
Liquid Biopsy in Glioblastoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Pitfalls and Limitations of Current Techniques for GBM Diagnosis and Follow-Up
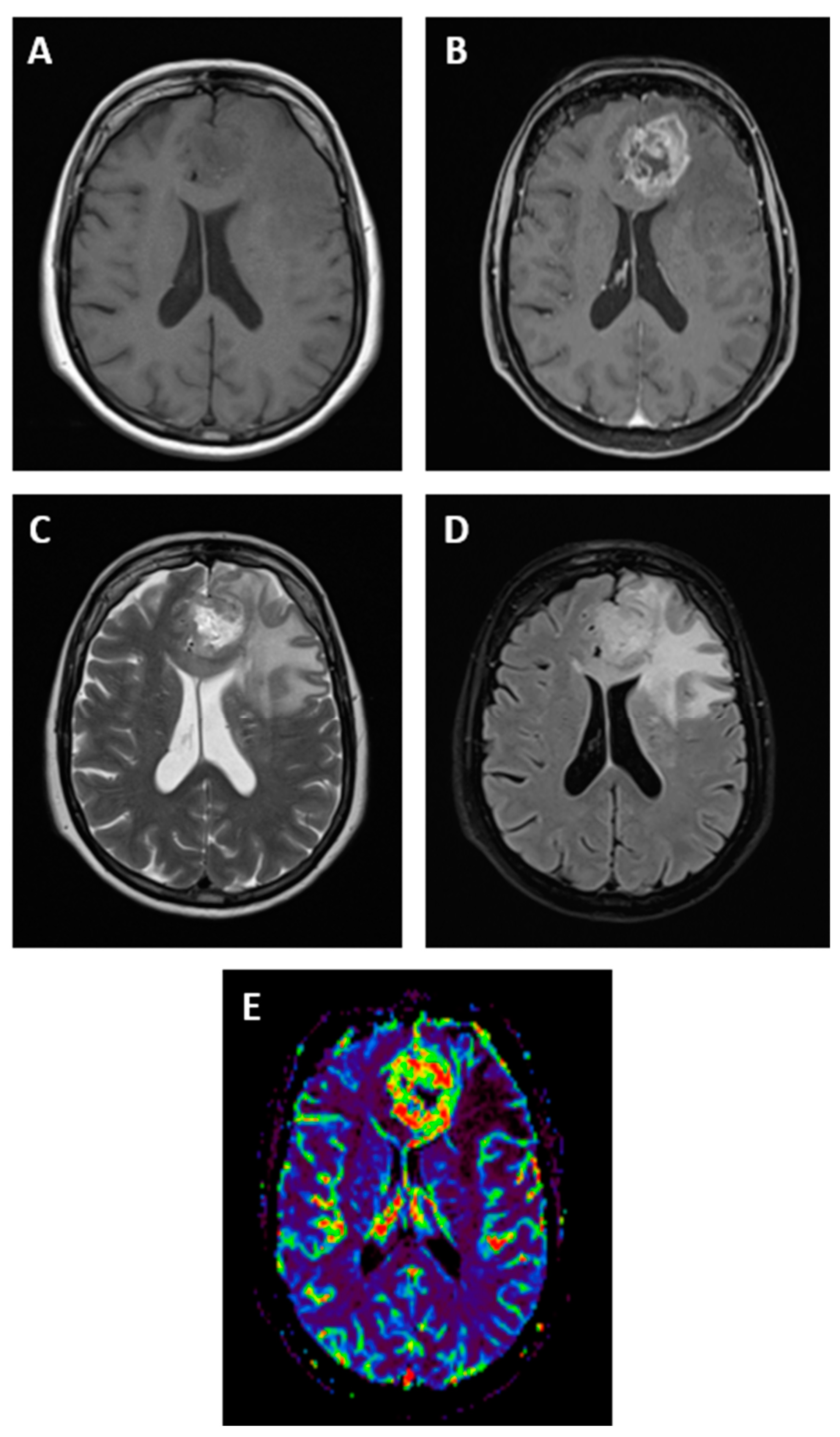
2.1. Neuroimaging
2.2. Tissue Biopsies
3. Circulating Biomarkers in GBM
3.1. Circulating Tumor DNA
3.2. Circulating microRNAs
3.3. Circulating Tumor Cells
3.4. Extracellular Vesicles
3.5. Circulating Nucleosome-Associated Histone Modifications
4. Conclusions and Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Kim, H.J.; Park, J.W.; Lee, J.H. Genetic Architectures and Cell-of-Origin in Glioblastoma. Front. Oncol. 2020, 10, 615400. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.D.; Ostrom, Q.T.; Kruchko, C.; Patil, N.; Tihan, T.; Cioffi, G.; Fuchs, H.E.; Waite, K.A.; Jemal, A.; Siegel, R.L.; et al. Brain and other central nervous system tumor statistics, 2021. CA Cancer J. Clin. 2021, 71, 381–406. [Google Scholar] [CrossRef] [PubMed]
- Kyritsis, A.P.; Bondy, M.L.; Rao, J.S.; Sioka, C. Inherited predisposition to glioma. Neuro-Oncology 2010, 12, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez Castro, L.N.; Wesseling, P. The cIMPACT-NOW updates and their significance to current neuro-oncology practice. Neurooncol. Pract. 2021, 8, 4–10. [Google Scholar] [CrossRef]
- Weller, M.; van den Bent, M.; Preusser, M.; Le Rhun, E.; Tonn, J.C.; Minniti, G.; Bendszus, M.; Balana, C.; Chinot, O.; Dirven, L.; et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021, 18, 170–186. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. S5), S2–S8. [Google Scholar] [CrossRef] [Green Version]
- Müller Bark, J.; Kulasinghe, A.; Chua, B.; Day, B.W.; Punyadeera, C. Circulating biomarkers in patients with glioblastoma. Br. J. Cancer 2020, 122, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Delgado-López, P.D.; Riñones-Mena, E.; Corrales-García, E.M. Treatment-related changes in glioblastoma: A review on the controversies in response assessment criteria and the concepts of true progression, pseudoprogression, pseudoresponse and radionecrosis. Clin. Transl. Oncol. 2018, 20, 939–953. [Google Scholar] [CrossRef]
- Ahmed, R.; Oborski, M.J.; Hwang, M.; Lieberman, F.S.; Mountz, J.M. Malignant gliomas: Current perspectives in diagnosis, treatment, and early response assessment using advanced quantitative imaging methods. Cancer Manag. Res. 2014, 6, 149–170. [Google Scholar] [PubMed] [Green Version]
- Wirsching, H.G.; Galanis, E.; Weller, M. Glioblastoma. Handb. Clin. Neurol. 2016, 134, 381–397. [Google Scholar] [PubMed]
- Lundy, P.; Domino, J.; Ryken, T.; Fouke, S.; McCracken, D.J.; Ormond, D.R.; Olson, J.J. The role of imaging for the management of newly diagnosed glioblastoma in adults: A systematic review and evidence-based clinical practice guideline update. J. Neurooncol. 2020, 150, 95–120. [Google Scholar] [CrossRef]
- Pope, W.B.; Brandal, G. Conventional and advanced magnetic resonance imaging in patients with high-grade glioma. Q. J. Nucl. Med. Mol. Imaging 2018, 62, 239–253. [Google Scholar] [CrossRef]
- Van Dijken, B.R.J.; van Laar, P.J.; Holtman, G.A.; van der Hoorn, A. Diagnostic accuracy of magnetic resonance imaging techniques for treatment response evaluation in patients with high-grade glioma, a systematic review and meta-analysis. Eur. Radiol. 2017, 27, 4129–4144. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-López, E.; Calatayud-Pérez, J.; Castells-Yus, I.; Gimeno-Peribáñez, M.J.; Mendoza-Calvo, N.; Morcillo, M.; Schuhmacher, A.J. Diagnosis of Glioblastoma by Immuno-Positron Emission Tomography. Cancers 2021, 14, 74. [Google Scholar] [CrossRef] [PubMed]
