
Primary and Acquired Resistance against Immune Check Inhibitors in Non-Small Cell Lung Cancer

 ,
, {kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
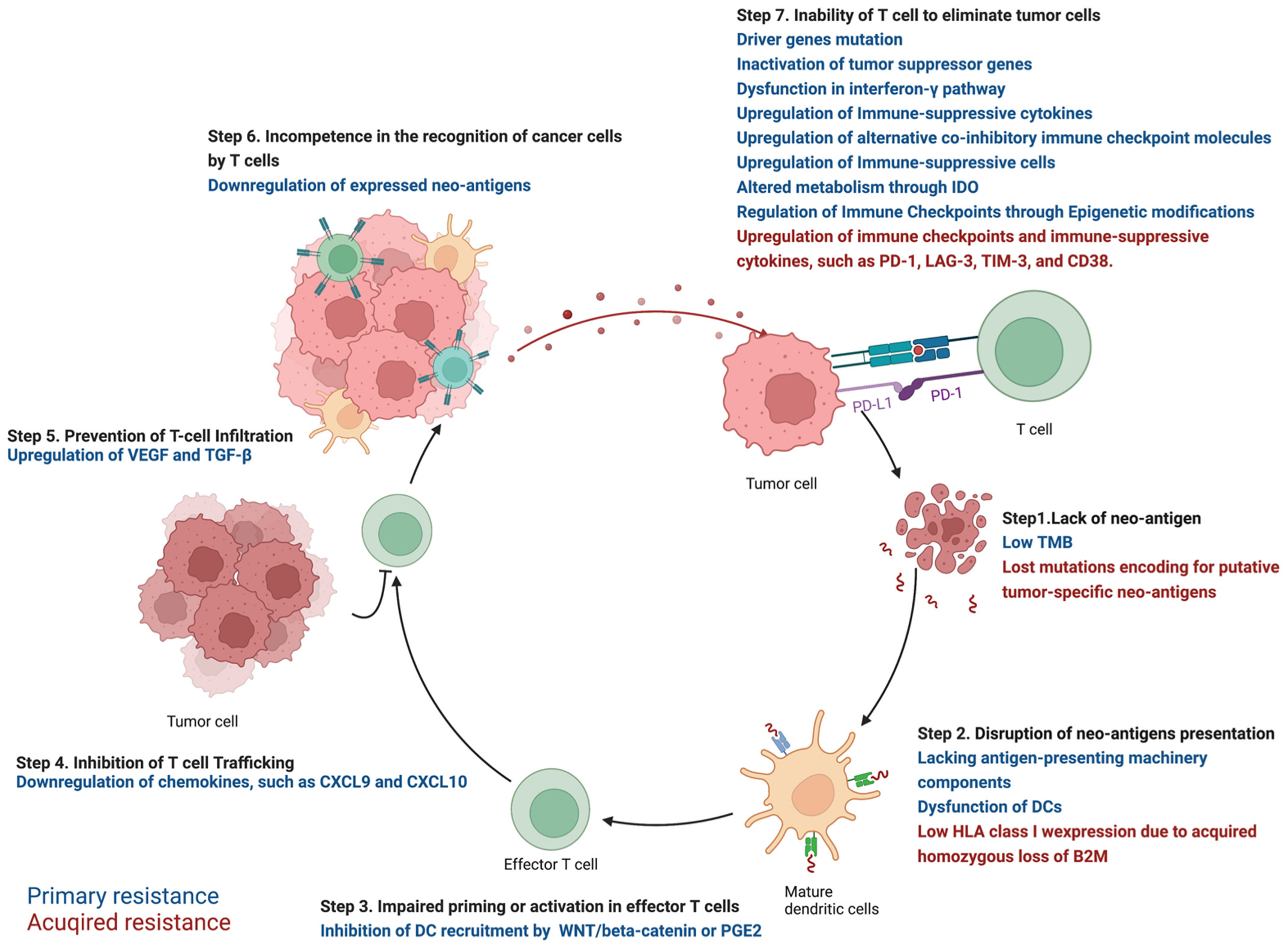
2. Primary Resistance against ICIs in NSCLC
2.1. Step 1. Lack of Neo-Antigen Due to Low TMB
2.2. Step 2. Disruption of Neo-Antigens Presentation
2.3. Step 3. Impaired Priming or Activation in Effector T Cells
2.4. Step 4. Inhibition of T Cell Trafficking
2.5. Step 5. Prevention of T-Cell Infiltration
2.6. Step 6. Incompetence in the Recognition of Cancer Cells by T Cells
2.7. Step 7. Inability of T Cell to Eliminate Tumor Cells
2.7.1. Driver Genes Mutation
2.7.2. Inactivation of Tumor Suppressor Genes
2.7.3. Dysfunction in Interferon-γ Pathway
2.7.4. Immune-Suppressive Cytokines
2.7.5. Upregulation of Alternative Co-Inhibitory Immune Checkpoint Molecules
2.7.6. Immune-Suppressive Cells
2.7.7. Altered Metabolism through Indoleamine 2,3-Dioxygenase (IDO)
2.7.8. Regulation of Immune Checkpoints through Epigenetic Modifications
3. Acquired Resistance in NSCLC
4. Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Abedi, S.; Janbabaei, G.; Afshari, M.; Moosazadeh, M.; Rashidi Alashti, M.; Hedayatizadeh-Omran, A.; Alizadeh-Navaei, R.; Abedini, E. Estimating the Survival of Patients with Lung Cancer: What Is the Best Statistical Model? J. Prev. Med. Public Health 2019, 52, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.K.; Horn, L. How I Treat Non-Small Cell Lung Cancer Refractory to Immunotherapy. Cancer J. 2020, 26, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Hellmann, M.D.; Rizvi, N.A.; Carcereny, E.; Leighl, N.B.; Ahn, M.J.; Eder, J.P.; Balmanoukian, A.S.; Aggarwal, C.; Horn, L.; et al. Five-Year Overall Survival for Patients with Advanced NonSmall-Cell Lung Cancer Treated with Pembrolizumab: Results from the Phase I KEYNOTE-001 Study. J. Clin. Oncol. 2019, 37, 2518–2527. [Google Scholar] [CrossRef] [PubMed]
- Gettinger, S.; Horn, L.; Jackman, D.; Spigel, D.; Antonia, S.; Hellmann, M.; Powderly, J.; Heist, R.; Sequist, L.V.; Smith, D.C.; et al. Five-Year Follow-Up of Nivolumab in Previously Treated Advanced Non-Small-Cell Lung Cancer: Results from the CA209-003 Study. J. Clin. Oncol. 2018, 36, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Koustas, E.; Sarantis, P.; Papavassiliou, A.G.; Karamouzis, M.V. The Resistance Mechanisms of Checkpoint Inhibitors in Solid Tumors. Biomolecules 2020, 10, 666. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565–594. [Google Scholar] [CrossRef] [Green Version]
- Liechtenstein, T.; Dufait, I.; Bricogne, C.; Lanna, A.; Pen, J.; Breckpot, K.; Escors, D. PD-L1/PD-1 Co-Stimulation, a Brake for T cell Activation and a T cell Differentiation Signal. J. Clin. Cell Immunol. 2012, S12, 6. [Google Scholar] [CrossRef]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Sabari, J.K.; Leonardi, G.C.; Shu, C.A.; Umeton, R.; Montecalvo, J.; Ni, A.; Chen, R.; Dienstag, J.; Mrad, C.; Bergagnini, I.; et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann. Oncol. 2018, 29, 2085–2091. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, M.; Yi, M.; Li, N.; Luo, S.; Wu, K. Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. Exp. Hematol. Oncol. 2021, 10, 18. [Google Scholar] [CrossRef]
