
Inhibition of RNA Binding in SND1 Increases the Levels of miR-1-3p and Sensitizes Cancer Cells to Navitoclax

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culturing
2.2. ShRNA Constructs, Lentivirus Production and Stable Cell Lines
2.3. CRISPR-Cas9 Knockout Cell Lines
2.4. Western Blot and Antibodies
2.5. Drug Sensitivity Testing and Drug Responses
2.6. Chemotherapeutic Drugs and Cell Viability Assay
2.7. MiRNA qPCR Panel
2.8. MiRNA qPCR
2.9. Protein Expression
2.10. Protein Purification
2.11. Fluorescence Polarization Assay
2.12. Screening Assay Development
2.13. Inhibitor Screening and Potency Measurements
2.14. Electrophoretic Mobility Shift Assay
2.15. Isothermal Titration Calorimetry
2.16. Differential Scanning Fluorimetry
2.17. Molecular Docking
2.18. Statistical Analysis
3. Results
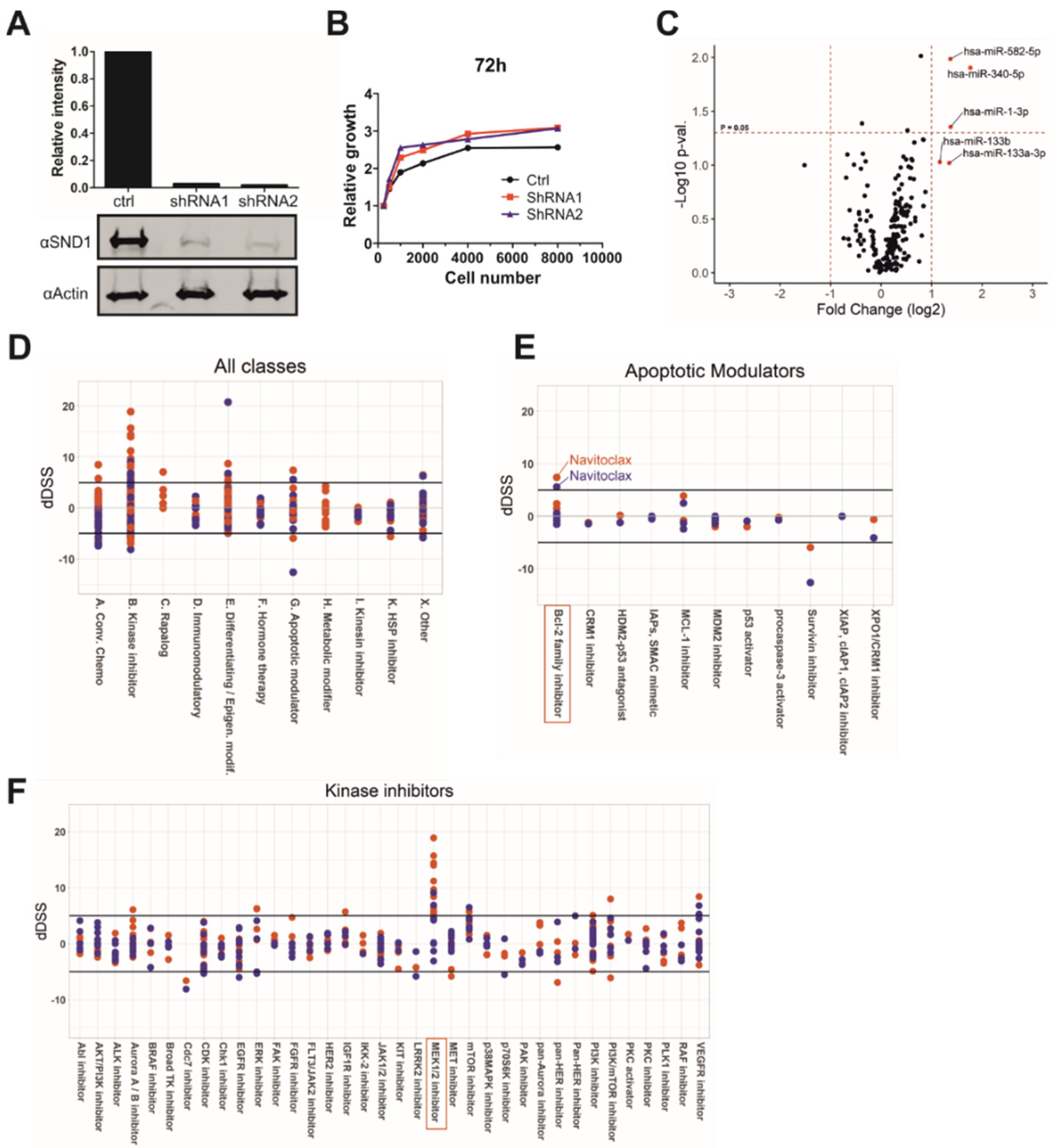
3.1. Silencing of SND1 Increases the Levels of Tumor Suppressor miRNAs
3.2. SND1 Is Involved in MEK-ERK-Pathway and Bcl-2 Related Apoptotic Pathway
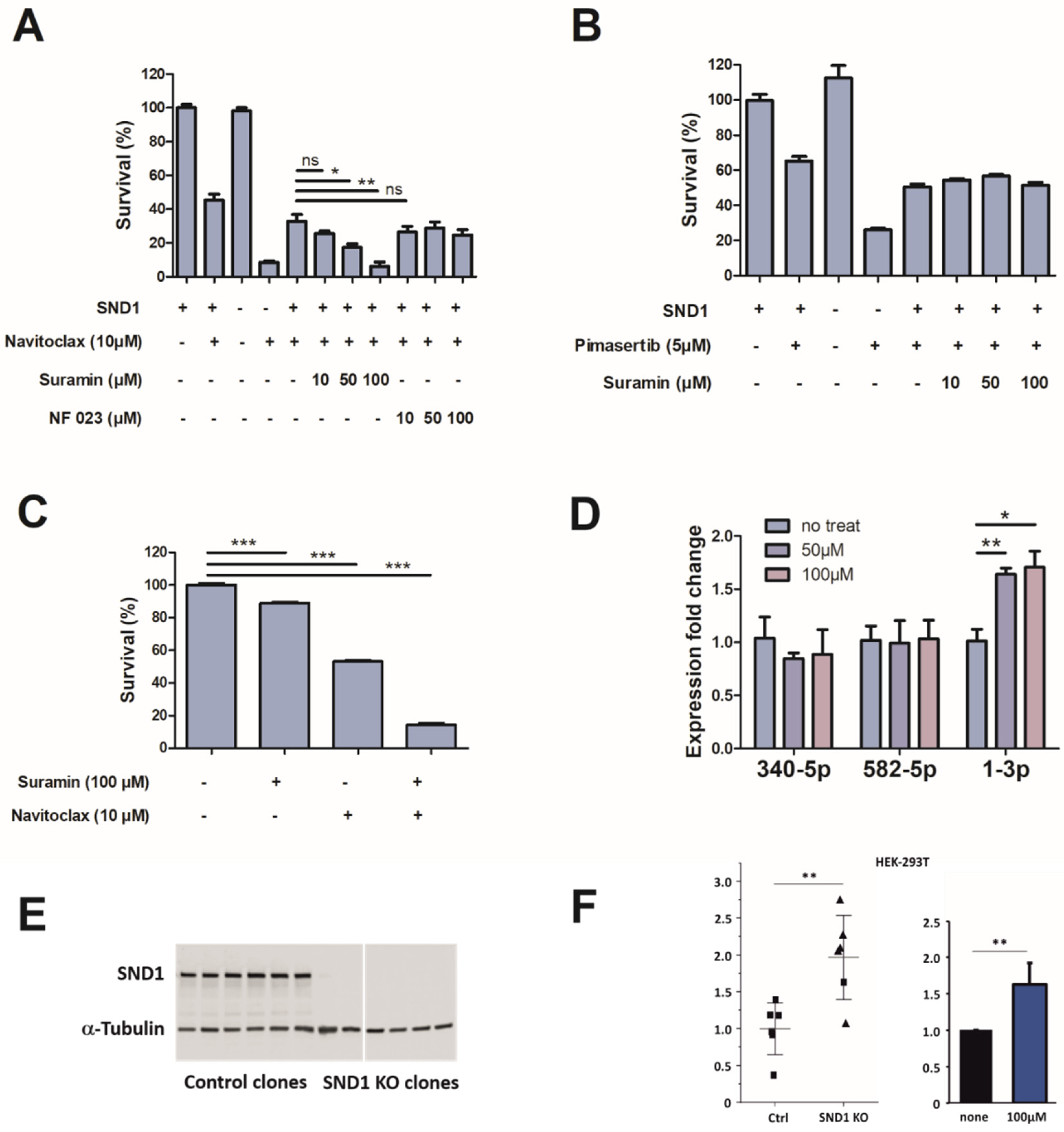
3.3. Discovery of Small Molecule SND1 Inhibitor
3.4. Characteristics of Suramin Binding to SND1
3.5. Suramin Treatment Increases the Expression of miR-1-3p