- Fathi Kazerooni, A.; Bakas, S.; Saligheh Rad, H.; Davatzikos, C. Imaging signatures of glioblastoma molecular characteristics: A radiogenomics review. J. Magn. Reson. Imaging 2020, 52, 54–69. [Google Scholar] [CrossRef]
- Vagvala, S.; Guenette, J.P.; Jaimes, C.; Huang, R.Y. Imaging diagnosis and treatment selection for brain tumors in the era of molecular therapeutics. Cancer Imaging 2022, 22, 19. [Google Scholar] [CrossRef]
- Brandes, A.A.; Franceschi, E.; Tosoni, A.; Blatt, V.; Pession, A.; Tallini, G.; Bertorelle, R.; Bartolini, S.; Calbucci, F.; Andreoli, A.; et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol. 2008, 26, 2192–2197. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.C.; Kim, C.Y.; Han, J.H.; Choe, G.Y.; Kim, J.H.; Kim, J.H.; Kim, I.A. Pseudoprogression in patients with malignant gliomas treated with concurrent temozolomide and radiotherapy: Potential role of p53. J. Neurooncol. 2011, 102, 157–162. [Google Scholar] [CrossRef]
- Qian, X.; Tan, H.; Zhang, J.; Liu, K.; Yang, T.; Wang, M.; Debinskie, W.; Zhao, W.; Chan, M.D.; Zhou, X. Identification of biomarkers for pseudo and true progression of GBM based on radiogenomics study. Oncotarget 2016, 7, 55377–55394. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, D.; Stalpers, L.; Taal, W.; Sminia, P.; van den Bent, M.J. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 2008, 9, 453–461. [Google Scholar] [CrossRef]
- Topkan, E.; Topuk, S.; Oymak, E.; Parlak, C.; Pehlivan, B. Pseudoprogression in patients with glioblastoma multiforme after concurrent radiotherapy and temozolomide. Am. J. Clin. Oncol. 2012, 35, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Ma, Y.; Sun, M.; Jiang, W.; Yuan, T.; Tong, D. Meta-analysis of the diagnostic performance of diffusion magnetic resonance imaging with apparent diffusion coefficient measurements for differentiating glioma recurrence from pseudoprogression. Medicine 2020, 99, e20270. [Google Scholar] [CrossRef]
- van Dijken, B.R.J.; van Laar, P.J.; Smits, M.; Dankbaar, J.W.; Enting, R.H.; van der Hoorn, A. Perfusion MRI in treatment evaluation of glioblastomas: Clinical relevance of current and future techniques. J. Magn. Reson. Imaging 2019, 49, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Langen, K.J.; Galldiks, N.; Hattingen, E.; Shah, N.J. Advances in neuro-oncology imaging. Nat. Rev. Neurol. 2017, 13, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Prather, K.Y.; O’Neal, C.M.; Westrup, A.M.; Tullos, H.J.; Hughes, K.L.; Conner, A.K.; Glenn, C.A.; Battiste, J.D. A systematic review of amino acid PET in assessing treatment response to temozolomide in glioma. Neurooncol. Adv. 2022, 4, vdac008. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, A.; Proietti, G.; Sica, G.; Scicchitano, B.M. Pathological and Molecular Features of Glioblastoma and Its Peritumoral Tissue. Cancers (Basel) 2019, 11, 469. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Balaj, L.; Stott, S.L.; Nahed, B.; Carter, B.S. Liquid biopsy for brain tumors. Expert Rev. Mol. Diagn. 2017, 17, 943–947. [Google Scholar] [CrossRef]
- Riche, M.; Amelot, A.; Peyre, M.; Capelle, L.; Carpentier, A.; Mathon, B. Complications after frame-based stereotactic brain biopsy: A systematic review. Neurosurg. Rev. 2021, 44, 301–307. [Google Scholar] [CrossRef]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; Liesche-Starnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, V.E.; Solheim, O.; Salvesen, Ø.; Torp, S.H. The histological representativeness of glioblastoma tissue samples. Acta Neurochir 2021, 163, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Gatto, L.; Franceschi, E.; Di Nunno, V.; Tosoni, A.; Lodi, R.; Brandes, A.A. Liquid Biopsy in Glioblastoma Management: From Current Research to Future Perspectives. Oncologist 2021, 26, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Mandel, P.; Metais, P. Nuclear Acids In Human Blood Plasma. C. R. Seances Soc. Biol. Fil. 1948, 142, 241–243. [Google Scholar] [PubMed]