- Carbone, D.P.; Reck, M.; Paz-Ares, L.; Creelan, B.; Horn, L.; Steins, M.; Felip, E.; van den Heuvel, M.M.; Ciuleanu, T.E.; Badin, F.; et al. First-Line Nivolumab in Stage IV or Recurrent Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2415–2426. [Google Scholar] [CrossRef] [PubMed]
- Ichinokawa, K.; Nakanishi, Y.; Hida, Y.; Tsuchikawa, T.; Kato, T.; Itoh, T.; Kaji, M.; Kaga, K.; Hirano, S. Downregulated expression of human leukocyte antigen class I heavy chain is associated with poor prognosis in non-small-cell lung cancer. Oncol. Lett. 2019, 18, 117–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef] [Green Version]
- Roche, P.A.; Cresswell, P. Antigen Processing and Presentation Mechanisms in Myeloid Cells. Microbiol. Spectr. 2016, 4, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Liu, C.; Liu, J.; Li, G. Resistance Mechanism of PD-1/PD-L1 Blockade in the Cancer-Immunity Cycle. Onco Targets Ther. 2020, 13, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Martins, A.; Han, J.; Kim, S.O. The multifaceted effects of granulocyte colony-stimulating factor in immunomodulation and potential roles in intestinal immune homeostasis. IUBMB Life 2010, 62, 611–617. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Udayanga, K.G.; Totsuka, N.; Weinberg, J.B.; Nunez, G.; Shibuya, A. Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host. Microbe. 2014, 15, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Pinato, D.J.; Howlett, S.; Ottaviani, D.; Urus, H.; Patel, A.; Mineo, T.; Brock, C.; Power, D.; Hatcher, O.; Falconer, A.; et al. Association of Prior Antibiotic Treatment with Survival and Response to Immune Checkpoint Inhibitor Therapy in Patients with Cancer. JAMA Oncol. 2019, 5, 1774–1778. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B. IFNgamma: Signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiang, Y.; Li, F.; Yin, C.; Li, B.; Ke, X. WNT/beta-Catenin Signaling Pathway Regulating T Cell-Inflammation in the Tumor Microenvironment. Front. Immunol. 2019, 10, 2293. [Google Scholar] [CrossRef] [Green Version]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Langenkamp, E.; Georganaki, M.; Loskog, A.; Fuchs, P.F.; Dieterich, L.C.; Kreuger, J.; Dimberg, A. VEGF suppresses T-lymphocyte infiltration in the tumor microenvironment through inhibition of NF-kappaB-induced endothelial activation. FASEB J. 2015, 29, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, G.; Sisi, M.; Andrini, E.; Palladini, A.; Giunchi, F.; Lollini, P.L.; Ardizzoni, A.; Gelsomino, F. The Mechanisms of PD-L1 Regulation in Non-Small-Cell Lung Cancer (NSCLC): Which Are the Involved Players? Cancers 2020, 12, 3129. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Chen, X.; Su, C.; Ren, S.; Zhou, C.; Jiang, T. Pan-cancer analysis of KEAP1 mutations as biomarkers for immunotherapy outcomes. Ann. Transl. Med. 2020, 8, 141. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanic, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J.; et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelhamed, S.; Ogura, K.; Yokoyama, S.; Saiki, I.; Hayakawa, Y. AKT-STAT3 Pathway as a Downstream Target of EGFR Signaling to Regulate PD-L1 Expression on NSCLC cells. J. Cancer 2016, 7, 1579–1586. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Ota, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; Kage, M.; et al. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann. Oncol. 2014, 25, 1935–1940. [Google Scholar] [CrossRef]
- Suda, K.; Rozeboom, L.; Rivard, C.J.; Yu, H.; Ellison, K.; Melnick, M.A.C.; Hinz, T.K.; Chan, D.; Heasley, L.E.; Politi, K.; et al. Therapy-induced E-cadherin downregulation alters expression of programmed death ligand-1 in lung cancer cells. Lung Cancer 2017, 109, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ota, K.; Azuma, K.; Kawahara, A.; Hattori, S.; Iwama, E.; Tanizaki, J.; Harada, T.; Matsumoto, K.; Takayama, K.; Takamori, S.; et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2015, 21, 4014–4021. [Google Scholar] [CrossRef] [Green Version]
- Coelho, M.A.; de Carne Trecesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Chen, N.; Fang, W.; Zhan, J.; Liu, Q.; Kang, S.; He, X.; Liu, L.; Zhou, T.; Huang, J.; et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology 2016, 5, e1094598. [Google Scholar] [CrossRef] [PubMed]
- Demuth, C.; Andersen, M.N.; Jakobsen, K.R.; Madsen, A.T.; Sorensen, B.S. Increased PD-L1 expression in erlotinib-resistant NSCLC cells with MET gene amplification is reversed upon MET-TKI treatment. Oncotarget 2017, 8, 68221–68229. [Google Scholar] [CrossRef] [Green Version]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef] [Green Version]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Aref, A.R.; Skoulidis, F.; Herter-Sprie, G.S.; Buczkowski, K.A.; Liu, Y.; Awad, M.M.; Denning, W.L.; et al. STK11/LKB1 Deficiency Promotes Neutrophil Recruitment and Proinflammatory Cytokine Production to Suppress T-cell Activity in the Lung Tumor Microenvironment. Cancer Res. 2016, 76, 999–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Ivanova, E.; Guo, S.; Yoshida, R.; Campisi, M.; Sundararaman, S.K.; Tange, S.; Mitsuishi, Y.; Thai, T.C.; Masuda, S.; et al. Suppression of STING Associated with LKB1 Loss in KRAS-Driven Lung Cancer. Cancer Discov. 2019, 9, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Biton, J.; Mansuet-Lupo, A.; Pecuchet, N.; Alifano, M.; Ouakrim, H.; Arrondeau, J.; Boudou-Rouquette, P.; Goldwasser, F.; Leroy, K.; Goc, J.; et al. TP53, STK11, and EGFR Mutations Predict Tumor Immune Profile and the Response to Anti-PD-1 in Lung Adenocarcinoma. Clin. Cancer Res. 2018, 24, 5710–5723. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yang, J.; Cai, Y.; Fu, S.; Zhang, N.; Fu, X.; Li, L. IFN-gamma-mediated inhibition of lung cancer correlates with PD-L1 expression and is regulated by PI3K-AKT signaling. Int. J. Cancer 2018, 143, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Giaj Levra, M.; Cotte, F.E.; Corre, R.; Calvet, C.; Gaudin, A.F.; Penrod, J.R.; Grumberg, V.; Jouaneton, B.; Jolivel, R.; Assie, J.B.; et al. Immunotherapy rechallenge after nivolumab treatment in advanced non-small cell lung cancer in the real-world setting: A national data base analysis. Lung Cancer 2020, 140, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Li, S.D.; Ma, M.; Li, H.; Waluszko, A.; Sidorenko, T.; Schadt, E.E.; Zhang, D.Y.; Chen, R.; Ye, F. Cancer gene profiling in non-small cell lung cancers reveals activating mutations in JAK2 and JAK3 with therapeutic implications. Genome Med. 2017, 9, 89. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [Green Version]
- Eser, P.O.; Janne, P.A. TGFbeta pathway inhibition in the treatment of non-small cell lung cancer. Pharmacol. Ther. 2018, 184, 112–130. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Guo, G.; Beckley, N.; Zhang, Y.; Yang, X.; Sharma, M.; Habib, A.A. Tumor necrosis factor in lung cancer: Complex roles in biology and resistance to treatment. Neoplasia 2021, 23, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Wang, S.; Zhou, Q. The Resistance Mechanisms of Lung Cancer Immunotherapy. Front. Oncol. 2020, 10, 568059. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance Mechanisms of Anti-PD1/PDL1 Therapy in Solid Tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Suda, K.; Ren, S.; Wu, C.; Hou, L.; et al. LAG-3 Protein Expression in Non-Small Cell Lung Cancer and Its Relationship with PD-1/PD-L1 and Tumor-Infiltrating Lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.S.; Kim, A.; Shin, J.Y.; Kim, Y.T. Comprehensive analysis of the tumor immune micro-environment in non-small cell lung cancer for efficacy of checkpoint inhibitor. Sci. Rep. 2018, 8, 14576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Botticelli, A.; Cerbelli, B.; Lionetto, L.; Zizzari, I.; Salati, M.; Pisano, A.; Federica, M.; Simmaco, M.; Nuti, M.; Marchetti, P. Can IDO activity predict primary resistance to anti-PD-1 treatment in NSCLC? J. Transl. Med. 2018, 16, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocher, F.; Amann, A.; Zimmer, K.; Geisler, S.; Fuchs, D.; Pichler, R.; Wolf, D.; Kurz, K.; Seeber, A.; Pircher, A. High indoleamine-2,3-dioxygenase 1 (IDO) activity is linked to primary resistance to immunotherapy in non-small cell lung cancer (NSCLC). Transl. Lung Cancer Res. 2021, 10, 304–313. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Lou, D.; Gao, Y.; Yu, J.; Kong, D.; Zhang, Q.; Jia, Y.; Zhang, H.; Wang, Z. Sodium arsenite exposure inhibits histone acetyltransferase p300 for attenuating H3K27ac at enhancers in mouse embryonic fibroblast cells. Toxicol. Appl. Pharm. 2018, 357, 70–79. [Google Scholar] [CrossRef]
- Marwitz, S.; Scheufele, S.; Perner, S.; Reck, M.; Ammerpohl, O.; Goldmann, T. Epigenetic modifications of the immune-checkpoint genes CTLA4 and PDCD1 in non-small cell lung cancer results in increased expression. Clin. Epigenetics 2017, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Asgarova, A.; Asgarov, K.; Godet, Y.; Peixoto, P.; Nadaradjane, A.; Boyer-Guittaut, M.; Galaine, J.; Guenat, D.; Mougey, V.; Perrard, J.; et al. PD-L1 expression is regulated by both DNA methylation and NF-kB during EMT signaling in non-small cell lung carcinoma. Oncoimmunology 2018, 7, e1423170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrangle, J.; Wang, W.; Koch, A.; Easwaran, H.; Mohammad, H.P.; Vendetti, F.; Vancriekinge, W.; Demeyer, T.; Du, Z.; Parsana, P.; et al. Alterations of immune response of Non-Small Cell Lung Cancer with Azacytidine. Oncotarget 2013, 4, 2067–2079. [Google Scholar] [CrossRef] [Green Version]