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Olejniczak, M.; Kotowska-Zimmer, A.; Krzyzosiak, W. Stress-induced changes in miRNA biogenesis and functioning. Cell. Mol. Life Sci. 2018, 75, 177–191. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Jariwala, N.; Rajasekaran, D.; Mendoza, R.G.; Shen, X.N.; Siddiq, A.; Akiel, M.A.; Robertson, C.L.; Subler, M.A.; Windle, J.J.; Fisher, P.B.; et al. Oncogenic Role of SND1 in Development and Progression of Hepatocellular Carcinoma. Cancer Res. 2017, 77, 3306–3316. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.; Wang, Y.; Bedford, M.T. The Role of the PRMT5-SND1 Axis in Hepatocellular Carcinoma. Epigenomes 2021, 5, 2. [Google Scholar] [CrossRef]
- Yoo, B.K.; Santhekadur, P.K.; Gredler, R.; Chen, D.; Emdad, L.; Bhutia, S.; Pannell, L.; Fisher, P.B.; Sarkar, D. Increased RNA-induced silencing complex (RISC) activity contributes to hepatocellular carcinoma. Hepatology 2011, 53, 1538–1548. [Google Scholar] [CrossRef] [Green Version]
- Kuruma, H.; Kamata, Y.; Takahashi, H.; Igarashi, K.; Kimura, T.; Miki, K.; Miki, J.; Sasaki, H.; Hayashi, N.; Egawa, S. Staphylococcal nuclease domain-containing protein 1 as a potential tissue marker for prostate cancer. Am. J. Pathol. 2009, 174, 2044–2050. [Google Scholar] [CrossRef]
