
Possible Metastatic Stage-Dependent ILC2 Activation Induces Differential Functions of MDSCs through IL-13/IL-13Rα1 Signaling during the Progression of Breast Cancer Lung Metastasis

 ,
,  ,
,  , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Development of the Mouse 4T1/LM4 Breast Cancer Model
2.3. Isolation of Leukocytes from the Lung
2.4. Flow Cytometric Aanalysis
2.5. RNA Isolation and Real-Time Quantitative PCR
2.6. Immunofluorescence Staining
2.7. ELISA
2.8. Statistical Analysis
3. Results
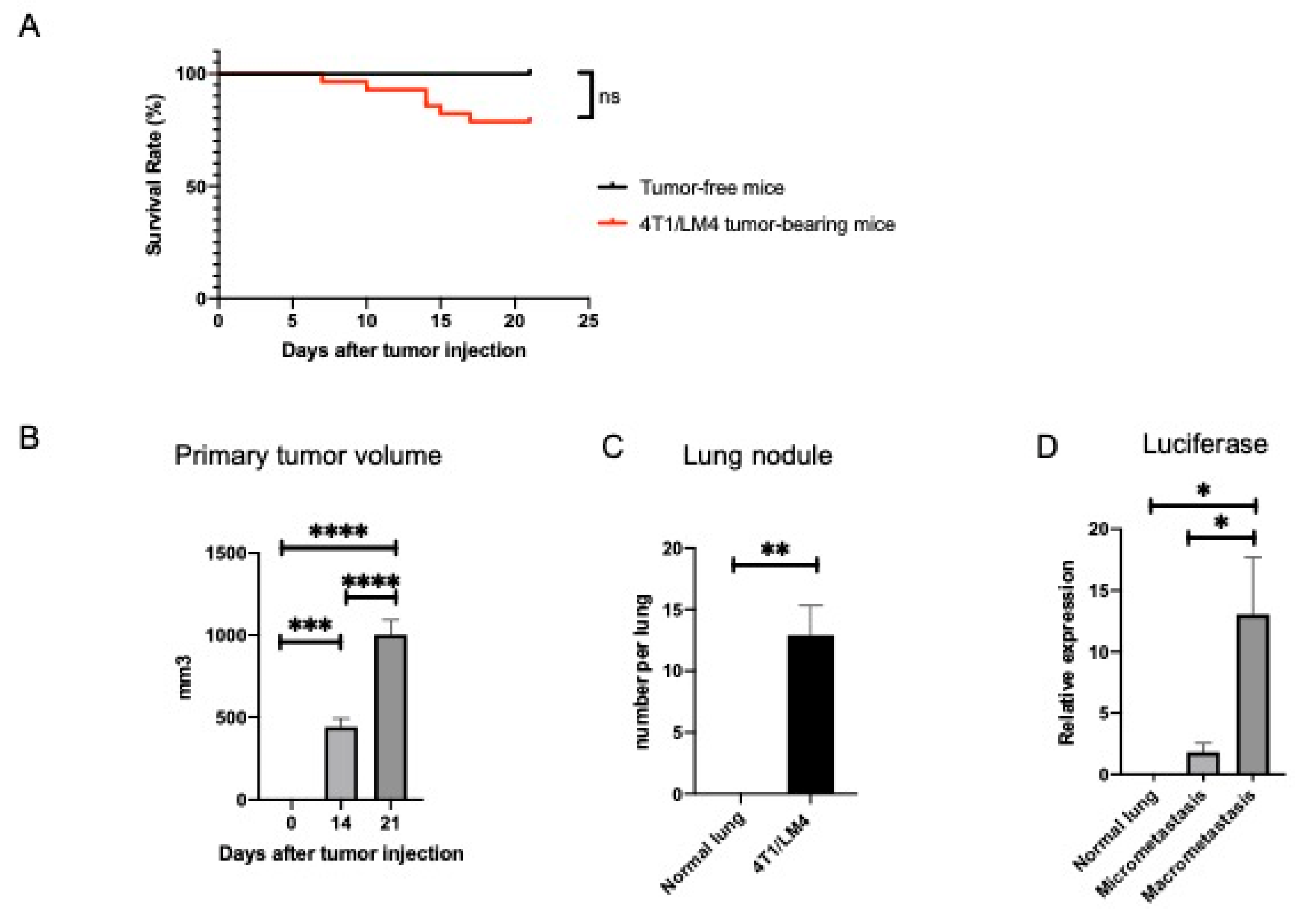
3.1. Separation of Metastases-Bearing Lung Tumor Samples into Micrometastatic and Macrometastatic Regions Using the 4T1/LM4 TNBC Mouse Model
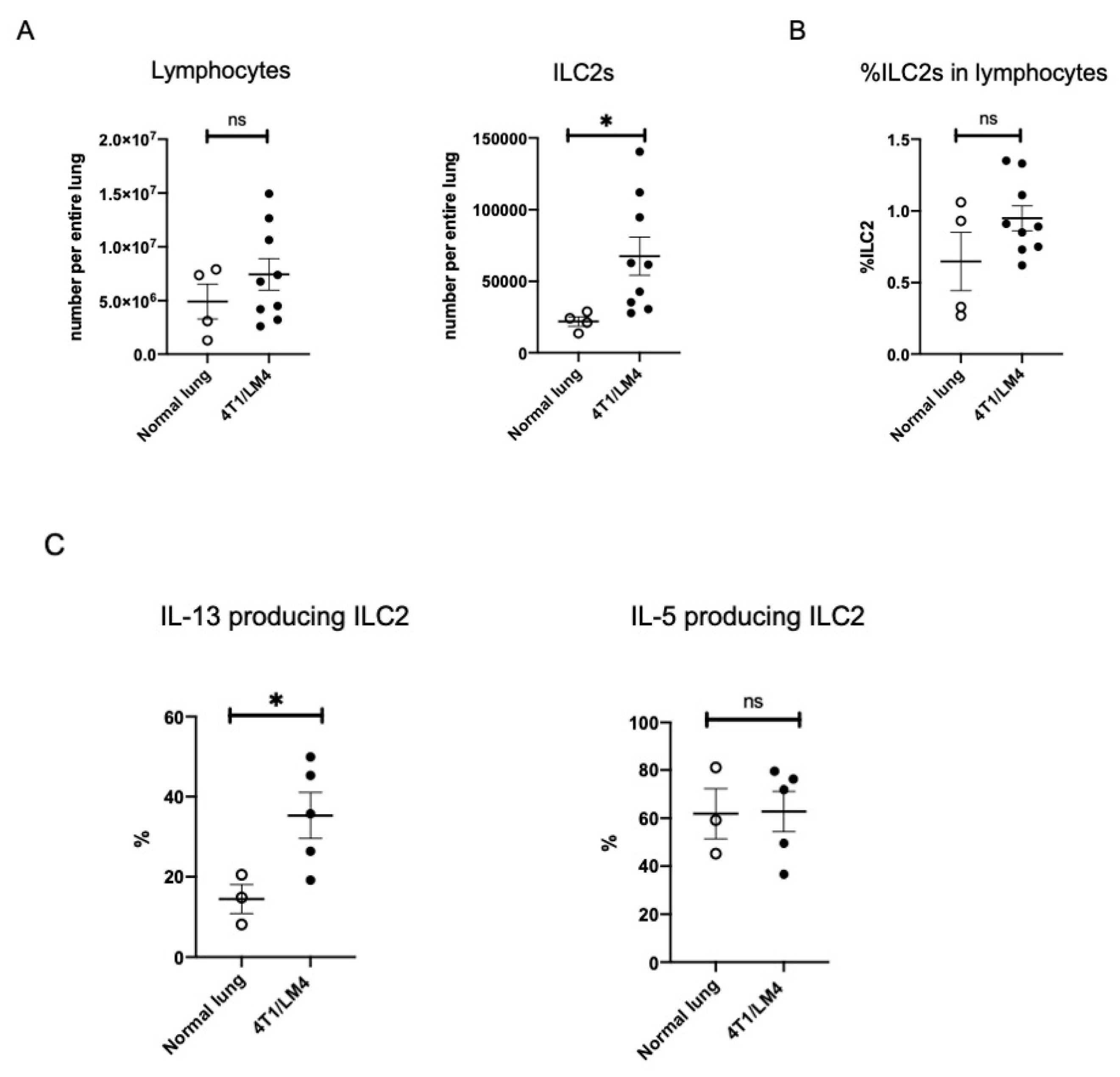
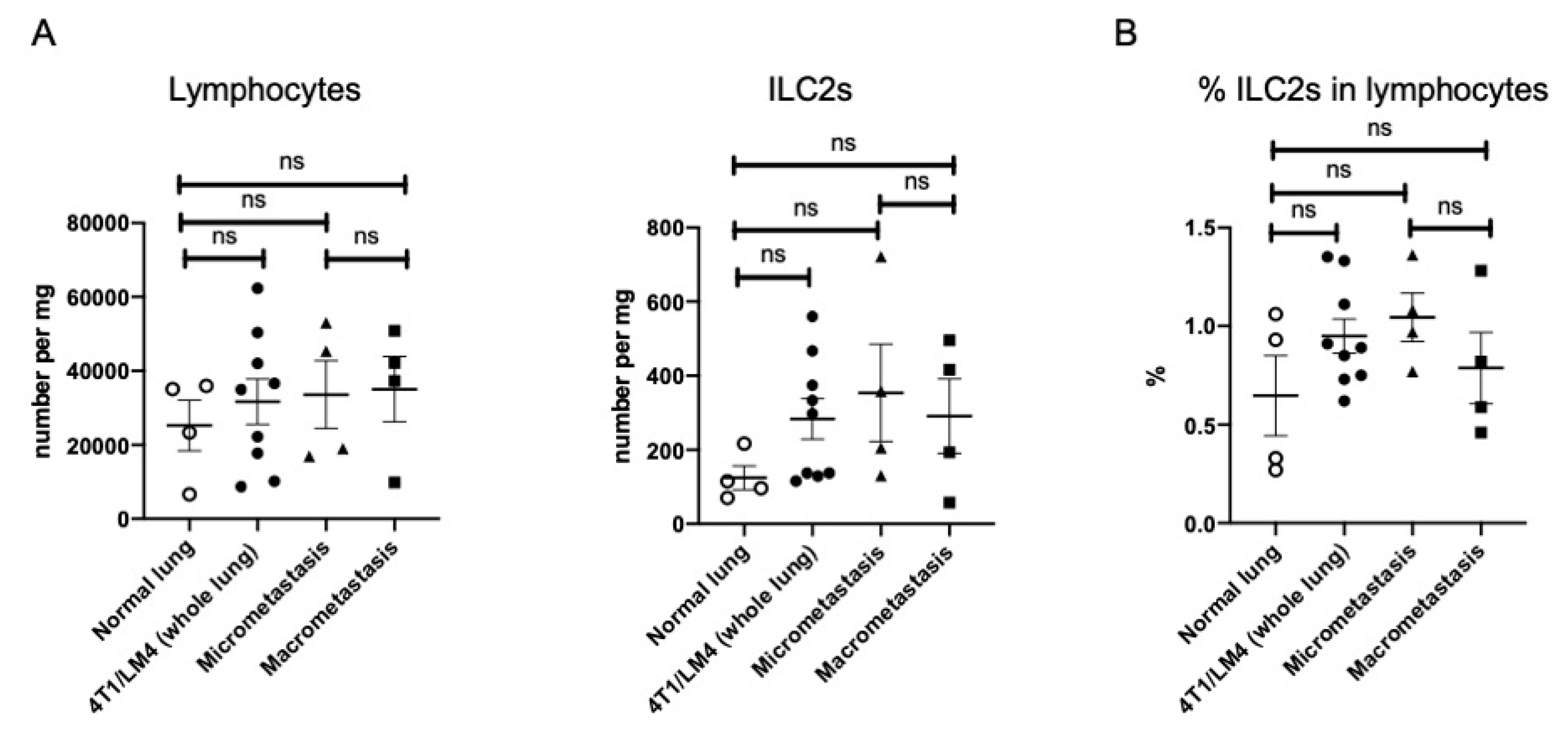
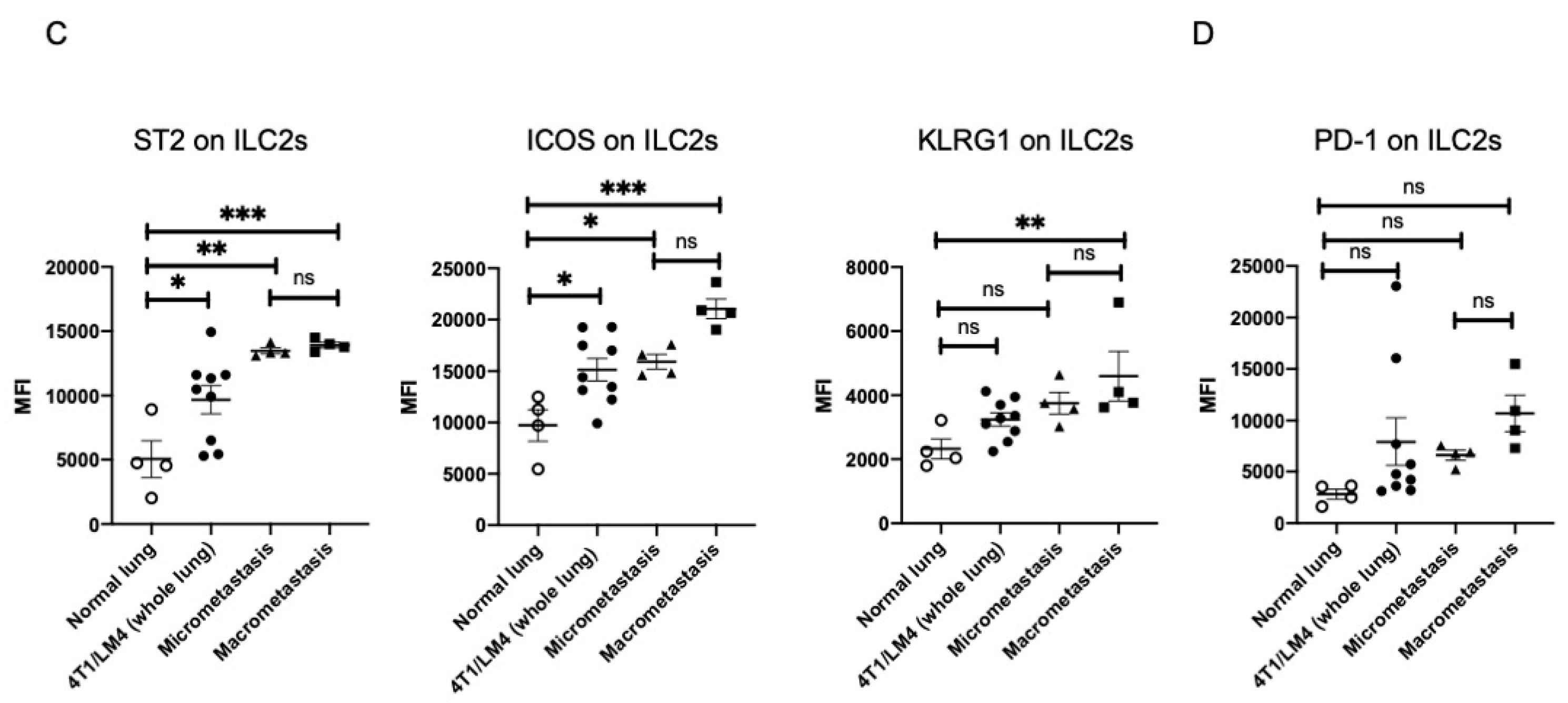
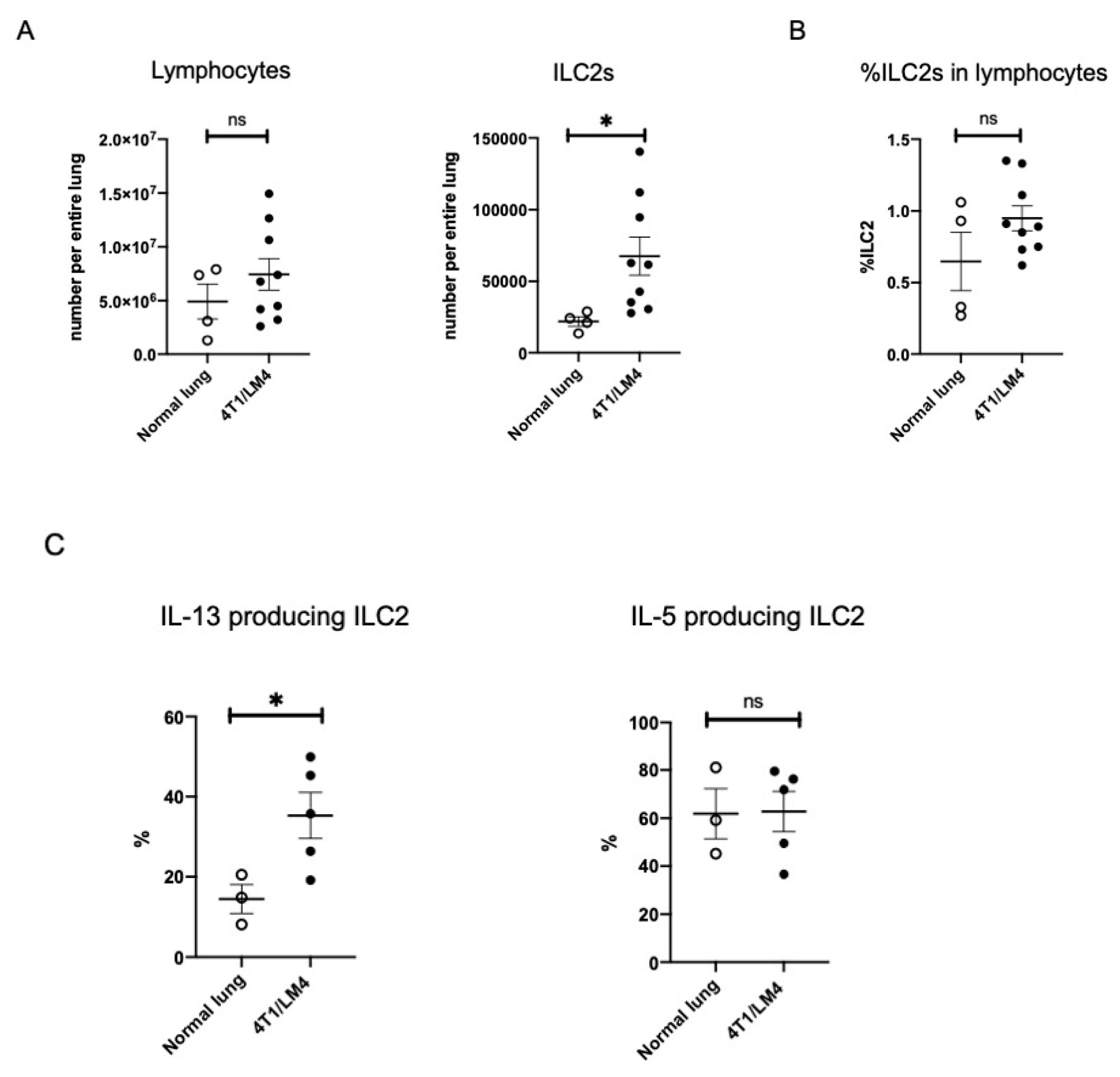
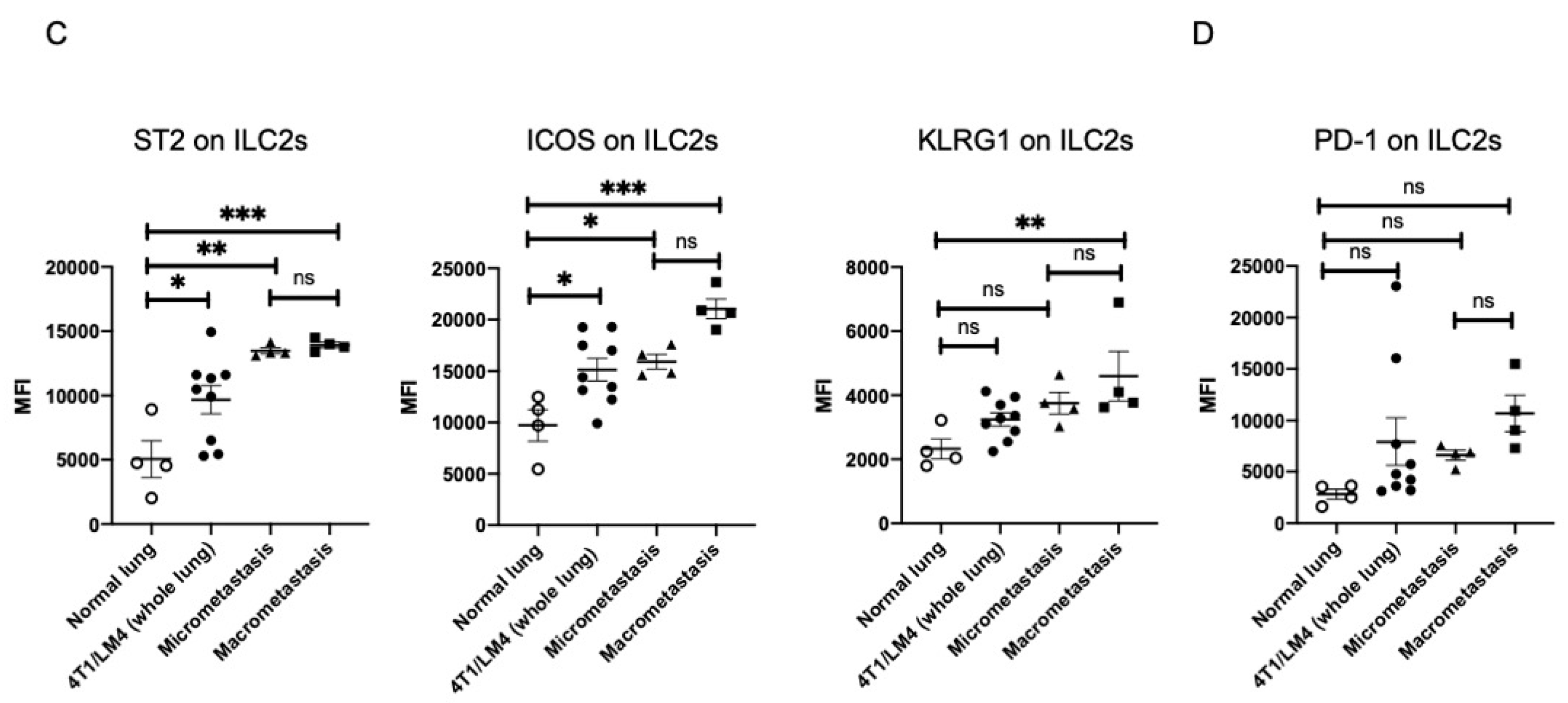
3.2. Lung ILC2s Remained Activated throughout the Metastatic Cascades, Thereby Producing IL-13
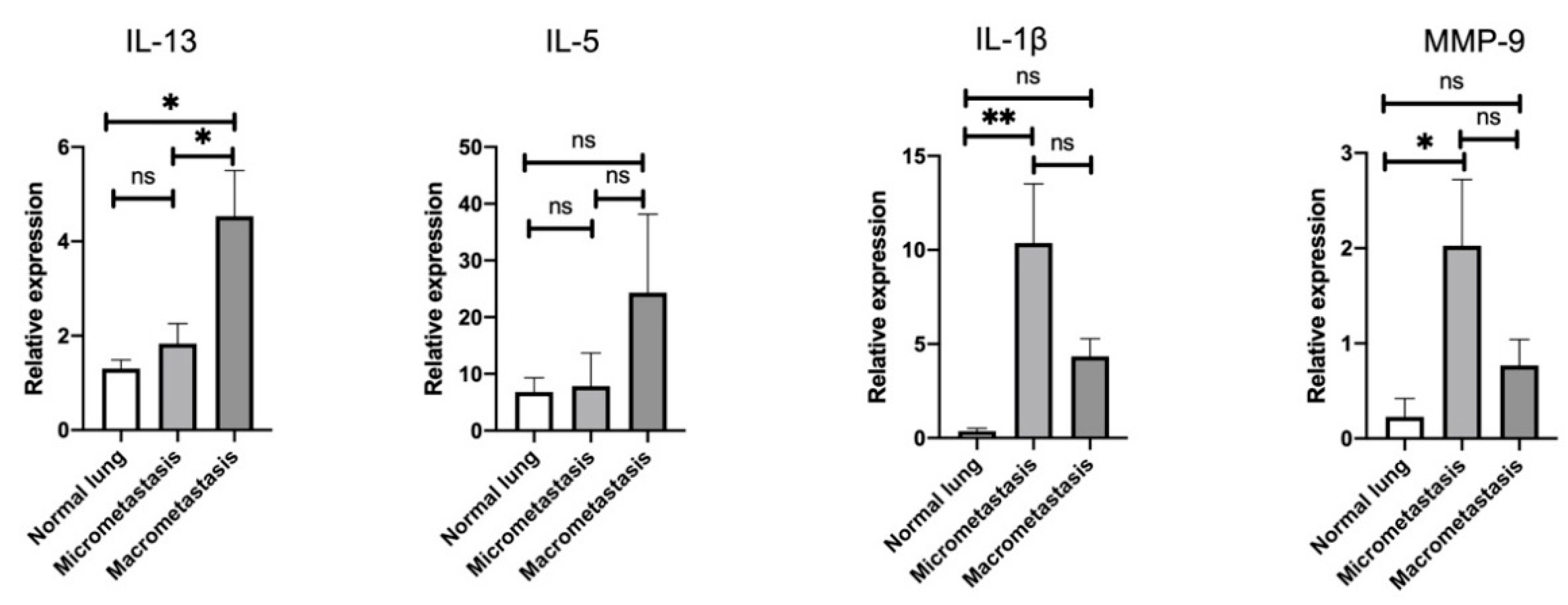
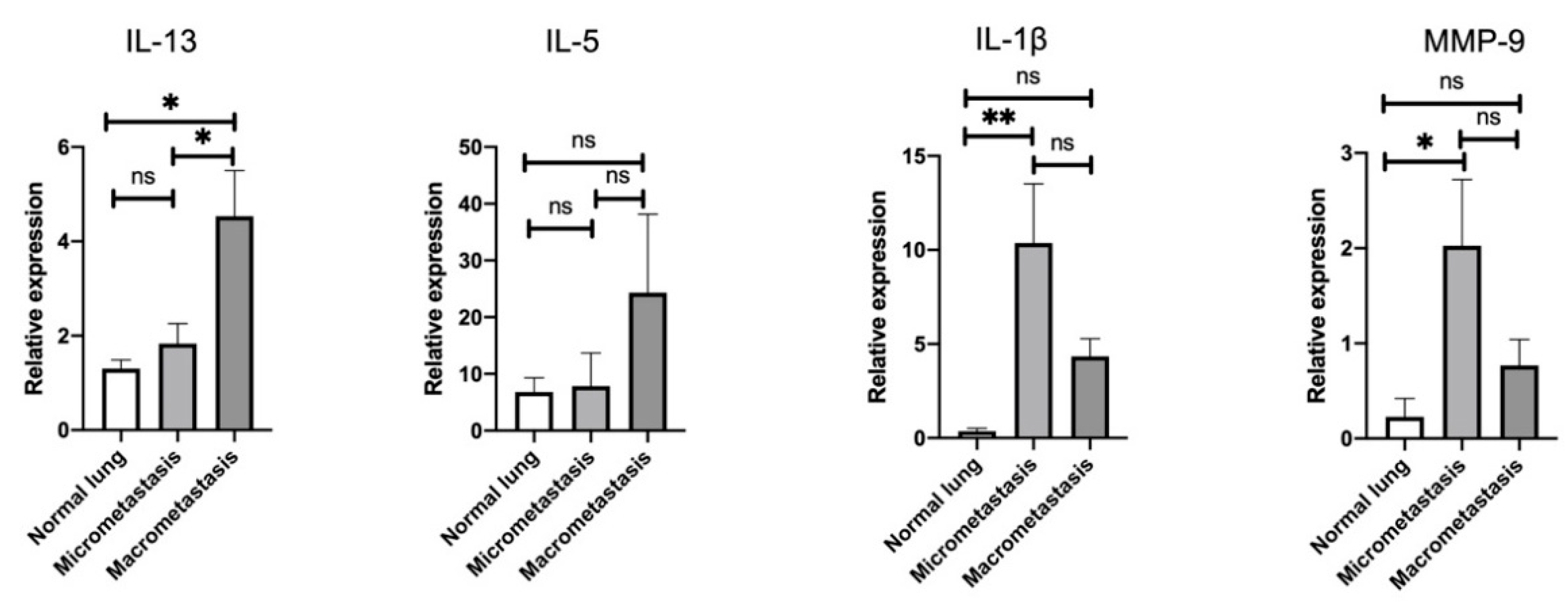
3.3. Stage-Specific Gene Signatures in the Pulmonary TME throughout the Metastatic Cascades
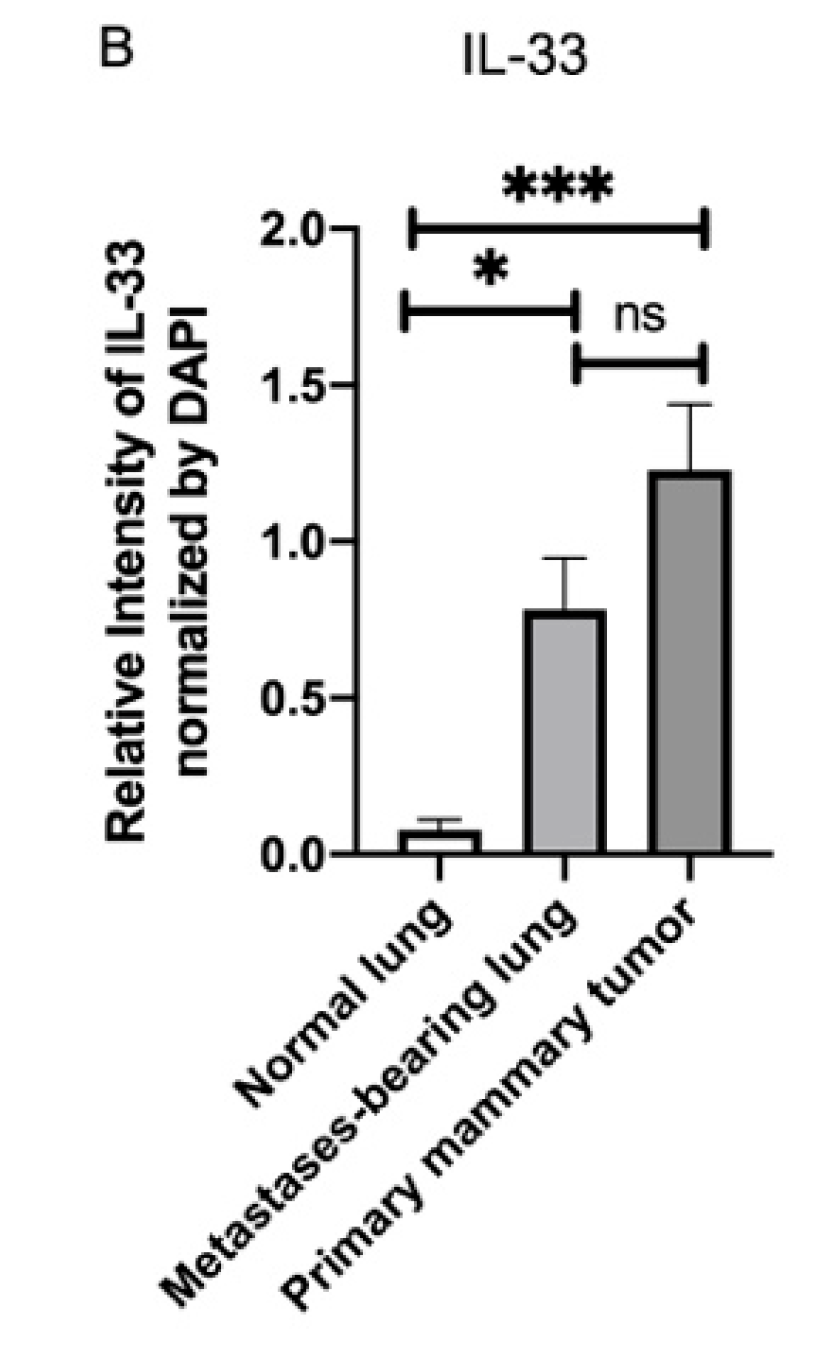
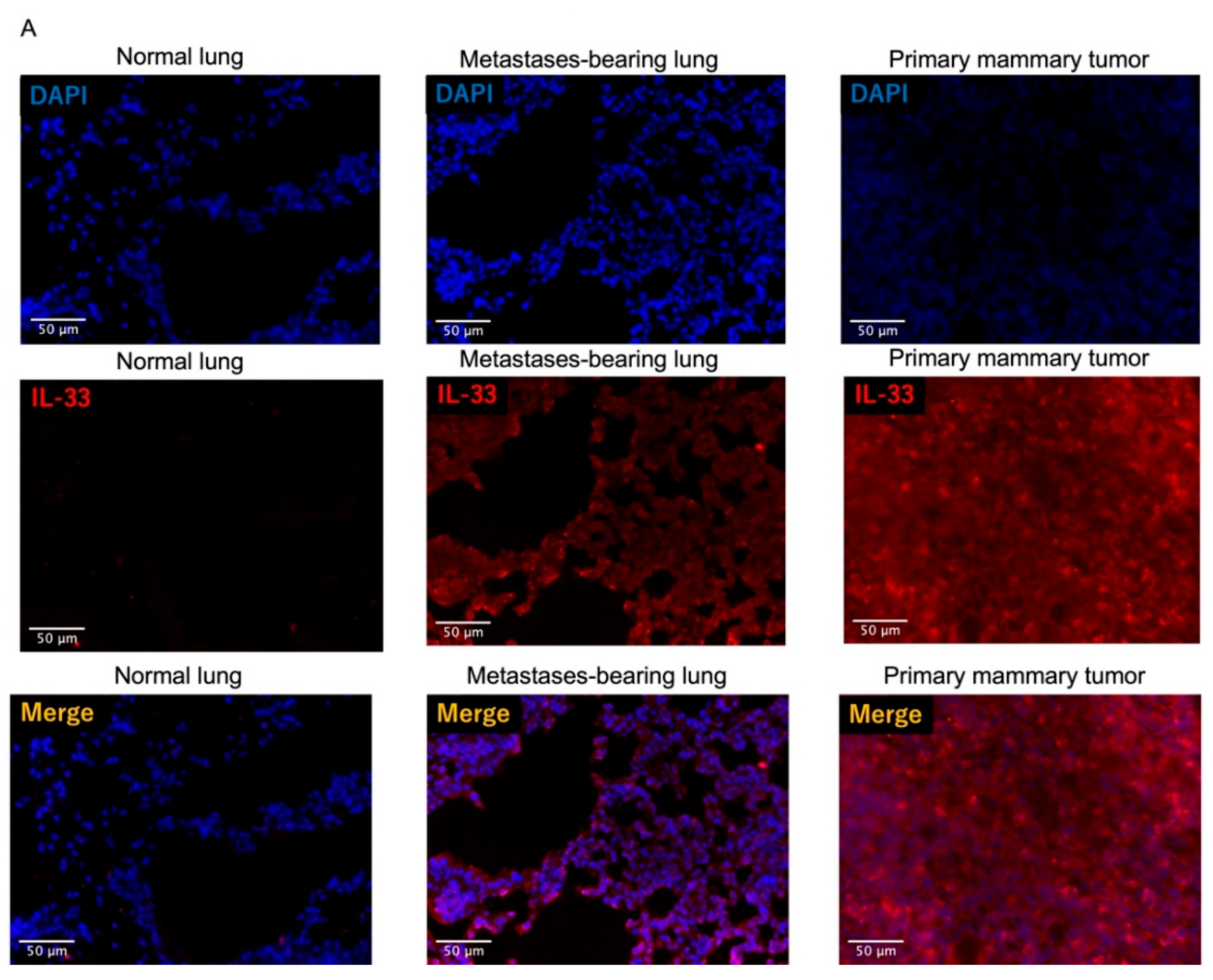
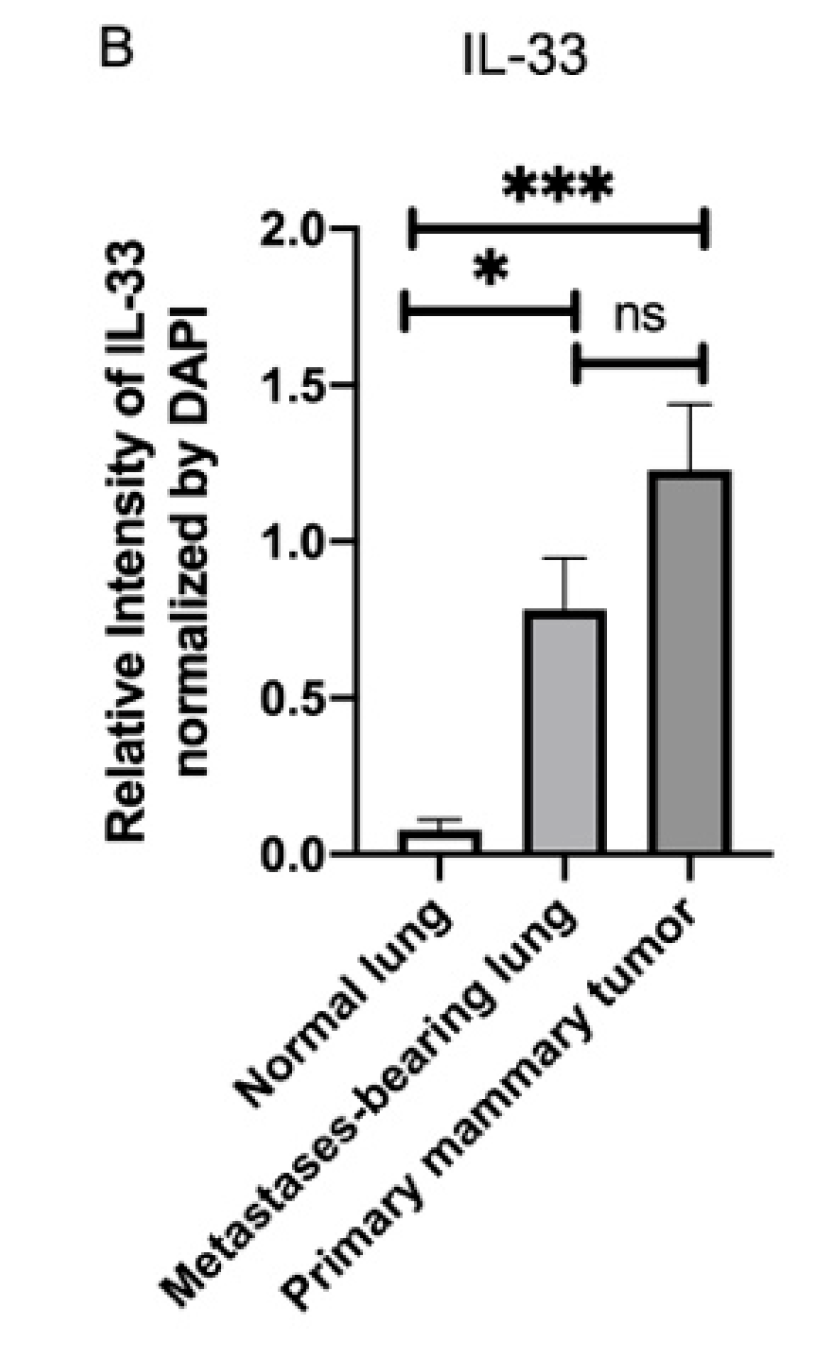
3.4. Fluorescence Intensity of IL-33 Was Elevated Not Only in the Metastatses-Bearing Lungs but Also in the Primary Mammary Tumors
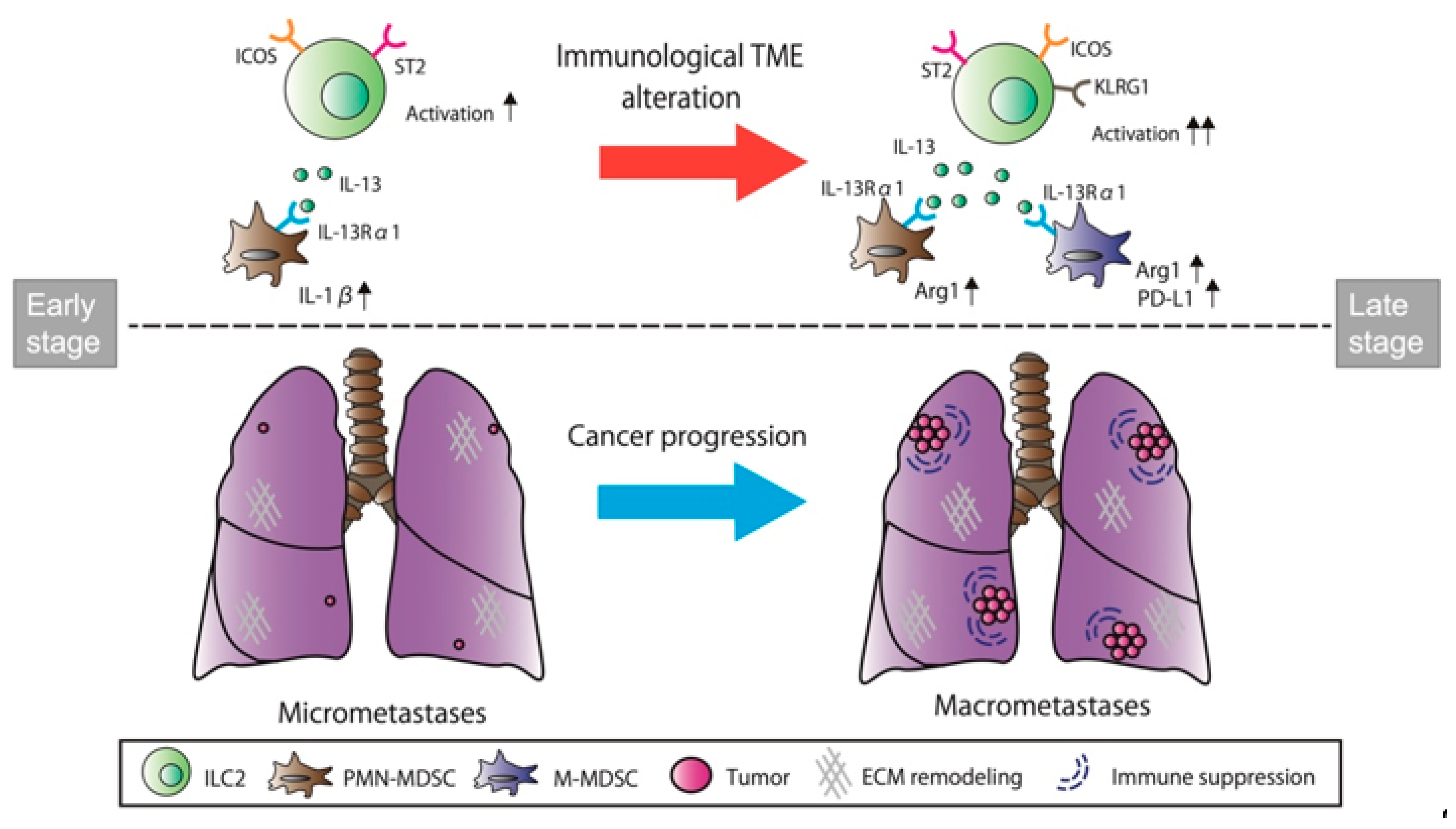
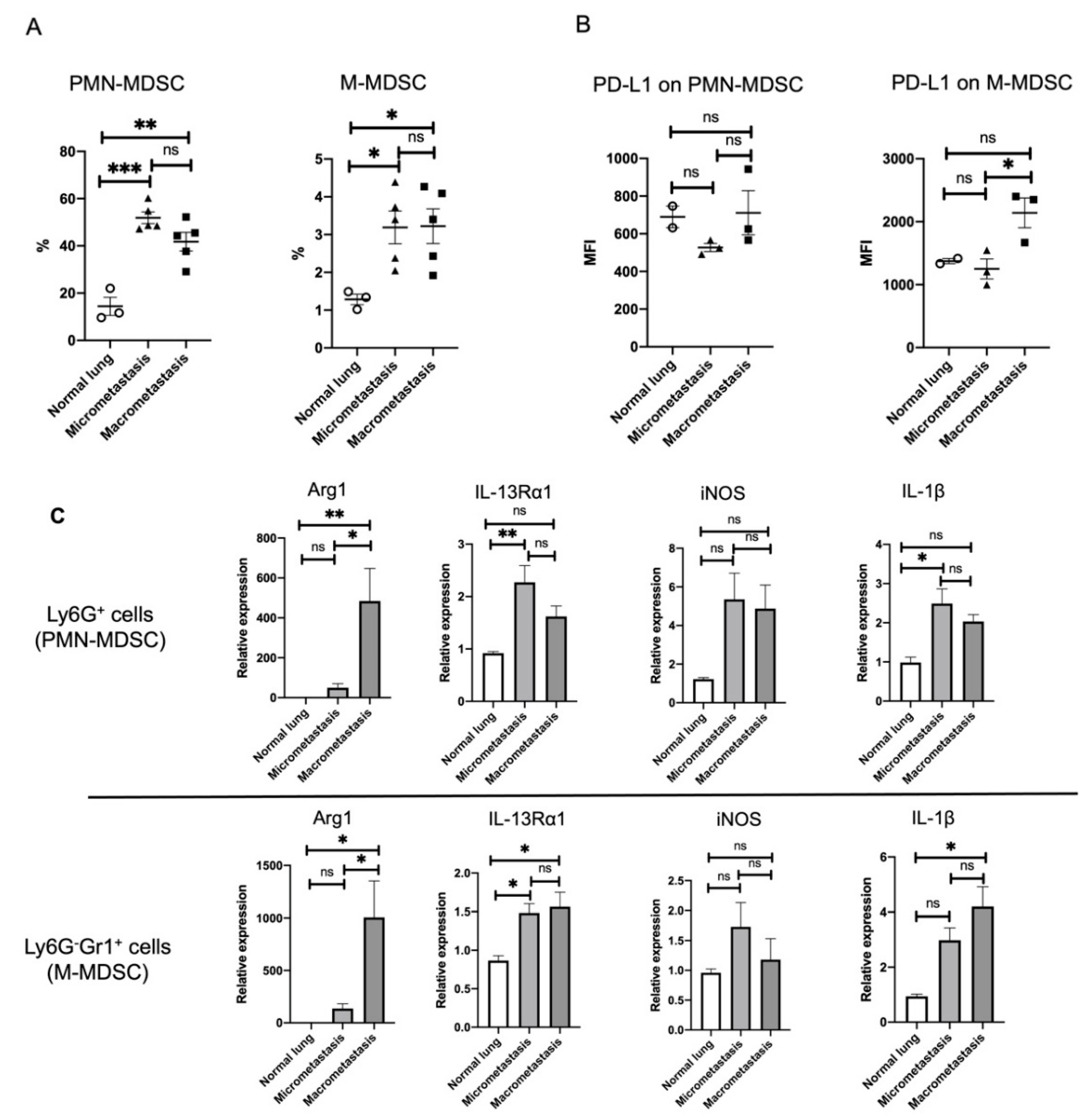
3.5. ILC2 Activation Is Associated with the Induction of Differential MDSC Functions during Cancer Progression from the Micro- to the Macrometastatic Stage
4. Discussion
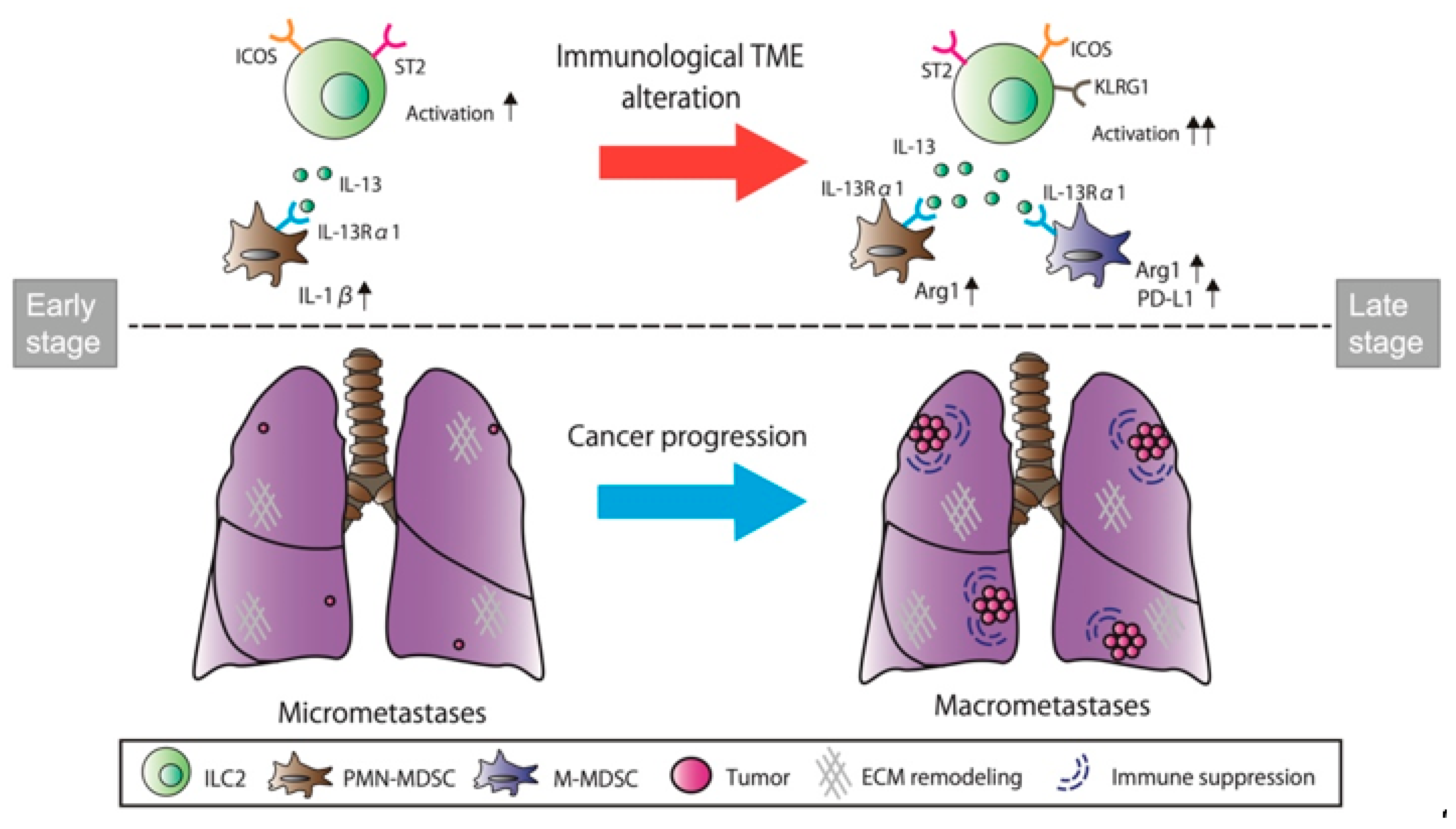
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hess, K.R.; Varadhachary, G.R.; Taylor, S.H.; Wei, W.; Raber, M.N.; Lenzi, R.; Abbruzzese, J.L. Metastatic patterns in adenocarcinoma. Cancer 2006, 106, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.; Allan, A.L. Molecular Mechanisms of Breast Cancer Metastasis to the Lung: Clinical and Experimental Perspectives. Int. J. Mol. Sci. 2019, 20, 2272. [Google Scholar] [CrossRef] [Green Version]
