
Changes of Immune Cell Fractions in Patients Treated with Immune Checkpoint Inhibitors

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
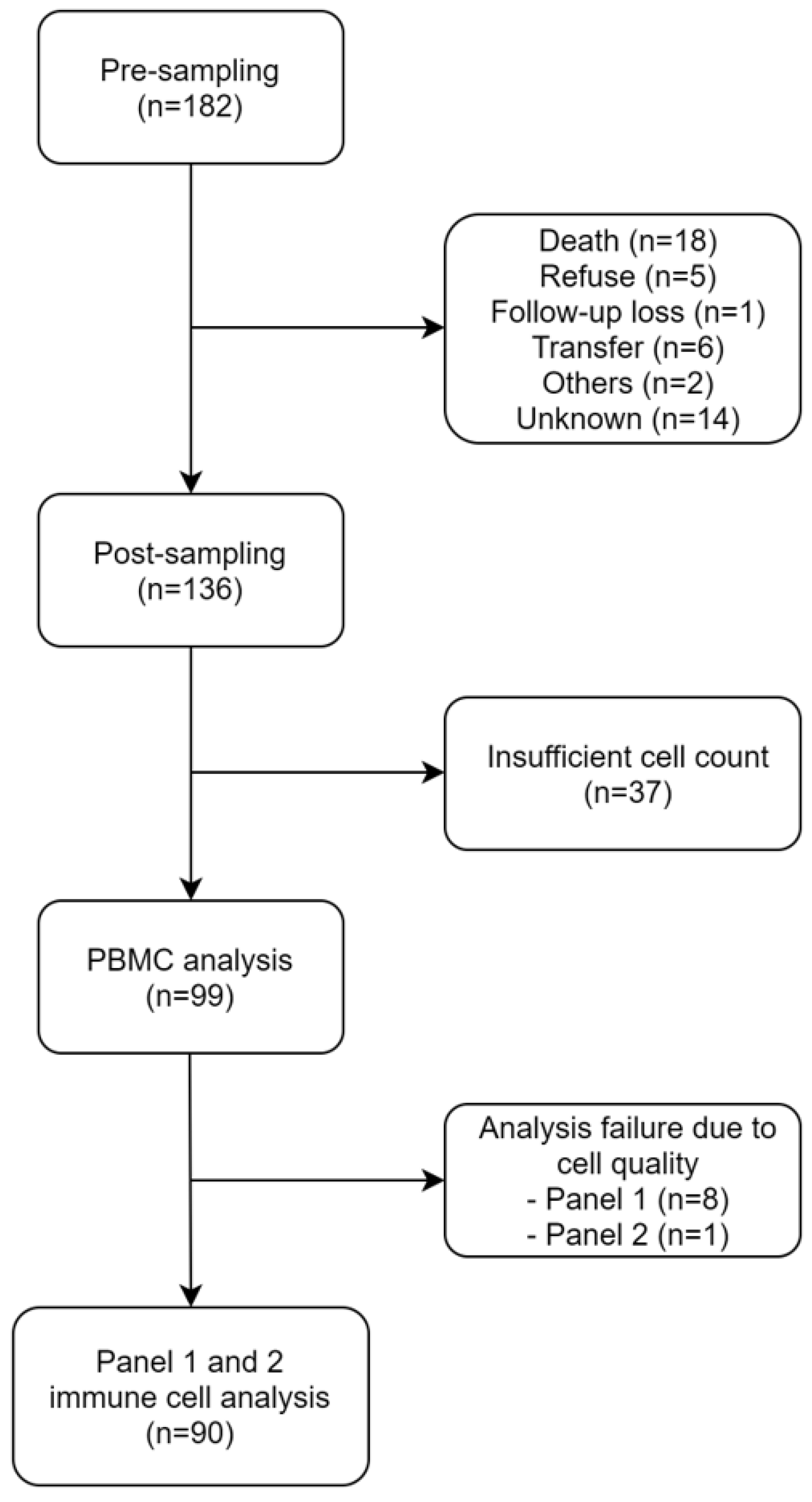
2.1. Patients
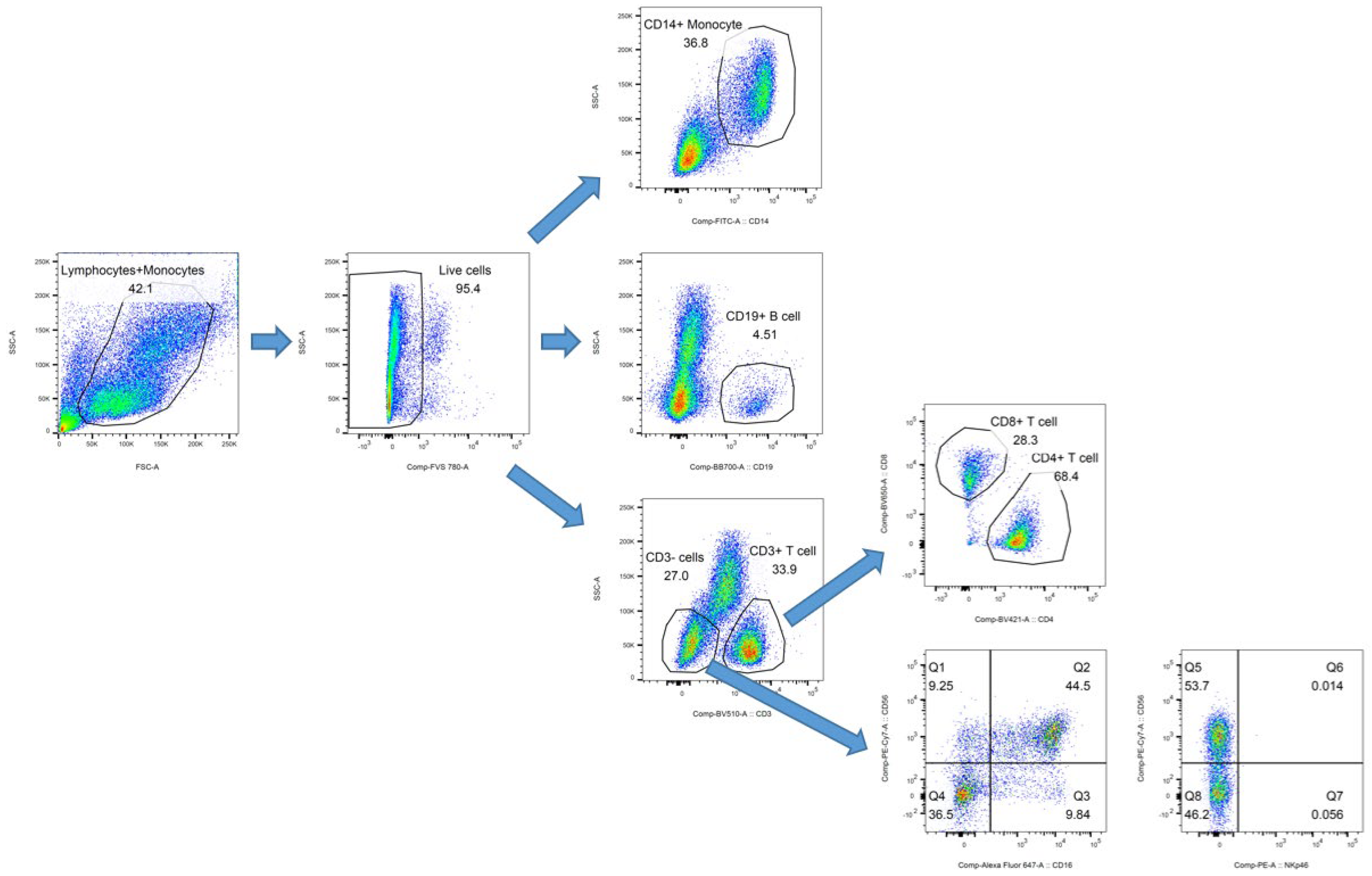
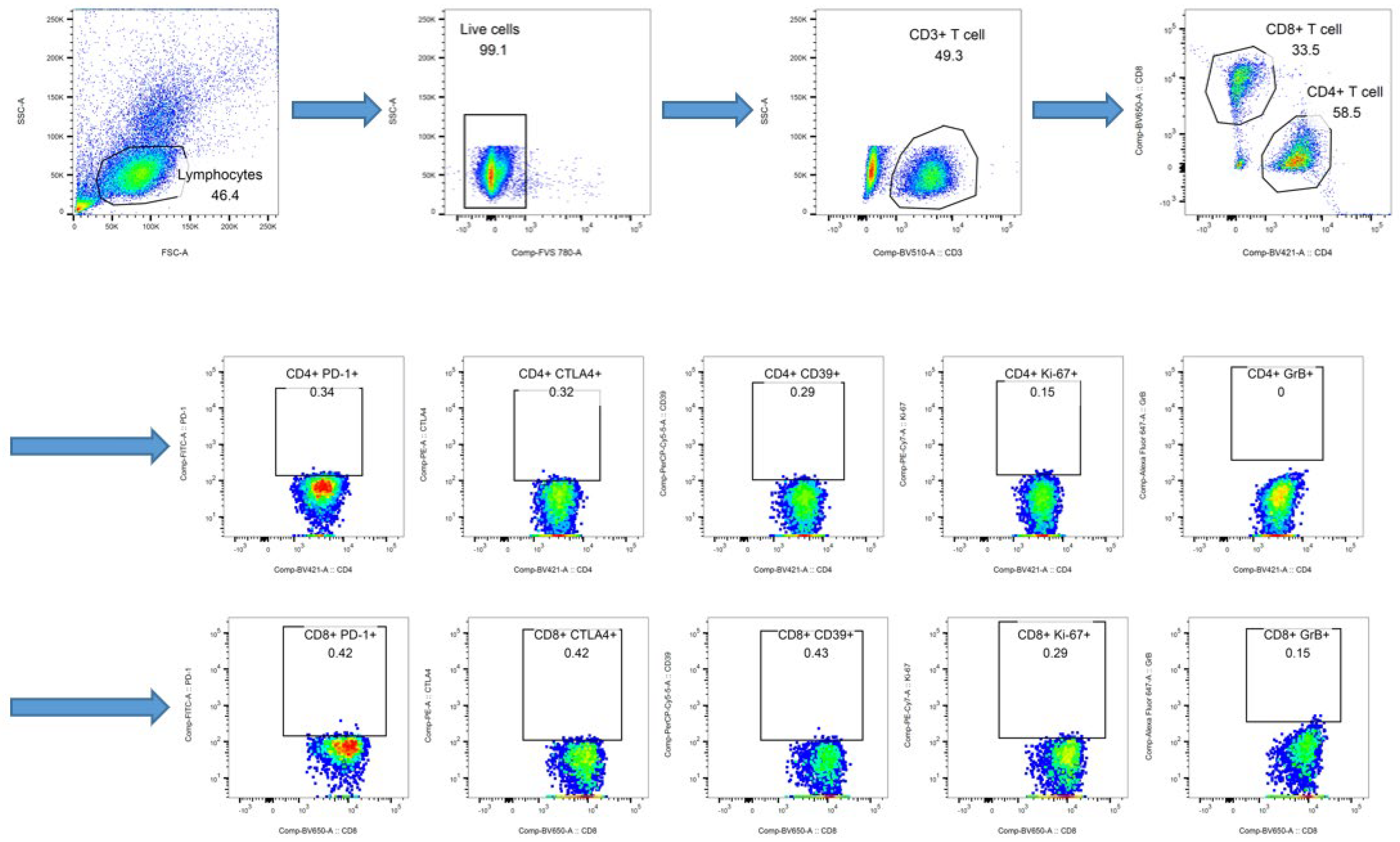
2.2. Immune Cell Analysis
2.3. Response and Survival Analysis
3. Results
3.1. Patients and Immune Cell Analysis
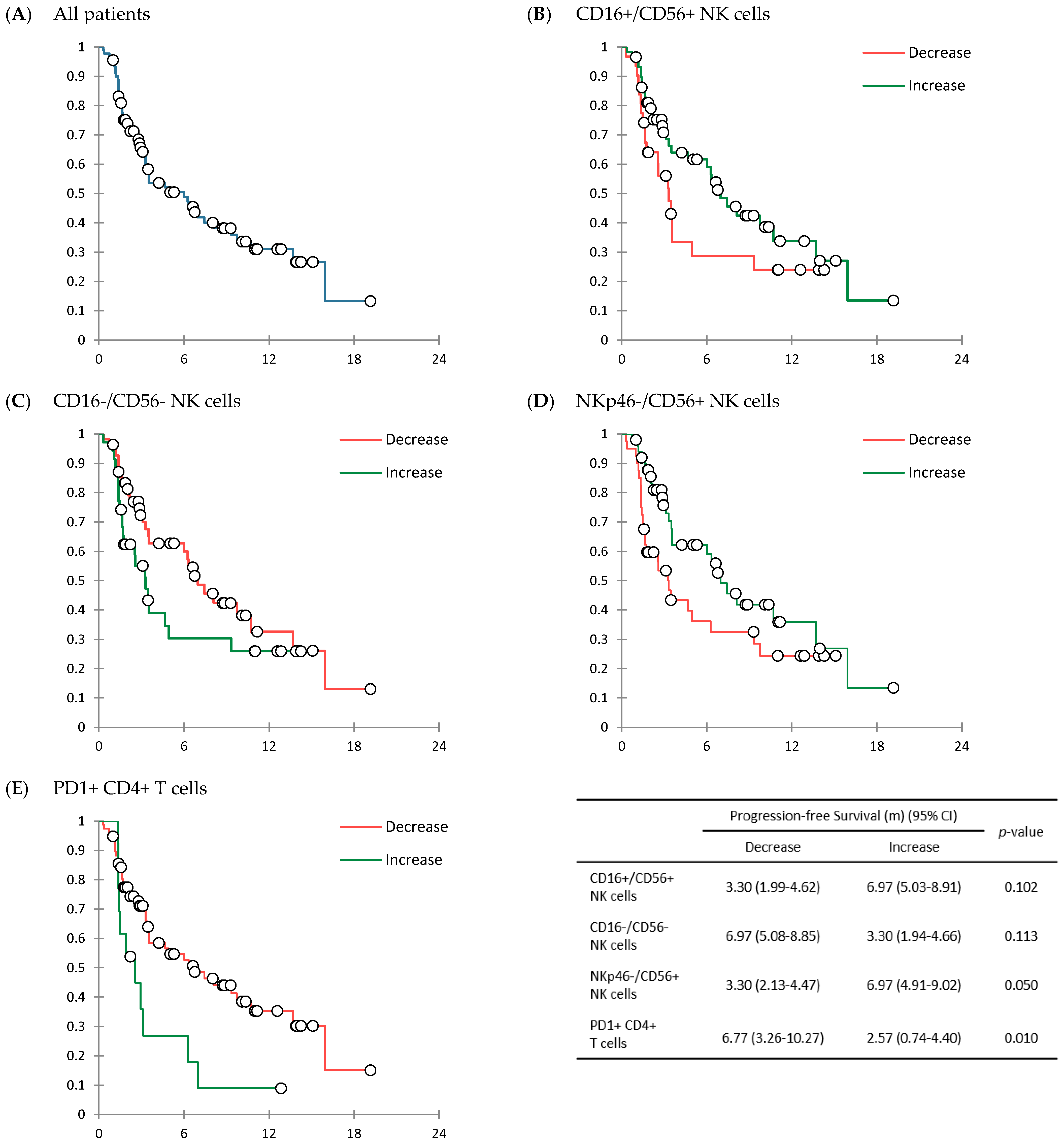
3.2. Response to Treatment and Survival Outcomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody Name | Catalog No. | Antibody Volume (µL) | |
|---|---|---|---|
| Panel 1 | CD4-BV421 | 562,424 | 0.3 (T4) |
| CD3-BV510 | 564,713 | 0.6 (T3) | |
| CD8-BV650 | 563,821 | 0.3 (T4) | |
| CD14-FITC | 347,493 | 0.2 (1:100) | |
| NKp46-PE | 557,991 | 5 (T2) | |
| CD19-BB700 | 566,397 | 0.6 (T3) | |
| CD56-PE-Cy7 | 557,747 | 1.2 (T2) | |
| CD16-AF647 | 557,710 | 0.05 (1:100) | |
| Panel 2 | CD4-BV421 | 562,424 | 0.3 (T4) |
| CD3-BV510 | 564,713 | 0.6 (T3) | |
| CD8-BV650 | 563,821 | 0.3 (T4) | |
| PD-1-FITC | 557,860 | 2 (T1) | |
| CTLA4-PE | 555,853 | 5 (T2) | |
| CD39-PerCP-Cy5.5 | 564,899 | 1.2 (T2) | |
| Ki-67-PE-Cy7 | 561,283 | 1.2 (T2) | |
| GranzymeB-AF647 | 560,212 | 0.1 (1:50) |
| Panel 1 | Surface Stains | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| FVS 780 (565,388) | CD3 BV510 (564,713) | CD4 BV421 (562,424) | CD8 BV650 (563,821) | CD14 FITC (347,493) | NKp46 PE (557,991) | CD19 BB700 (566,396) | CD56 PE-Cy7 (557,747) | CD16 AF647 (557,710) | |