- Leon, S.A.; Shapiro, B.; Sklaroff, D.M.; Yaros, M.J. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977, 37, 646–650. [Google Scholar] [PubMed]
- Stroun, M.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Beljanski, M. Neoplastic characteristics of the DNA found in the plasma of cancer patients. Oncology 1989, 46, 318–322. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hsieh, J.S.; Chang, M.Y.; Huang, T.J.; Chen, F.M.; Cheng, T.L.; Alexandersen, K.; Huang, Y.-S.; Tzou, W.-S.; Lin, S.-R. Molecular detection of APC, K- ras, and p53 mutations in the serum of colorectal cancer patients as circulating biomarkers. World J. Surg. 2004, 28, 721–726. [Google Scholar] [CrossRef]
- Fujiwara, K.; Fujimoto, N.; Tabata, M.; Nishii, K.; Matsuo, K.; Hotta, K.; Kozuki, T.; Aoe, M.; Kiura, K.; Ueoka, H.; et al. Identification of epigenetic aberrant promoter methylation in serum DNA is useful for early detection of lung cancer. Clin. Cancer Res. 2005, 11, 1219–1225. [Google Scholar] [CrossRef]
- Shaw, J.A.; Smith, B.M.; Walsh, T.; Johnson, S.; Primrose, L.; Slade, M.J.; Walker , R.A.; Coombes , R.C. Microsatellite alterations plasma DNA of primary breast cancer patients. Clin. Cancer Res. 2000, 6, 1119–1124. [Google Scholar]
- Cheng, F.; Su, L.; Qian, C. Circulating tumor DNA: A promising biomarker in the liquid biopsy of cancer. Oncotarget 2016, 7, 48832–48841. [Google Scholar] [CrossRef] [Green Version]
- Leary, R.J.; Sausen, M.; Kinde, I.; Papadopoulos, N.; Carpten, J.D.; Craig, D.; O’Shaughnessy, J.; Kinzler, K.W.; Parmigiani, G.; Vogelstein, B.; et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci. Transl. Med. 2012, 4, 162ra54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.-F.; Kingsbury, Z.; Wong, A.S.C.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Frio, T.R.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 2015, 9, 783–790. [Google Scholar] [CrossRef]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating liquid biopsies into the management of cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.D.; Knippers, R. DNA fragments in the blood plasma of cancer patients: Quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001, 61, 1659–1665. [Google Scholar] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Bettegowda, C. Applications of DNA-Based Liquid Biopsy for Central Nervous System Neoplasms. J. Mol. Diagn. 2017, 19, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.Y.; Guo, Q.R.; Wang, F.H.; Adhikari, R.; Zhu, Z.Y.; Zhang, H.Y.; Zhou, W.-M.; Yu, H.; Li, J.-Q.; Zhang, J.-Y. Cell-Free DNA: Hope and Potential Application in Cancer. Front. Cell Dev. Biol. 2021, 9, 639233. [Google Scholar] [CrossRef]
- Touat, M.; Duran-Peña, A.; Alentorn, A.; Lacroix, L.; Massard, C.; Idbaih, A. Emerging circulating biomarkers in glioblastoma: Promises and challenges. Expert Rev. Mol. Diagn. 2015, 15, 1311–1323. [Google Scholar] [CrossRef]