- Markmiller, S.; Soltanieh, S.; Server, K.L.; Mak, R.; Jin, W.; Fang, M.Y.; Luo, E.C.; Krach, F.; Yang, D.; Sen, A.; et al. Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell 2018, 172, 590–604.e513. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Beltran, E.; Moschou, P.N.; Smertenko, A.P.; Bozhkov, P.V. Tudor staphylococcal nuclease links formation of stress granules and processing bodies with mRNA catabolism in Arabidopsis. Plant Cell 2015, 27, 926–943. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Beltran, E.; Elander, P.H.; Dalman, K.; Dayhoff, G.W., 2nd; Moschou, P.N.; Uversky, V.N.; Crespo, J.L.; Bozhkov, P.V. Tudor staphylococcal nuclease is a docking platform for stress granule components and is essential for SnRK1 activation in Arabidopsis. EMBO J. 2021, 40, e105043. [Google Scholar] [CrossRef]
- Gao, X.; Ge, L.; Shao, J.; Su, C.; Zhao, H.; Saarikettu, J.; Yao, X.; Yao, Z.; Silvennoinen, O.; Yang, J. Tudor-SN interacts with and co-localizes with G3BP in stress granules under stress conditions. FEBS Lett. 2010, 584, 3525–3532. [Google Scholar] [CrossRef] [Green Version]
- Saarikettu, J.M.; Valanne, S. The RNA-binding Protein Snd1/Tudor-SN is Required for Adaptation to Hypoxia in Mouse and Drosophila. FASEB J. 2021, submitted.