- Oliver, A.J.; Lau, P.K.H.; Unsworth, A.S.; Loi, S.; Darcy, P.K.; Kershaw, M.H.; Slaney, C.Y. Tissue-Dependent Tumor Microenvironments and Their Impact on Immunotherapy Responses. Front. Immunol. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, S.; Wei, M.; Wang, S.; Dong, J.; Wei, J. Pan-Cancer Analysis of Immune Cell Infiltration Identifies a Prognostic Immune-Cell Characteristic Score (ICCS) in Lung Adenocarcinoma. Front. Immunol. 2020, 11, 1218. [Google Scholar] [CrossRef]
- Oliver, A.J.; Davey, A.S.; Keam, S.P.; Mardiana, S.; Chan, J.D.; von Scheidt, B.; Beavis, P.A.; House, I.G.; Van Audernaerde, J.R.; Darcy, P.K.; et al. Tissue-specific tumor microenvironments influence responses to immunotherapies. Clin. Transl. Immunol. 2019, 8, e1094. [Google Scholar] [CrossRef] [Green Version]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.; Baker, D.; Ten Dijke, P. TGF-β-Mediated Epithelial-Mesenchymal Transition and Cancer Metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef] [Green Version]
- Raja, R.; Pandey, A.; Kumar, P. Epithelial to mesenchymal plasticity: Role in cancer progression. Front. Biosci. 2020, 25, 838–873. [Google Scholar] [CrossRef]
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adh. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [Green Version]
- Chrabaszcz, K.; Jasztal, A.; Smęda, M.; Zieliński, B.; Blat, A.; Diem, M.; Chlopicki, S.; Malek, K.; Marzec, K.M. Label-free FTIR spectroscopy detects and visualizes the early stage of pulmonary micrometastasis seeded from breast carcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3574–3584. [Google Scholar] [CrossRef]
- Rosen, P.P.; Saigo, P.E.; Braun, D.W.; Weathers, E.; Fracchia, A.A.; Kinne, D.W. Axillary micro- and macrometastases in breast cancer: Prognostic significance of tumor size. Ann. Surg. 1981, 194, 585–591. [Google Scholar] [CrossRef]