- Jung, M.; Klotzek, S.; Lewandowski, M.; Fleischhacker, M.; Jung, K. Changes in concentration of DNA in serum and plasma during storage of blood samples. Clin. Chem. 2003, 49 Pt 1, 1028–1029. [Google Scholar] [CrossRef] [Green Version]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.; Laramy, J.K.; et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Nguyen, H.; Drummond, K.; Morokoff, A. Circulating Biomarkers for Glioma: A Review. Neurosurgery 2021, 88, E221–E230. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Schwaederle, M.; Husain, H.; Fanta, P.T.; Piccioni, D.E.; Kesari, S.; Schwab, R.B.; Banks, K.C.; Lanman, R.B.; Talasaz, A.; Parker, B.A.; et al. Detection rate of actionable mutations in diverse cancers using a biopsy-free (blood) circulating tumor cell DNA assay. Oncotarget 2016, 7, 9707–9717. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; David, R.; Gandara, M.D.; Philip, C.; Justin, I.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [Green Version]
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical Utility of Plasma Cell-Free DNA in Adult Patients with Newly Diagnosed Glioblastoma: A Pilot Prospective Study. Clin. Cancer Res. 2020, 26, 397–407. [Google Scholar] [CrossRef]
- Cordova, C.; Syeda, M.M.; Corless, B.; Wiggins, J.M.; Patel, A.; Kurz, S.C.; Delara, M.; Sawaged, Z.; Utate, M.; Placantonakis, D.; et al. Plasma cell-free circulating tumor DNA (ctDNA) detection in longitudinally followed glioblastoma patients using TERT promoter mutation-specific droplet digital PCR assays. J. Clin. Oncol. 2019, 37 (Suppl. S15), 2026. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, W.; Wang, Y.; Guo, Y.; Cong, Z.; Du, F.; Song, B. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed Rep. 2015, 3, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Juratli, T.A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M.A.; Thowe, R.T.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin. Cancer Res. 2018, 24, 5282–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Juratli, M.A.; Thowe, R.T.; Hennig, S.; Makina, M.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C.G.; Su, J.; Morris, J.; Watts, C.; Brindle, K.M.; Rosenfeld, N. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 2018, 10, e9323. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Ramon y Cajal, S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Felekkis, K.; Touvana, E.; Stefanou, C.; Deltas, C. microRNAs: A newly described class of encoded molecules that play a role in health and disease. Hippokratia 2010, 14, 236–240. [Google Scholar]
- Roth, P.; Wischhusen, J.; Happold, C.; Chandran, P.A.; Hofer, S.; Eisele, G.; Weller, M.; Keller, A. A specific miRNA signature in the peripheral blood of glioblastoma patients. J. Neurochem. 2011, 118, 449–457. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Li, P.; Li, A.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma specific miRNAs as predictive biomarkers for diagnosis and prognosis of glioma. J. Exp. Clin. Cancer Res. 2012, 31, 97. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, C.; Chen, X.; Chen, S.; Zhang, Y.; Zhi, F.; Wang, J.; Li, L.; Zhou, X.; Li, N.; et al. Identification of seven serum microRNAs from a genome-wide serum microRNA expression profile as potential noninvasive biomarkers for malignant astrocytomas. Int. J. Cancer 2013, 132, 116–127. [Google Scholar] [CrossRef]
- Sun, J.; Liao, K.; Wu, X.; Huang, J.; Zhang, S.; Lu, X. Serum microRNA-128 as a biomarker for diagnosis of glioma. Int. J. Clin. Exp. Med. 2015, 8, 456–463. [Google Scholar] [PubMed]