| 1. Unstained PBMC compensation control | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 2. Killed PBMC FVS780 viability compensation control | 1:1000 | NA | NA | NA | NA | NA | NA | NA | NA |
| 3. NKp46 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 10 µL * | NA | 0.6 µL | 1.2 µL | 2.5 µL * |
| 4. CD16 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 10 µL * | 5 µL | 0.6 µL | 1.2 µL | NA * |
| 5. CD56 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 10 µL * | 5 µL | 0.6 µL | NA | 2.5 µL * |
| 6. All Stains | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 10 µL * | 5 µL | 0.6 µL | 1.2 µL | 2.5 µL * |
| Panel 2 | Surface Stains | Intracellular Stains | |||||||
|---|---|---|---|---|---|---|---|---|---|
| FVS 780 (565,388) | CD3 BV510 (564,713) | CD4 BV421 (562,424) | CD8 BV650 (563,821) | PD-1 FITC (557,860) | CD39-PerCP-Cy5.5 (564,899) | CTLA4 PE (555,853) | Ki-67-PE-Cy7 (561,283) | GranzymeB-AF647 (560,212) | |
| 1. Unstained PBMC compensation control | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| 2. Killed PBMC FVS780 viability compensation control | 1:1000 | NA | NA | NA | NA | NA | NA | NA | NA |
| 3. PD-1 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | NA | 1.2 µL | 5 µL | 1.2 µL | 5 µL * |
| 4. CD39 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 2 µL | NA | 5 µL | 1.2 µL | 5 µL * |
| 5. CTLA4 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 2 µL | 1.2 µL | NA | 1.2 µL | 5 µL * |
| 6. Ki-67 FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 2 µL | 1.2 µL | 5 µL | NA | 5 µL * |