- Aramaki, N.; Ishii, G.; Yamada, E.; Morise, M.; Aokage, K.; Kojima, M.; Hishida, T.; Yoshida, J.; Ikeda, N.; Tsuboi, M.; et al. Drastic morphological and molecular differences between lymph node micrometastatic tumors and macrometastatic tumors of lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 37–46. [Google Scholar] [CrossRef]
- Wei, R.; Liu, S.; Zhang, S.; Min, L.; Zhu, S. Cellular and Extracellular Components in Tumor Microenvironment and Their Application in Early Diagnosis of Cancers. Anal. Cell. Pathol. 2020, 2020, 6283796. [Google Scholar] [CrossRef] [Green Version]
- Teleanu, R.I.; Chircov, C.; Grumezescu, A.M.; Teleanu, D.M. Tumor Angiogenesis and Anti-Angiogenic Strategies for Cancer Treatment. J. Clin. Med. 2019, 9, 84. [Google Scholar] [CrossRef] [Green Version]
- Shani, O.; Raz, Y.; Monteran, L.; Scharff, Y.; Levi-Galibov, O.; Megides, O.; Shacham, H.; Cohen, N.; Silverbush, D.; Avivi, C.; et al. Evolution of fibroblasts in the lung metastatic microenvironment is driven by stage-specific transcriptional plasticity. eLife 2021, 10, e60745. [Google Scholar] [CrossRef]
- Quesnel, B. Tumor dormancy and immunoescape. Apmis 2008, 116, 685–694. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, I.P.; Pejnovic, N.N.; Radosavljevic, G.D.; Pantic, J.M.; Milovanovic, M.Z.; Arsenijevic, N.N.; Lukic, M.L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int. J. Cancer 2014, 134, 1669–1682. [Google Scholar] [CrossRef] [PubMed]
- Ercolano, G.; Falquet, M.; Vanoni, G.; Trabanelli, S.; Jandus, C. ILC2s: New Actors in Tumor Immunity. Front. Immunol 2019, 10, 2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef] [PubMed]
- Bie, Q.; Zhang, P.; Su, Z.; Zheng, D.; Ying, X.; Wu, Y.; Yang, H.; Chen, D.; Wang, S.; Xu, H. Polarization of ILC2s in peripheral blood might contribute to immunosuppressive microenvironment in patients with gastric cancer. J. Immunol. Res. 2014, 2014, 923135. [Google Scholar] [CrossRef]