- Ivo D’Urso, P.; Fernando D’Urso, O.; Damiano Gianfreda, C.; Mezzolla, V.; Storelli, C.; Marsigliante, S. miR-15b and miR-21 as Circulating Biomarkers for Diagnosis of Glioma. Curr. Genom. 2015, 16, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, N.S.; Wu, D.G.; Fang, X.G.; Lin, Y.C.; Chen, S.S.; Li, Z.B.; Xu, S.-S. Serum microRNA-210 as a potential noninvasive biomarker for the diagnosis and prognosis of glioma. Br. J. Cancer 2015, 112, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Shao, N.; Wang, L.; Xue, L.; Wang, R.; Lan, Q. Plasma miR-454-3p as a potential prognostic indicator in human glioma. Neurol. Sci. 2015, 36, 309–313. [Google Scholar] [CrossRef]
- Regazzo, G.; Terrenato, I.; Spagnuolo, M.; Carosi, M.; Cognetti, G.; Cicchillitti, L.; Sperati, F.; Villani, V.; Carapella, C.; Piaggio, G.; et al. A restricted signature of serum miRNAs distinguishes glioblastoma from lower grade gliomas. J. Exp. Clin. Cancer Res. 2016, 35, 124. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Zhang, L.; Song, Z.; Guo, C.; Zhu, J.; Li, Z.; Zhu, S. Potential Diagnostic and Prognostic Value of Plasma Circulating MicroRNA-182 in Human Glioma. Med. Sci. Monit. 2016, 22, 855–862. [Google Scholar] [CrossRef]
- Yue, X.; Lan, F.; Hu, M.; Pan, Q.; Wang, Q.; Wang, J. Downregulation of serum microRNA-205 as a potential diagnostic and prognostic biomarker for human glioma. J. Neurosurg. 2016, 124, 122–128. [Google Scholar] [CrossRef]
- Swellam, M.; Ezz El Arab, L.; Al-Posttany, A.S.; Said, S.B. Clinical impact of circulating oncogenic MiRNA-221 and MiRNA-222 in glioblastoma multiform. J. Neurooncol. 2019, 144, 545–551. [Google Scholar] [CrossRef]
- Zhi, F.; Shao, N.; Wang, R.; Deng, D.; Xue, L.; Wang, Q.; Zhang, Y.; Shi, Y.; Xia, X.; Wang, S.; et al. Identification of 9 serum microRNAs as potential noninvasive biomarkers of human astrocytoma. Neuro-Oncology 2015, 17, 383–391. [Google Scholar] [CrossRef]
- Morokoff, A.; Jones, J.; Nguyen, H.; Ma, C.; Lasocki, A.; Gaillard, F.; Bennett, I.; Luwor, R.; Stylli, S.; Paradiso, L.; et al. Serum microRNA is a biomarker for post-operative monitoring in glioma. J. Neurooncol. 2020, 149, 391–400. [Google Scholar] [CrossRef]
- Siegal, T.; Charbit, H.; Paldor, I.; Zelikovitch, B.; Canello, T.; Benis, A.; Wong, M.L.; Morokoff, A.P.; Kaye, A.H.; Lavon, I.; et al. Dynamics of circulating hypoxia-mediated miRNAs and tumor response in patients with high-grade glioma treated with bevacizumab. J. Neurosurg. 2016, 125, 1008–1015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashworth, T.R. A Case of Cancer in Which Cells Similar to Those in the Tumours Were Seen in the Blood after Death. Med. J. Aust. 1869, 14, 146–147. [Google Scholar]
- Pantel, K.; Speicher, M.R. The biology of circulating tumor cells. Oncogene 2016, 35, 1216–1224. [Google Scholar] [CrossRef]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Müller, C.; Holtschmidt, J.; Auer, M.; Heitzer, E.; Lamszus, K.; Schulte, A.; Matschke, J.; Langer-Freitag, S.; Gasch, C.; Stoupiec, M.; et al. Hematogenous dissemination of glioblastoma multiforme. Sci. Transl. Med. 2014, 6, 247ra101. [Google Scholar] [CrossRef]
- Fonkem, E.; Lun, M.; Wong, E.T. Rare phenomenon of extracranial metastasis of glioblastoma. J. Clin. Oncol. 2011, 29, 4594–4595. [Google Scholar] [CrossRef]