- Jeong, C.Y.; Lee, W.J.; Truong, H.A.; Trinh, C.S.; Jin, J.Y.; Kim, S.; Hwang, K.Y.; Kang, C.S.; Moon, J.K.; Hong, S.W.; et al. Dual role of SND1 facilitates efficient communication between abiotic stress signalling and normal growth in Arabidopsis. Sci. Rep. 2018, 8, 10114. [Google Scholar] [CrossRef]
- Valineva, T.; Yang, J.; Palovuori, R.; Silvennoinen, O. The transcriptional co-activator protein p100 recruits histone acetyltransferase activity to STAT6 and mediates interaction between the CREB-binding protein and STAT6. J. Biol. Chem. 2005, 280, 14989–14996. [Google Scholar] [CrossRef] [Green Version]
- Paukku, K.; Yang, J.; Silvennoinen, O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol. Endocrinol. 2003, 17, 1805–1814. [Google Scholar] [CrossRef]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Shen, M.; Wei, Y.; Kim, H.; Wan, L.; Jiang, Y.Z.; Hang, X.; Raba, M.; Remiszewski, S.; Rowicki, M.; Wu, C.G.; et al. Small-molecule inhibitors that disrupt the MTDH-SND1 complex suppress breast cancer progression and metastasis. Nat. Cancer 2022, 3, 43–59. [Google Scholar] [CrossRef]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.C.; Zalmas, L.P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef] [Green Version]
- Cappellari, M.; Bielli, P.; Paronetto, M.P.; Ciccosanti, F.; Fimia, G.M.; Saarikettu, J.; Silvennoinen, O.; Sette, C. The transcriptional co-activator SND1 is a novel regulator of alternative splicing in prostate cancer cells. Oncogene 2014, 33, 3794–3802. [Google Scholar] [CrossRef] [Green Version]
- Paukku, K.; Kalkkinen, N.; Silvennoinen, O.; Kontula, K.K.; Lehtonen, J.Y. p100 increases AT1R expression through interaction with AT1R 3’-UTR. Nucleic Acids Res. 2008, 36, 4474–4487. [Google Scholar] [CrossRef]
- Scadden, A.D. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nat. Struct. Mol. Biol. 2005, 12, 489–496. [Google Scholar] [CrossRef]
- Baquero-Perez, B.; Antanaviciute, A.; Yonchev, I.D.; Carr, I.M.; Wilson, S.A.; Whitehouse, A. The Tudor SND1 protein is an m(6)A RNA reader essential for replication of Kaposi’s sarcoma-associated herpesvirus. eLife 2019, 8, e47261. [Google Scholar] [CrossRef]
- Martin, S.E.; Gan, H.; Toomer, G.; Sridhar, N.; Sztuba-Solinska, J. The m(6)A landscape of polyadenylated nuclear (PAN) RNA and its related methylome in the context of KSHV replication. RNA 2021, 27, 1102–1125. [Google Scholar] [CrossRef]
- Lan, Q.; Liu, P.Y.; Bell, J.L.; Wang, J.Y.; Hüttelmaier, S.; Zhang, X.D.; Zhang, L.; Liu, T. The Emerging Roles of RNA m(6)A Methylation and Demethylation as Critical Regulators of Tumorigenesis, Drug Sensitivity, and Resistance. Cancer Res. 2021, 81, 3431–3440. [Google Scholar] [CrossRef]
- Caudy, A.A.; Ketting, R.F.; Hammond, S.M.; Denli, A.M.; Bathoorn, A.M.; Tops, B.B.; Silva, J.M.; Myers, M.M.; Hannon, G.J.; Plasterk, R.H. A micrococcal nuclease homologue in RNAi effector complexes. Nature 2003, 425, 411–414. [Google Scholar] [CrossRef]
- Elbarbary, R.A.; Miyoshi, K.; Myers, J.R.; Du, P.; Ashton, J.M.; Tian, B.; Maquat, L.E. Tudor-SN-mediated endonucleolytic decay of human cell microRNAs promotes G1/S phase transition. Science 2017, 356, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, S.; Zarrabi, A.; Hashemi, F.; Zabolian, A.; Saleki, H.; Ranjbar, A.; Seyed Saleh, S.H.; Bagherian, M.; Sharifzadeh, S.O.; Hushmandi, K.; et al. Regulation of Nuclear Factor-KappaB (NF-κB) signaling pathway by non-coding RNAs in cancer: Inhibiting or promoting carcinogenesis? Cancer Lett. 2021, 509, 63–80. [Google Scholar] [CrossRef]