- Chevalier, M.F.; Trabanelli, S.; Racle, J.; Salomé, B.; Cesson, V.; Gharbi, D.; Bohner, P.; Domingos-Pereira, S.; Dartiguenave, F.; Fritschi, A.S.; et al. ILC2-modulated T cell-to-MDSC balance is associated with bladder cancer recurrence. J. Clin. Investig. 2017, 127, 2916–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Ealey, K.N.; Tetsu, H.; Kiniwa, T.; Motomura, Y.; Moro, K.; Koyasu, S. Tumor-Derived Lactic Acid Contributes to the Paucity of Intratumoral ILC2s. Cell Rep. 2020, 30, 2743–2757.e2745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquelot, N.; Seillet, C.; Wang, M.; Pizzolla, A.; Liao, Y.; Hediyeh-Zadeh, S.; Grisaru-Tal, S.; Louis, C.; Huang, Q.; Schreuder, J.; et al. Blockade of the co-inhibitory molecule PD-1 unleashes ILC2-dependent antitumor immunity in melanoma. Nat. Immunol. 2021, 22, 851–864. [Google Scholar] [CrossRef]
- Zhao, N.; Zhu, W.; Wang, J.; Liu, W.; Kang, L.; Yu, R.; Liu, B. Group 2 innate lymphoid cells promote TNBC lung metastasis via the IL-13-MDSC axis in a murine tumor model. Int. Immunopharmacol. 2021, 99, 107924. [Google Scholar] [CrossRef] [PubMed]
- Hiraga, T.; Williams, P.J.; Ueda, A.; Tamura, D.; Yoneda, T. Zoledronic acid inhibits visceral metastases in the 4T1/luc mouse breast cancer model. Clin. Cancer Res. 2004, 10, 4559–4567. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Zhang, Q.; Shao, N.; Shan, Z.; Cheang, T.; Zhang, Z.; Su, Q.; Wang, S.; Lin, Y. Respiratory hyperoxia reverses immunosuppression by regulating myeloid-derived suppressor cells and PD-L1 expression in a triple-negative breast cancer mouse model. Am. J. Cancer Res. 2019, 9, 529–545. [Google Scholar]
- Bausero, M.A.; Page, D.T.; Osinaga, E.; Asea, A. Surface expression of Hsp25 and Hsp72 differentially regulates tumor growth and metastasis. Tumour. Biol. 2004, 25, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Samant, R.S.; Debies, M.T.; Hurst, D.R.; Moore, B.P.; Shevde, L.A.; Welch, D.R. Suppression of murine mammary carcinoma metastasis by the murine ortholog of breast cancer metastasis suppressor 1 (Brms1). Cancer Lett. 2006, 235, 260–265. [Google Scholar] [CrossRef]