- Macarthur, K.M.; Kao, G.D.; Chandrasekaran, S.; Alonso-Basanta, M.; Chapman, C.; Lustig, R.A.; Wileyto, E.P.; Hahn, S.M.; Dorsey, J.F. Detection of brain tumor cells in the peripheral blood by a telomerase promoter-based assay. Cancer Res. 2014, 74, 2152–2159. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.P.; Nahed, B.V.; Madden, M.W.; Oliveira, S.M.; Springer, S.; Bhere, D.; Chi, A.S.; Wakimoto, H.; Rothenberg, S.M.; Sequist, L.V.; et al. Brain tumor cells in circulation are enriched for mesenchymal gene expression. Cancer Discov. 2014, 4, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Cui, Y.; Jiang, H.; Sui, D.; Wang, Y.; Jiang, Z.; Zhao, J.; Lin, S. Circulating tumor cell is a common property of brain glioma and promotes the monitoring system. Oncotarget 2016, 7, 71330–71340. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of circulating tumour cell clusters in human glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, M.; Chanteloup, G.; Isambert, N.; Seigneuric, R.; Fumoleau, P.; Garrido, C.; Gobbo, J. Exosomes in cancer theranostic: Diamonds in the rough. Cell Adhes. Migr. 2017, 11, 151–163. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Zhao, C.; Wang, H.; Xiong, C.; Liu, Y. Hypoxic glioblastoma release exosomal VEGF-A induce the permeability of blood-brain barrier. Biochem. Biophys. Res. Commun. 2018, 502, 324–331. [Google Scholar] [CrossRef]
- Minciacchi, V.R.; Freeman, M.R.; Di Vizio, D. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Semin. Cell Dev. Biol. 2015, 40, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Koch, C.J.; Lustig, R.A.; Yang, X.Y.; Jenkins, W.T.; Wolf, R.L.; Martinez-Lage, M.; Desai, A.; Williams, D.; Evans, S.M. Microvesicles as a Biomarker for Tumor Progression versus Treatment Effect in Radiation/Temozolomide-Treated Glioblastoma Patients. Transl. Oncol. 2014, 7, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.M.; Putt, M.; Yang, X.Y.; Lustig, R.A.; Martinez-Lage, M.; Williams, D.; Desai, A.; Wolf, R.; Brem, S.; Koch, C.J. Initial evidence that blood-borne microvesicles are biomarkers for recurrence and survival in newly diagnosed glioblastoma patients. J. Neurooncol. 2016, 127, 391–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Osti, D.; Del Bene, M.; Rappa, G.; Santos, M.; Matafora, V.; Richichi, C.; Faletti, S.; Beznoussenko, G.V.; Mironov, A.; Bachi, A.; et al. Clinical Significance of Extracellular Vesicles in Plasma from Glioblastoma Patients. Clin. Cancer Res. 2019, 25, 266–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André-Grégoire, G.; Bidère, N.; Gavard, J. Temozolomide affects Extracellular Vesicles Released by Glioblastoma Cells. Biochimie 2018, 155, 11–15. [Google Scholar] [CrossRef]
- Indira Chandran, V.; Welinder, C.; Månsson, A.S.; Offer, S.; Freyhult, E.; Pernemalm, M.; Sigrid, M.L.; Pedersen, S.; Lehtiö, J.; Marko-Varga, G.; et al. Ultrasensitive Immunoprofiling of Plasma Extracellular Vesicles Identifies Syndecan-1 as a Potential Tool for Minimally Invasive Diagnosis of Glioma. Clin. Cancer Res. 2019, 25, 3115–3127. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Qing, Q.; Pan, Q.; Hu, M.; Yu, H.; Yue, X. Serum exosomal miR-301a as a potential diagnostic and prognostic biomarker for human glioma. Cell Oncol. 2018, 41, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimkhani, S.; Vafaee, F.; Hallal, S.; Wei, H.; Lee, M.Y.T.; Young, P.E.; Satgunaseelan, L.; Beadnall, H.; Barnett, M.H.; Shivalingam, B.; et al. Deep sequencing of circulating exosomal microRNA allows non-invasive glioblastoma diagnosis. NPJ Precis. Oncol. 2018, 2, 28. [Google Scholar] [CrossRef] [Green Version]
- Manterola, L.; Guruceaga, E.; Gállego Pérez-Larraya, J.; González-Huarriz, M.; Jauregui, P.; Tejada, S.; Diez-Valle, R.; Segura, V.; Samprón, N.; Barrena, C.; et al. A small noncoding RNA signature found in exosomes of GBM patient serum as a diagnostic tool. Neuro-Oncology 2014, 16, 520–527. [Google Scholar] [CrossRef]