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Nigita, G.; Acunzo, M.; Romano, G.; Veneziano, D.; Laganà, A.; Vitiello, M.; Wernicke, D.; Ferro, A.; Croce, C.M. microRNA editing in seed region aligns with cellular changes in hypoxic conditions. Nucleic Acids Res. 2016, 44, 6298–6308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederschain, D.; Wee, S.; Chen, L.; Loo, A.; Yang, G.; Huang, A.; Chen, Y.; Caponigro, G.; Yao, Y.M.; Lengauer, C.; et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009, 8, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Regan, S.N.; Xia, Y.; Oostrom, L.A.; Cowan, C.A.; Musunuru, K. Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 2013, 12, 393–394. [Google Scholar] [CrossRef] [Green Version]
- Saarikettu, J.; Ovod, V.; Vuoksio, M.; Gronholm, J.; Yang, J.; Silvennoinen, O. Monoclonal antibodies against human Tudor-SN. Hybridoma (Larchmt) 2010, 29, 231–236. [Google Scholar] [CrossRef]
- Pemovska, T.; Kontro, M.; Yadav, B.; Edgren, H.; Eldfors, S.; Szwajda, A.; Almusa, H.; Bespalov, M.M.; Ellonen, P.; Elonen, E.; et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013, 3, 1416–1429. [Google Scholar] [CrossRef] [Green Version]
- Yadav, B.; Pemovska, T.; Szwajda, A.; Kulesskiy, E.; Kontro, M.; Karjalainen, R.; Majumder, M.M.; Malani, D.; Murumagi, A.; Knowles, J.; et al. Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci. Rep. 2014, 4, 5193. [Google Scholar] [CrossRef] [Green Version]
- Peltier, H.J.; Latham, G.J. Normalization of microRNA expression levels in quantitative RT-PCR assays: Identification of suitable reference RNA targets in normal and cancerous human solid tissues. RNA 2008, 14, 844–852. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [Google Scholar] [CrossRef] [Green Version]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Dart, D.A.; Owen, S.; Wen, X.; Ji, J.; Jiang, W. Insights into roles of the miR-1, -133 and -206 family in gastric cancer (Review). Oncol. Rep. 2016, 36, 1191–1198. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xia, L.; Chen, M.; Lin, C.; Wu, H.; Zhang, Y.; Pan, S.; Li, X. miR-133b, a particular member of myomiRs, coming into playing its unique pathological role in human cancer. Oncotarget 2017, 8, 50193–50208. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wong, K.Y.; Calin, G.A.; Chng, W.J.; Chan, G.C.; Chim, C.S. Epigenetic silencing of miR-340-5p in multiple myeloma: Mechanisms and prognostic impact. Clin. Epigenet. 2019, 11, 71. [Google Scholar] [CrossRef]
- Liu, J.; Liu, S.; Deng, X.; Rao, J.; Huang, K.; Xu, G.; Wang, X. MicroRNA-582-5p suppresses non-small cell lung cancer cells growth and invasion via downregulating NOTCH1. PLoS ONE 2019, 14, e0217652. [Google Scholar] [CrossRef] [Green Version]
- Cuatrecasas, P.; Fuchs, S.; Anfinsen, C.B. Catalytic properties and specificity of the extracellular nuclease of Staphylococcus aureus. J. Biol. Chem. 1967, 242, 1541–1547. [Google Scholar] [CrossRef]