- Cok, S.J.; Acton, S.J.; Morrison, A.R. The proximal region of the 3'-untranslated region of cyclooxygenase-2 is recognized by a multimeric protein complex containing HuR, TIA-1, TIAR, and the heterogeneous nuclear ribonucleoprotein U. J. Biol. Chem. 2003, 278, 36157–36162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akama, Y.; Park, E.J.; Satoh-Takayama, N.; Gaowa, A.; Ito, A.; Kawamoto, E.; Darkwah, S.; Appiah, M.G.; Myint, P.K.; Ohno, H.; et al. Sepsis Induces Deregulation of IL-13 Production and PD-1 Expression in Lung Group 2 Innate Lymphoid Cells. Shock 2021, 55, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef]
- Akama, Y.; Satoh-Takayama, N.; Kawamoto, E.; Ito, A.; Gaowa, A.; Park, E.J.; Imai, H.; Shimaoka, M. The Role of Innate Lymphoid Cells in the Regulation of Immune Homeostasis in Sepsis-Mediated Lung Inflammation. Diagnostics 2020, 10, 808. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Juedes, A.E.; Temann, U.A.; Shresta, S.; Allison, J.P.; Ruddle, N.H.; Flavell, R.A. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001, 409, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Maazi, H.; Patel, N.; Sankaranarayanan, I.; Suzuki, Y.; Rigas, D.; Soroosh, P.; Freeman, G.J.; Sharpe, A.H.; Akbari, O. ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 2015, 42, 538–551. [Google Scholar] [CrossRef] [Green Version]
- Tata, A.; Dodard, G.; Fugère, C.; Leget, C.; Ors, M.; Rossi, B.; Vivier, E.; Brossay, L. Combination blockade of KLRG1 and PD-1 promotes immune control of local and disseminated cancers. Oncoimmunology 2021, 10, 1933808. [Google Scholar] [CrossRef]
- Yousef, E.M.; Tahir, M.R.; St-Pierre, Y.; Gaboury, L.A. MMP-9 expression varies according to molecular subtypes of breast cancer. BMC Cancer 2014, 14, 609. [Google Scholar] [CrossRef] [Green Version]