- Santangelo, A.; Imbrucè, P.; Gardenghi, B.; Belli, L.; Agushi, R.; Tamanini, A.; Munari, S.; Bossi, A.M.; Scambi, I.; Benati, D.; et al. A microRNA signature from serum exosomes of patients with glioma as complementary diagnostic biomarker. J. Neurooncol. 2018, 136, 51–62. [Google Scholar] [CrossRef]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589 Pt A, 2914–2922. [Google Scholar] [CrossRef] [Green Version]
- Peterson, C.L.; Laniel, M.A. Histones and histone modifications. Curr. Biol. 2004, 14, R546–R551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.Z. Altered histone modifications in gliomas. Brain Tumor Res. Treat. 2014, 2, 7–21. [Google Scholar] [CrossRef] [Green Version]
- McAnena, P.; Brown, J.A.; Kerin, M.J. Circulating Nucleosomes and Nucleosome Modifications as Biomarkers in Cancer. Cancers 2017, 9, 5. [Google Scholar] [CrossRef]
- Holdenrieder, S.; Von Pawel, J.; Nagel, D.; Stieber, P. Long-term stability of circulating nucleosomes in serum. Anticancer Res. 2010, 30, 1613–1615. [Google Scholar]
- Rahier, J.F.; Druez, A.; Faugeras, L.; Martinet, J.P.; Géhénot, M.; Josseaux, E.; Herzog, M.; Micallef, J.; George, F.; Delos, M.; et al. Circulating nucleosomes as new blood-based biomarkers for detection of colorectal cancer. Clin. Epigenetics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Romani, M.; Pistillo, M.P.; Banelli, B. Epigenetic Targeting of Glioblastoma. Front. Oncol. 2018, 8, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Huang, B.S.; Xiong, Y.Y.; Yang, L.J.; Wu, L.X. 4,5-Dimethoxycanthin-6-one is a novel LSD1 inhibitor that inhibits proliferation of glioblastoma cells and induces apoptosis and pyroptosis. Cancer Cell Int. 2022, 22, 32. [Google Scholar] [CrossRef]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.L.; Cheng, J.X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2888–2896. [Google Scholar] [CrossRef] [Green Version]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| Study | Patients (n) | Biofluid | Method | ctDNA Detection Rate |
|---|---|---|---|---|
| Schwaederle et al. [55] | 33 | Plasma | NGS | 27% |
| Piccioni et al. [56] | 222 | Plasma | NGS | 55% |
| Zill et al. [57] | 107 | Plasma | NGS | 51% |
| Bagley et al. [58] | 20 | Plasma | NGS | 55% |
| Cordova et al. [59] | 13 | Plasma | ddPCR | 46% |
| Wang et al. [60] | 19 | Serum, CSF | Methylation specific PCR assay | 37% (Serum), 61% (CSF) |
| Juratli et al. [61] | 38 | Plasma, CSF | Nested PCR | 8% (Plasma), 92% (CSF) |
| Wang et al. [62] | 11 | CSF | WGS | 100% |
| Mouliere et al. [63] | 10 | CSF | WGS | 50% |
| Martínez-Ricarte et al. [64] | 9 | CSF | ddPCR | 100% |
| Miller et al. [65] | 46 | CSF | NGS | 59% |
| Study | No. of Patients (Cases/Controls) | Controls Type | Biofluid | miRNA | Upregulation or Downregulation | Method |
|---|---|---|---|---|---|---|
| Roth et al. [68] | 20/20 | Healthy | Blood | miR-128 miR-342-3p | Upregulation Downregulation | qRT-PCR |
| Wang et al. [69] | 10/10 | Healthy | Plasma | miR-21 miR-128 miR-342-3p | Upregulation Downregulation Downregulation | qRT-PCR |
| Yang et al. [70] | 33/80 | Healthy | Serum | miR-15b, miR-23a, miR-133a, miR-150, miR-197, miR-497 and miR-548b-5p | Downregulation | qRT-PCR |
| Sun et al. [71] | 61/53 | Healthy | Serum | miR-128 | Downregulation | qRT-PCR |
| D’Urso et al. [72] | 16/30 | Neurologic disorders | Serum | miR-16 | Downregulation | qRT-PCR |
| Lai et al. [73] | 42/50 | Healthy | Serum | miR-210 | Upregulation | qRT-PCR |
| Shao et al. [74] | 22/70 | Healthy | Plasma | miR-454-3p | Upregulation | qRT-PCR |
| Regazzo et al. [75] | 10/15 | Healthy | Serum | miR-497 | Downregulation | qRT-PCR |
| Xiao et al. [76] | 39/54 | Healthy | Plasma | miR-182 | Upregulation | qRT-PCR |
