.; et al. Polycomb complexes associate with enhancers and promote oncogenic transcriptional programs in cancer through multiple mechanisms. Nat. Commun. 2018, 9, 3377. [Google Scholar] [CrossRef] [Green Version]
- Clermont, P.L.; Sun, L.; Crea, F.; Thu, K.L.; Zhang, A.; Parolia, A.; Lam, W.L.; Helgason, C.D. Genotranscriptomic meta-analysis of the Polycomb gene CBX2 in human cancers: Initial evidence of an oncogenic role. Br. J. Cancer 2014, 111, 1663–1672. [Google Scholar] [CrossRef]
- Clermont, P.L.; Crea, F.; Chiang, Y.T.; Lin, D.; Zhang, A.; Wang, J.Z.; Parolia, A.; Wu, R.; Xue, H.; Wang, Y.; et al. Identification of the epigenetic reader CBX2 as a potential drug target in advanced prostate cancer. Clin. Epigenetics 2016, 8, 16. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.K.; Lin, H.Y.; Chen, C.F.; Zeng, D. Prognostic values of distinct CBX family members in breast cancer. Oncotarget 2017, 8, 92375–92387. [Google Scholar] [CrossRef]
- Hu, F.F.; Chen, H.; Duan, Y.; Lan, B.; Liu, C.J.; Hu, H.; Dong, X.; Zhang, Q.; Cheng, Y.M.; Liu, M.; et al. CBX2 and EZH2 cooperatively promote the growth and metastasis of lung adenocarcinoma. Mol. Ther. Nucleic Acids 2022, 27, 670–684. [Google Scholar] [CrossRef]
- Li, J.; Xu, Z.; Zhou, L.; Hu, K. Expression profile and prognostic values of Chromobox family members in human glioblastoma. Aging (Albany NY) 2022, 14, 1910. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Li, B.; Yang, L.; Guan, Q. CBX2 depletion inhibits the proliferation, invasion and migration of gastric cancer cells by inactivating the YAP/beta-catenin pathway. Mol. Med. Rep. 2021, 23, 137. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Zhang, X.Y.; Liu, T.; Liu, Y.; Zhao, Y.S.; Pang, D. Chromobox homolog 2 protein: A novel biomarker for predicting prognosis and Taxol sensitivity in patients with breast cancer. Oncol. Lett. 2017, 13, 1149–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pique, D.G.; Montagna, C.; Greally, J.M.; Mar, J.C. A novel approach to modelling transcriptional heterogeneity identifies the oncogene candidate CBX2 in invasive breast carcinoma. Br. J. Cancer 2019, 120, 746–753. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Lv, P.; Su, J.; Miao, K.; Xu, H.; Li, M. Overexpression of CBX2 in breast cancer promotes tumor progression through the PI3K/AKT signaling pathway. Am. J. Transl. Res. 2019, 11, 1668–1682. [Google Scholar] [PubMed]
- Iqbal, M.A.; Siddiqui, S.; Ur Rehman, A.; Siddiqui, F.A.; Singh, P.; Kumar, B.; Saluja, D. Multiomics integrative analysis reveals antagonistic roles of CBX2 and CBX7 in metabolic reprogramming of breast cancer. Mol. Oncol. 2021, 15, 1450–1465. [Google Scholar] [CrossRef]
- Gaughan, L.; Logan, I.R.; Cook, S.; Neal, D.E.; Robson, C.N. Tip60 and histone deacetylase 1 regulate androgen receptor activity through changes to the acetylation status of the receptor. J. Biol. Chem. 2002, 277, 25904–25913. [Google Scholar] [CrossRef] [Green Version]
- Wade, M.A.; Jones, D.; Wilson, L.; Stockley, J.; Coffey, K.; Robson, C.N.; Gaughan, L. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 2015, 43, 196–207. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.; Wilson, M.D.; Spyrou, C.; Brown, G.D.; Hadfield, J.; Odom, D.T. ChIP-seq: Using high-throughput sequencing to discover protein-DNA interactions. Methods 2009, 48, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, T.; Machida, S.; Kurumizaka, H.; Tagami, H.; Nakayama, J.I. Phosphorylation of CBX2 controls its nucleosome-binding specificity. J. Biochem. 2017, 162, 343–355. [Google Scholar] [CrossRef]