- Mondal, S.; Adhikari, N.; Banerjee, S.; Amin, S.A.; Jha, T. Matrix metalloproteinase-9 (MMP-9) and its inhibitors in cancer: A minireview. Eurr. J. Med. Chem. 2020, 194, 112260. [Google Scholar] [CrossRef]
- Lin, C.C.; Kuo, C.T.; Cheng, C.Y.; Wu, C.Y.; Lee, C.W.; Hsieh, H.L.; Lee, I.T.; Yang, C.M. IL-1 beta promotes A549 cell migration via MAPKs/AP-1- and NF-kappaB-dependent matrix metalloproteinase-9 expression. Cell Signal. 2009, 21, 1652–1662. [Google Scholar] [CrossRef]
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1β activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuyama, A.; Hosokawa, T.; Mochitate, K. Interleukin-1beta and tumor necrosis factor-alpha have opposite effects on fibroblasts and epithelial cells during basement membrane formation. Matrix Biol. 2008, 27, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Trabanelli, S.; Chevalier, M.F.; Martinez-Usatorre, A.; Gomez-Cadena, A.; Salomé, B.; Lecciso, M.; Salvestrini, V.; Verdeil, G.; Racle, J.; Papayannidis, C.; et al. Tumour-derived PGD2 and NKp30-B7H6 engagement drives an immunosuppressive ILC2-MDSC axis. Nat. Commun. 2017, 8, 593. [Google Scholar] [CrossRef] [Green Version]
- Chaves, K.C.; Costa, E.M.; Teixeira, L.F.; Bellini, M.H. Impact of endostatin gene therapy on myeloid-derived suppressor cells from a metastatic renal cell carcinoma. Exp. Oncol. 2018, 40, 24–32. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Jayaraman, P.; Parikh, F.; Lopez-Rivera, E.; Hailemichael, Y.; Clark, A.; Ma, G.; Cannan, D.; Ramacher, M.; Kato, M.; Overwijk, W.W.; et al. Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J. Immunol. 2012, 188, 5365–5376. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Lüthi, A.U.; Cullen, S.P.; McNeela, E.A.; Duriez, P.J.; Afonina, I.S.; Sheridan, C.; Brumatti, G.; Taylor, R.C.; Kersse, K.; Vandenabeele, P.; et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009, 31, 84–98. [Google Scholar] [CrossRef]