| 7. Granzyme B FMO | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 2 µL | 1.2 µL | 5 µL | 1.2 µL | NA * |
| 8. All Stains | 1:1000 | 0.6 µL | 0.3 µL | 0.3 µL | 2 µL | 1.2 µL | 5 µL | 1.2 µL | 5 µL * |

References
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.; Johnson, D.H.; The Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. N. Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current Challenges in Cancer Treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzone, L.; Salomone, S.; Libra, M. Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Front. Pharmacol. 2018, 9, 1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Fu, Y.; Zhu, B.; Wang, J.; Zhang, B. Predictive Biomarkers of Immune Checkpoint Inhibitors-Related Toxicities. Front. Immunol. 2020, 11, 2023. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Liu, B.; Song, Y.; Liu, D. Recent development in clinical applications of PD-1 and PD-L1 antibodies for cancer immunotherapy. J. Hematol. Oncol. 2017, 10, 174. [Google Scholar] [CrossRef] [Green Version]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef]
- Wang, C.; Yu, X.; Wang, W. A meta-analysis of efficacy and safety of antibodies targeting PD-1/PD-L1 in treatment of advanced nonsmall cell lung cancer. Medicine 2016, 95, e5539. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, F.R.; McElhinny, A.; Stanforth, D.; Ranger-Moore, J.; Jansson, M.; Kulangara, K.; Richardson, W.; Towne, P.; Hanks, D.; Vennapusa, B.; et al. PD-L1 Immunohistochemistry Assays for Lung Cancer: Results from Phase 1 of the Blueprint PD-L1 IHC Assay Comparison Project. J. Thorac. Oncol. 