- Wang, S.; Alpsoy, A.; Sood, S.; Ordonez-Rubiano, S.C.; Dhiman, A.; Sun, Y.; Jiao, G.; Krusemark, C.J.; Dykhuizen, E.C. A Potent, Selective CBX2 Chromodomain Ligand and Its Cellular Activity during Prostate Cancer Neuroendocrine Differentiation. Chembiochem 2021, 22, 2335–2344. [Google Scholar] [CrossRef]
- Zou, Z.; Tao, T.; Li, H.; Zhu, X. mTOR signaling pathway and mTOR inhibitors in cancer: Progress and challenges. Cell Biosci. 2020, 10, 31. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Sadasivam, S.; DeCaprio, J.A. The DREAM complex: Master coordinator of cell cycle-dependent gene expression. Nat. Rev. Cancer 2013, 13, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litovchick, L.; Sadasivam, S.; Florens, L.; Zhu, X.; Swanson, S.K.; Velmurugan, S.; Chen, R.; Washburn, M.P.; Liu, X.S.; DeCaprio, J.A. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 2007, 26, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ren, B.; Zhuang, H.; Zhong, Y.; Nan, Y. CBX2 Induces Glioma Cell Proliferation and Invasion through the Akt/PI3K Pathway. Technol. Cancer Res. Treat. 2021, 20, 15330338211045831. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, L.J.; Watson, Z.L.; Qamar, L.; Yamamoto, T.M.; Post, M.D.; Berning, A.A.; Spillman, M.A.; Behbakht, K.; Bitler, B.G. CBX2 identified as driver of anoikis escape and dissemination in high grade serous ovarian cancer. Oncogenesis 2018, 7, 92. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Tian, Y.; Wang, C.; Jiang, K.; Li, R.; Yao, Y.; Zhang, R.; Sun, D.; Liang, R.; Gao, Z.; et al. CBX2 Regulates Proliferation and Apoptosis via the Phosphorylation of YAP in Hepatocellular Carcinoma. J. Cancer 2019, 10, 2706–2719. [Google Scholar] [CrossRef] [Green Version]
- Mallela, K.; Kumar, A. Role of TSC1 in physiology and diseases. Mol. Cell. Biochem. 2021, 476, 2269–2282. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Cao, W.; Li, J.; Hao, Q.; Vadgama, J.V.; Wu, Y. AMP-activated protein kinase: A potential therapeutic target for triple-negative breast cancer. Breast Cancer Res. 2019, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef]
- Claudio, P.P.; Howard, C.M.; Baldi, A.; De Luca, A.; Fu, Y.; Condorelli, G.; Sun, Y.; Colburn, N.; Calabretta, B.; Giordano, A. p130/pRb2 has growth suppressive properties similar to yet distinctive from those of retinoblastoma family members pRb and p107. Cancer Res. 1994, 54, 5556–5560. [Google Scholar] [PubMed]
- Baldi, A.; Esposito, V.; De Luca, A.; Howard, C.M.; Mazzarella, G.; Baldi, F.; Caputi, M.; Giordano, A. Differential expression of the retinoblastoma gene family members pRb/p105, p107, and pRb2/p130 in lung cancer. Clin. Cancer Res. 1996, 2, 1239–1245. [Google Scholar] [PubMed]
- Susini, T.; Massi, D.; Paglierani, M.; Masciullo, V.; Scambia, G.; Giordano, A.; Amunni, G.; Massi, G.; Taddei, G.L. Expression of the retinoblastoma-related gene Rb2/p130 is downregulated in atypical endometrial hyperplasia and adenocarcinoma. Hum. Pathol. 2001, 32, 360–367. [Google Scholar] [CrossRef] [PubMed]
- D’Andrilli, G.; Masciullo, V.; Bagella, L.; Tonini, T.; Minimo, C.; Zannoni, G.F.; Giuntoli, R.L., 2nd; Carlson, J.A., Jr.; Soprano, D.R.; Soprano, K.J.; et al. Frequent loss of pRb2/p130 in human ovarian carcinoma. Clin. Cancer Res. 2004, 10, 3098–3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, F.; Khan, T.; Ali, N.; Malik, F.A.; Kayani, M.A.; Shah, S.T.; Saeed, M. Promoter Methylation Status Modulate the Expression of Tumor Suppressor (RbL2/p130) Gene in Breast Cancer. PLoS ONE 2015, 10, e0134687. [Google Scholar] [CrossRef] [PubMed]