- Xiao, P.; Wan, X.; Cui, B.; Liu, Y.; Qiu, C.; Rong, J.; Zheng, M.; Song, Y.; Chen, L.; He, J.; et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1063772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimen Bozkus, C.; Elzey, B.D.; Crist, S.A.; Ellies, L.G.; Ratliff, T.L. Expression of Cationic Amino Acid Transporter 2 Is Required for Myeloid-Derived Suppressor Cell-Mediated Control of T Cell Immunity. J. Immunol. 2015, 195, 5237–5250. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Downs, L.C.; Thorpe, J.E.; Disch, B.C.; Bastian, A.; Hauser, P.J.; Farasyn, T.; Berry, W.L.; Hurst, R.E.; Ihnat, M.A. Development and characterization of a preclinical model of breast cancer lung micrometastatic to macrometastatic progression. PLoS ONE 2014, 9, e98624. [Google Scholar] [CrossRef] [PubMed]
- Pein, M.; Insua-Rodríguez, J.; Hongu, T.; Riedel, A.; Meier, J.; Wiedmann, L.; Decker, K.; Essers, M.A.G.; Sinn, H.P.; Spaich, S.; et al. Metastasis-initiating cells induce and exploit a fibroblast niche to fuel malignant colonization of the lungs. Nat. Commun. 2020, 11, 1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Liu, W.; Song, J.; Du, X.; Zhou, Y.; Li, Y.; Li, R.; Lyu, L.; He, Y.; Hao, J.; Ben, J.; et al. AKR1B10 (Aldo-keto reductase family 1 B10) promotes brain metastasis of lung cancer cells in a multi-organ microfluidic chip model. Acta Biomater. 2019, 91, 195–208. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Kubota, S.I.; Takahashi, K.; Nishida, J.; Morishita, Y.; Ehata, S.; Tainaka, K.; Miyazono, K.; Ueda, H.R. Whole-Body Profiling of Cancer Metastasis with Single-Cell Resolution. Cell Rep. 2017, 20, 236–250. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward 5′–3′ | Reverse 5′–3′ | Species |
|---|---|---|---|
| β-actin | CATCGTACTCCTGCTTGCTG | AGCGCAAGTACTCTGTGTGG | mouse |
| IL-13 | GCTTATTGAGGAGCTGAGCAAC | GGCCAGGTCCACACTCCATA | mouse |
| IL-5 | CGCTCACCGAGCTCTGTTG | CCAATGCATAGCTGGTGATTTT | mouse |