| Yue et al. [77] | 27/45 | Healthy | Serum | miR-205 | Downregulation | qRT-PCR |
| Swellam et al. [78] | 20/20 | Healthy | Serum | miR-221 and miR-222 | Upregulation | qRT-PCR |
| Study | Patients (n) | Biofluid | Method | CTCs Detection Rate |
|---|---|---|---|---|
| Müller et al. [87] | 141 | Peripheral blood | Density gradient centrifugation followed by immunostaining for GFAP | 21% |
| MacArthur et al. [89] | 11 | Peripheral blood | Density gradient centrifugation followed by telomerase-based test | 72% preradiotherapy 8% postradiotherapy |
| Sullivan et al. [90] | 33 | Peripheral blood | CTC–iCHIP technology; characterization using antibodies cocktail | 39% |
| Gao et al. [91] | 11 | Peripheral blood | Examination for aneuploidy of chromosome 8 by FISH | 82% |
| Krol et al. [92] | 13 | Peripheral blood | Parsortix microfluidic technology; characterization using antibodies cocktail | 54% |
| Diagnostic Method | Advantages | Disadvantages |
|---|---|---|
| MRI | Allows initial diagnosis and anatomic characterization of GBM with non-invasive procedure | Difficulty in discriminating GBM from other brain diseases and other concomitant pathological processes Difficulty in correlating MRI features with molecular features Difficulty in distinct actual tumor recurrence from PsP |
| Tissue biopsy | Allows histologic and molecular characterization of GBM | Highly invasive procedure with risks, limiting repeated sampling May not reflect the intra-tumoral heterogeneity Cannot evaluate the tumor activity in real-time |
| ctDNA | Higher levels than CTCs Very specific Quantity correlates with the disease stage Easier to collect than CTCs and established detection techniques | Short half-life (<2 h) Released mainly by apoptotic or necrotic cells and therefore represents only a subpopulation of tumor cells Sensitivity of detection limited |
| miRNAs | Relatively stable | No standardized methods for RNA extraction and sequencing Less specific than ctDNA |
| CTCs | Highly specific Can provide information on protein, DNA and RNA levels | Lack of standardized methods to isolate and characterize CTCs Low presence in blood May not represent the whole tumor |
| EVs | Can carry RNAs, proteins, and lipids which are protected from enzyme degradation Can cross an intact BBB Released by all cells, including cancer cells | Lack of standardized methods to isolate EVs released by non-neoplastic cells, so there is a background of nontumoral EVs in the blood |
| Circulating nucleosome-associated histone modifications | Highly stable Simple methods (ELISA, ChLIA) to detect them Epigenetics is a new intensive field of research | Low specificity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ronvaux, L.; Riva, M.; Coosemans, A.; Herzog, M.; Rommelaere, G.; Donis, N.; D’Hondt, L.; Douxfils, J. Liquid Biopsy in Glioblastoma. Cancers 2022, 14, 3394. https://doi.org/10.3390/cancers14143394
Ronvaux L, Riva M, Coosemans A, Herzog M, Rommelaere G, Donis N, D’Hondt L, Douxfils J. Liquid Biopsy in Glioblastoma. Cancers. 2022; 14(14):3394. https://doi.org/10.3390/cancers14143394
Chicago/Turabian StyleRonvaux, Lorian, Matteo Riva, An Coosemans, Marielle Herzog, Guillaume Rommelaere, Nathalie Donis, Lionel D’Hondt, and Jonathan Douxfils. 2022. "Liquid Biopsy in Glioblastoma" Cancers 14, no. 14: 3394. https://doi.org/10.3390/cancers14143394
APA StyleRonvaux, L., Riva, M., Coosemans, A., Herzog, M., Rommelaere, G., Donis, N., D’Hondt, L., & Douxfils, J. (2022). Liquid Biopsy in Glioblastoma. Cancers, 14(14), 3394. https://doi.org/10.3390/cancers14143394