2017, 12, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, R.; Longshore, J.W.; López-Ríos, F.; Merkelbach-Bruse, S.; Normanno, N.; Rouleau, E.; Penault-Llorca, F. Implementing TMB measurement in clinical practice: Considerations on assay requirements. ESMO Open 2019, 4, e000442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reck, M.; Rodriguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csoszi, T.; Fulop, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef] [Green Version]
- Muro, K.; Chung, H.C.; Shankaran, V.; Geva, R.; Catenacci, D.; Gupta, S.; Eder, J.P.; Golan, T.; Le, D.T.; Burtness, B.; et al. Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): A multicentre, open-label, phase 1b trial. Lancet Oncol. 2016, 17, 717–726. [Google Scholar] [CrossRef]
- Gadgeel, S.; Rodríguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Dómine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated Analysis From KEYNOTE-189: Pembrolizumab or Placebo Plus Pemetrexed and Platinum for Previously Untreated Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Park, S.; Ock, C.Y.; Kim, H.; Pereira, S.; Park, S.; Ma, M.; Choi, S.; Kim, S.; Shin, S.; Aum, B.J.; et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as Complementary Biomarker for Immune Checkpoint Inhibition in Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 1916–1928. [Google Scholar] [CrossRef]
- Teng, M.W.; Galon, J.; Fridman, W.H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Investig. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors in cancer therapy: A focus on T-regulatory cells. Immunol. Cell Biol. 2018, 96, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Sivori, S.; Vitale, M.; Morelli, L.; Sanseverino, L.; Augugliaro, R.; Bottino, C.; Moretta, L.; Moretta, A. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med. 1997, 186, 1129–1136. [Google Scholar] [CrossRef] [Green Version]
- Glasner, A.; Levi, A.; Enk, J.; Isaacson, B.; Viukov, S.; Orlanski, S.; Scope, A.; Neuman, T.; Enk, C.D.; Hanna, J.H.; et al. NKp46 Receptor-Mediated Interferon-γ Production by Natural Killer Cells Increases Fibronectin 1 to Alter Tumor Architecture and Control Metastasis. Immunity 2018, 48, 107.e4–119.e4. [Google Scholar] [CrossRef]
- Murad, S.; Michen, S.; Becker, A.; Fussel, M.; Schackert, G.; Tonn, T.; Momburg, F.; Temme, A. NKG2C+ NK Cells for Immunotherapy of Glioblastoma Multiforme. Int. J. Mol. Sci. 2022, 23, 5857. [Google Scholar] [CrossRef] [PubMed]
- Mushti, S.L.; Mulkey, F.; Sridhara, R. Evaluation of Overall Response Rate and Progression-Free Survival as Potential Surrogate Endpoints for Overall Survival in Immunotherapy Trials. Clin. Cancer Res. 2018, 24, 2268–2275. [Google Scholar] [CrossRef] [Green Version]

| Variables (n = 90) | No. of Patients (%) |
|---|---|
| Median age (years) (range) | 65.0 (26 to 84) |
| Sex | |
| Male | 55 (61.1%) |
| Female | 35 (38.9%) |
| ECOG performance status | |
| 0 | 10 (11.1%) |