| Arg1 | AACACGGCAGTGGCTTTAACC | GGTTTTCATGTGGCGCATTC | mouse |
| iNOS | CAGCTGGGCTGTACAAACCTT | CATTGGAAGTGAAGCGGTTCG | mouse |
| IL-13Rα1 | CATGGAGGGTACAAGTTGTTTCC | GTTTTGACTCTTACTCTGACTGTGTAGACA | mouse |
| IL-1β | GCCTTGGGCCTCAAAGGAAAGAATC | GGAAGACACAGATTCCATGGTGAAG | mouse |
| MMP9 | CTGGACAGCCAGACACTAAAG | CTCGCGGCAAGTCTTCAGAG | mouse |
| Luciferase | GCGCGGAGGAGTTGTGTT | TCTGATTTTTCTTGCGTCGAGTT | mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ito, A.; Akama, Y.; Satoh-Takayama, N.; Saito, K.; Kato, T.; Kawamoto, E.; Gaowa, A.; Park, E.J.; Takao, M.; Shimaoka, M. Possible Metastatic Stage-Dependent ILC2 Activation Induces Differential Functions of MDSCs through IL-13/IL-13Rα1 Signaling during the Progression of Breast Cancer Lung Metastasis. Cancers 2022, 14, 3267. https://doi.org/10.3390/cancers14133267
Ito A, Akama Y, Satoh-Takayama N, Saito K, Kato T, Kawamoto E, Gaowa A, Park EJ, Takao M, Shimaoka M. Possible Metastatic Stage-Dependent ILC2 Activation Induces Differential Functions of MDSCs through IL-13/IL-13Rα1 Signaling during the Progression of Breast Cancer Lung Metastasis. Cancers. 2022; 14(13):3267. https://doi.org/10.3390/cancers14133267
Chicago/Turabian StyleIto, Atsushi, Yuichi Akama, Naoko Satoh-Takayama, Kanako Saito, Takuma Kato, Eiji Kawamoto, Arong Gaowa, Eun Jeong Park, Motoshi Takao, and Motomu Shimaoka. 2022. "Possible Metastatic Stage-Dependent ILC2 Activation Induces Differential Functions of MDSCs through IL-13/IL-13Rα1 Signaling during the Progression of Breast Cancer Lung Metastasis" Cancers 14, no. 13: 3267. https://doi.org/10.3390/cancers14133267
APA StyleIto, A., Akama, Y., Satoh-Takayama, N., Saito, K., Kato, T., Kawamoto, E., Gaowa, A., Park, E. J., Takao, M., & Shimaoka, M. (2022). Possible Metastatic Stage-Dependent ILC2 Activation Induces Differential Functions of MDSCs through IL-13/IL-13Rα1 Signaling during the Progression of Breast Cancer Lung Metastasis. Cancers, 14(13), 3267. https://doi.org/10.3390/cancers14133267