- Farman, F.U.; Iqbal, M.; Azam, M.; Saeed, M. Nucleosomes positioning around transcriptional start site of tumor suppressor (Rbl2/p130) gene in breast cancer. Mol. Biol. Rep. 2018, 45, 185–194. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hallmark Gene Set | ||||
| NAME | SIZE | NES | NOM p-val | FDR q-val |
| HALLMARK_G2M_CHECKPOINT | 49 | −4.395 | <0.001 | <0.001 |
| HALLMARK_E2F_TARGETS | 45 | −4.221 | <0.001 | <0.001 |
| HALLMARK_XENOBIOTIC_METABOLISM | 48 | −2.373 | <0.001 | 0.005 |
| HALLMARK_ESTROGEN_RESPONSE_LATE | 48 | −2.173 | 0.004 | 0.018 |
| HALLMARK_APOPTOSIS | 46 | −2.106 | 0.006 | 0.023 |
| HALLMARK_MITOTIC_SPINDLE | 46 | −1.989 | 0.006 | 0.037 |
| HALLMARK_MTORC1_SIGNALING | 45 | −1.902 | 0.012 | 0.050 |
| HALLMARK_ESTROGEN_RESPONSE_EARLY | 56 | −1.866 | 0.010 | 0.054 |
| HALLMARK_UV_RESPONSE_UP | 33 | −1.864 | 0.008 | 0.048 |
| HALLMARK_MYC_TARGETS_V1 | 19 | −1.853 | 0.010 | 0.046 |
| Gene Ontology Gene Set | ||||
| NAME | SIZE | NES | NOM p-val | FDR q-val |
| GO_NEUROGENESIS | 288 | −3.522 | <0.001 | <0.001 |
| GO_CHROMOSOME_ORGANIZATION | 184 | −3.492 | <0.001 | <0.001 |
| GO_POSITIVE_REGULATION_OF_RESPONSE_TO_STIMULUS | 390 | −3.478 | <0.001 | <0.001 |
| GO_CELL_CYCLE | 233 | −3.416 | <0.001 | <0.001 |
| GO_REGULATION_OF_CELL_PROLIFERATION | 336 | −3.369 | <0.001 | <0.001 |
| GO_POSITIVE_REGULATION_OF_GENE_EXPRESSION | 347 | −3.362 | <0.001 | <0.001 |
| GO_CELL_PROLIFERATION | 134 | −3.360 | <0.001 | <0.001 |
| GO_NEGATIVE_REGULATION_OF_DEVELOPMENTAL_PROCESS | 168 | −3.295 | <0.001 | <0.001 |
| GO_POSITIVE_REGULATION_OF_CELL_PROLIFERATION | 185 | −3.286 | <0.001 | <0.001 |
| GO_CELL_DIVISION | 96 | −3.240 | <0.001 | <0.001 |
| Oncogenic Signature Gene Set | ||||
| NAME | SIZE | NES | NOM p-val | FDR q-val |
| RPS14_DN.V1_DN | 32 | −3.631 | <0.001 | <0.001 |
| E2F3_UP.V1_UP | 45 | −3.291 | <0.001 | <0.001 |
| PRC2_EZH2_UP.V1_DN | 46 | −2.782 | <0.001 | 0.002 |
| LEF1_UP.V1_UP | 47 | −2.593 | <0.001 | 0.005 |
| CSR_LATE_UP.V1_UP | 47 | −2.565 | <0.001 | 0.005 |
| NFE2L2.V2 | 87 | −2.331 | <0.001 | 0.015 |
| HOXA9_DN.V1_DN | 43 | −2.244 | <0.001 | 0.024 |
| BMI1_DN.V1_DN | 37 | −2.179 | 0.004 | 0.031 |
| CORDENONSI_YAP_CONSERVED_SIGNATURE | 20 | −2.140 | 0.002 | 0.035 |
| KRAS.600_UP.V1_DN | 53 | −2.099 | 0.004 | 0.040 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilton, L.J.; Warren, C.; Humphries, R.M.; Kalsi, S.; Waters, E.; Francis, T.; Dobrowinski, W.; Beltran-Alvarez, P.; Wade, M.A. The Epigenetic Regulatory Protein CBX2 Promotes mTORC1 Signalling and Inhibits DREAM Complex Activity to Drive Breast Cancer Cell Growth. Cancers 2022, 14, 3491. https://doi.org/10.3390/cancers14143491
Bilton LJ, Warren C, Humphries RM, Kalsi S, Waters E, Francis T, Dobrowinski W, Beltran-Alvarez P, Wade MA. The Epigenetic Regulatory Protein CBX2 Promotes mTORC1 Signalling and Inhibits DREAM Complex Activity to Drive Breast Cancer Cell Growth. Cancers. 2022; 14(14):3491. https://doi.org/10.3390/cancers14143491
Chicago/Turabian StyleBilton, Lucie J., Chloe Warren, Rebecca M. Humphries, Shannon Kalsi, Ella Waters, Thomas Francis, Wojtek Dobrowinski, Pedro Beltran-Alvarez, and Mark A. Wade. 2022. "The Epigenetic Regulatory Protein CBX2 Promotes mTORC1 Signalling and Inhibits DREAM Complex Activity to Drive Breast Cancer Cell Growth" Cancers 14, no. 14: 3491. https://doi.org/10.3390/cancers14143491