| 1 | 78 (86.7%) |
| 2 | 2 (2.2%) |
| Type of cancer | |
| Lung cancer | 45 (50.0%) |
| Others † | 45 (50.0%) |
| Previous lines of treatment | |
| 0 | 13 (14.4%) |
| 1 | 41 (45.6%) |
| 2 | 22 (24.4%) |
| ≥3 | 14 (15.6%) |
| Immune checkpoint inhibitor | |
| Pembrolizumab | 36 (40.0%) |
| Nivolumab | 40 (44.4%) |
| Atezolizumab | 14 (15.6%) |
| Durvalumab | - |
| Combined chemotherapy | |
| Yes ‡ | 2 (2.2%) |
| No | 88 (97.8%) |
| Type of Immune Cells | Median Value of Change (%) (Range) |
|---|---|
| Panel 1 | |
| CD4+ T cell | −1.95 (−36.20 to 31.60) |
| CD8+ T cell | 1.55 (−27.40 to 32.50) |
| CD14+ monocyte | −0.51 (−34.90 to 43.60) |
| CD19+ B cell | −0.27 (−6.90 to 10.38) |
| Q1: CD16−/CD56+ | −0.45 (−17.20 to 11.31) |
| Q2: CD16+/CD56+ | 4.05 (−37.10 to 48.58) |
| Q3: CD16+/CD56− | 0.33 (−18.80 to 11.17) |
| Q4: CD16−/CD56− | −4.00 (−49.60 to 42.80) |
| Q5: NKp46−/CD56+ | 0.30 (−24.80 to 20.50) |
| Q6: Kp46+/CD56+ | 1.45 (−28.14 to 33.90) |
| Q7: NKp46+/CD56− | 0.12 (−3.25 to 4.24) |
| Q8: NKp46−/CD56− | −4.60 (−51.60 to 43.10) |
| Panel 2 | |
| PD-1+ CD4+ | −5.49 (−30.60 to 24.77) |
| CTLA4+ CD4+ | 1.25 (−12.30 to 27.80) |
| CD39+ CD4+ | 0.29 (−6.50 to 12.71) |
| Ki-67+ CD4+ | 0.45 (−8.66 to 6.90) |
| GrB+ CD4+ | 0.96 (−26.82 to 24.30) |
| PD-1+ CD8+ | −5.31 (−31.47 to 42.57) |
| CTLA4+ CD8+ | 0.71 (−13.10 to 32.20) |
| CD39+ CD8+ | 0.37 (−19.67 to 17.11) |
| Ki-67+ CD8+ | 0.53 (−14.80 to 20.60) |
| Variables (n = 90) | Value |
|---|---|
| Best response | |
| CR | - |
| PR | 13 (14.4%) |
| SD | 32 (35.6%) |