- Toyokawa, G.; Takada, K.; Tagawa, T.; Hamamoto, R.; Yamada, Y.; Shimokawa, M.; Oda, Y.; Maehara, Y. A Positive Correlation Between the EZH2 and PD-L1 Expression in Resected Lung Adenocarcinomas. Ann. Thorac. Surg. 2019, 107, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108, djv303. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Anagnostou, V.; Smith, K.N.; Forde, P.M.; Niknafs, N.; Bhattacharya, R.; White, J.; Zhang, T.; Adleff, V.; Phallen, J.; Wali, N.; et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017, 7, 264–276. [Google Scholar] [CrossRef] [Green Version]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI Insight 2018, 3, e120360. [Google Scholar] [CrossRef] [Green Version]
- Limagne, E.; Richard, C.; Thibaudin, M.; Fumet, J.D.; Truntzer, C.; Lagrange, A.; Favier, L.; Coudert, B.; Ghiringhelli, F. Tim-3/galectin-9 pathway and mMDSC control primary and secondary resistances to PD-1 blockade in lung cancer patients. Oncoimmunology 2019, 8, e1564505. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, N.; Yokobori, T.; Kaira, K.; Turtoi, A.; Baatar, S.; Gombodorj, N.; Handa, T.; Tsukagoshi, M.; Ubukata, Y.; Kimura, A.; et al. High Stromal TGFBI in Lung Cancer and Intratumoral CD8-Positive T Cells were Associated with Poor Prognosis and Therapeutic Resistance to Immune Checkpoint Inhibitors. Ann. Surg. Oncol. 2020, 27, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.; Ofek, E.; Barshack, I.; Gottfried, T.; Zadok, O.; Kamer, I.; Urban, D.; Perelman, M.; Onn, A. Transformation to small cell lung cancer as a mechanism of resistance to immunotherapy in non-small cell lung cancer. Lung Cancer 2019, 138, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Pages, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Q.; Wei, X.; Wang, Z.; Zhu, Y.; Zhao, W.; Dong, Y. Primary and Acquired Resistance against Immune Check Inhibitors in Non-Small Cell Lung Cancer. Cancers 2022, 14, 3294. https://doi.org/10.3390/cancers14143294
Sun Q, Wei X, Wang Z, Zhu Y, Zhao W, Dong Y. Primary and Acquired Resistance against Immune Check Inhibitors in Non-Small Cell Lung Cancer. Cancers. 2022; 14(14):3294. https://doi.org/10.3390/cancers14143294
Chicago/Turabian StyleSun, Qinying, Xiangzhen Wei, Zhonglin Wang, Yan Zhu, Weiying Zhao, and Yuchao Dong. 2022. "Primary and Acquired Resistance against Immune Check Inhibitors in Non-Small Cell Lung Cancer" Cancers 14, no. 14: 3294. https://doi.org/10.3390/cancers14143294
APA StyleSun, Q., Wei, X., Wang, Z., Zhu, Y., Zhao, W., & Dong, Y. (2022). Primary and Acquired Resistance against Immune Check Inhibitors in Non-Small Cell Lung Cancer. Cancers, 14(14), 3294. https://doi.org/10.3390/cancers14143294