| PD | 34 (37.8%) |
| Unknown | 11 (12.2%) |
| ORR | 13 (14.4%) |
| CBR | 45 (50.0%) |
| Median treatment cycle (range) | 5 (1 to 28) |
| Median treatment duration (months) (IQR) | 2.40 (1.31 to 5.53) |
| Median PFS (months) (95% CI) | 6.00 (3.11 to 8.89) |
| Median OS (months) (95% CI) | 13.90 (5.62 to 22.19) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.R.; Kang, J.H.; Kim, S.H.; Kim, S.T.; Kim, I.; Min, Y.J.; Shin, S.H.; Oh, S.Y.; Lee, G.-W.; Lee, J.H.; et al. Changes of Immune Cell Fractions in Patients Treated with Immune Checkpoint Inhibitors. Cancers 2022, 14, 3440. https://doi.org/10.3390/cancers14143440
Kim HR, Kang JH, Kim SH, Kim ST, Kim I, Min YJ, Shin SH, Oh SY, Lee G-W, Lee JH, et al. Changes of Immune Cell Fractions in Patients Treated with Immune Checkpoint Inhibitors. Cancers. 2022; 14(14):3440. https://doi.org/10.3390/cancers14143440
Chicago/Turabian StyleKim, Hye Ryeon, Jung Hun Kang, Sung Hyun Kim, Seung Tae Kim, Ilhwan Kim, Young Joo Min, Seong Hoon Shin, Sung Yong Oh, Gyeong-Won Lee, Ji Hyun Lee, and et al. 2022. "Changes of Immune Cell Fractions in Patients Treated with Immune Checkpoint Inhibitors" Cancers 14, no. 14: 3440. https://doi.org/10.3390/cancers14143440
APA StyleKim, H. R., Kang, J. H., Kim, S. H., Kim, S. T., Kim, I., Min, Y. J., Shin, S. H., Oh, S. Y., Lee, G.-W., Lee, J. H., Ji, J. H., Huh, S. J., & Lee, S. (2022). Changes of Immune Cell Fractions in Patients Treated with Immune Checkpoint Inhibitors. Cancers, 14(14), 3440. https://doi.org/10.3390/cancers14143440