- Li, C.L.; Yang, W.Z.; Chen, Y.P.; Yuan, H.S. Structural and functional insights into human Tudor-SN, a key component linking RNA interference and editing. Nucleic Acids Res. 2008, 36, 3579–3589. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Wan, L.; Zheng, A.; Stanevich, V.; Wei, Y.; Satyshur, K.A.; Shen, M.; Lee, W.; Kang, Y.; Xing, Y. Structural insights into the tumor-promoting function of the MTDH-SND1 complex. Cell Rep. 2014, 8, 1704–1713. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, D.; Eide, P.W.; Eilertsen, I.A.; Danielsen, S.A.; Eknaes, M.; Hektoen, M.; Lind, G.E.; Lothe, R.A. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis 2013, 2, e71. [Google Scholar] [CrossRef]
- Wiedemar, N.; Hauser, D.A.; Mäser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, e01168-19. [Google Scholar] [CrossRef]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet. 2006, 38, 228–233. [Google Scholar] [CrossRef]
- Safa, A.; Bahroudi, Z.; Shoorei, H.; Majidpoor, J.; Abak, A.; Taheri, M.; Ghafouri-Fard, S. miR-1: A comprehensive review of its role in normal development and diverse disorders. Biomed. Pharmacother. 2020, 132, 110903. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, C.; Wu, Z.; Wang, Y.; Yin, W.; Lin, Y.; Zhou, L.; Lu, J. Upregulation of microRNA-1 inhibits proliferation and metastasis of breast cancer. Mol. Med. Rep. 2020, 22, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Y.; Ding, P.; He, L. miR-1 Targeted Downregulation of Bcl-2 Increases Chemosensitivity of Lung Cancer Cells. Genet. Test. Mol. Biomark. 2021, 25, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pu, N.; Su, W.; Yang, X.; Xing, C. Downregulation of miR-1 in colorectal cancer promotes radioresistance and aggressive phenotypes. J. Cancer 2020, 11, 4832–4840. [Google Scholar] [CrossRef]
- Chang, J.; Xu, W.; Du, X.; Hou, J. MALAT1 silencing suppresses prostate cancer progression by upregulating miR-1 and downregulating KRAS. OncoTargets Ther. 2018, 11, 3461–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Kim, H.K.; Rettig, R.L.; Kim, J.; Lee, E.T.; Aprelikova, O.; Choi, I.J.; Munroe, D.J.; Green, J.E. miRNA signature associated with outcome of gastric cancer patients following chemotherapy. BMC Med. Genom. 2011, 4, 79. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Construct | IC50 ± Std. Error (µM) | ||
|---|---|---|---|
| Suramin | NF 023 | PPNDS | |
| SND1 | 0.6 ± 1.0 | 1.6 ± 1.0 | 0.5 ± 1.0 |
| SN1-SN2 | 2.4 ± 1.1 | 1.2 ± 1.1 | 28.4 ± 1.1 |
| SN3-SN4-TSN | 7.5 ± 1.1 | 16.3 ± 1.3 | 16.5 ± 1.1 |
| Thermodynamic Parameters | SN1-SN2 | SN3-SN4-TSN |
|---|---|---|
| One-site binding model | ||
| K (M−1) | 8.24 ± 1.09 × 104 | 1.79 ± 0.09 × 105 |
| ΔH (kcal/mol) | −28.8 ± 1.1 | −14.8 ± 0.3 |
| −TΔS (kcal/mol) | 22.0 | 7.7 |
| ΔG (kcal/mol) | −6.8 | −7.1 |
| n | 1.5 | 1.9 |
| Two-site binding model | ||
| K1 (M−1) | 2.45 ± 0.33 × 106 | 7.04 ± 6.97 × 105 |
| ΔH1 (kcal/mol) | −16.2 ± 0.1 | −11.9 ± 0.1 |
| −TΔS1 (kcal/mol) | 7.5 | 3.9 |
| ΔG1 (kcal/mol) | −8.7 | −8.0 |
| n1 | 0.9 | 1.0 |
| K2 (M−1) | 5.96 ± 1.01 × 104 | 8.47 ± 4.08 × 104 |
| ΔH2 (kcal/mol) | −16.6 ± 0.3 | −18.0 ± 2.1 |
| −TΔS2 (kcal/mol) | 10.1 | 11.2 |
| ΔG2 (kcal/mol) | −6.5 | −6.8 |
| n2 | 1.8 | 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehmusvaara, S.; Haikarainen, T.; Saarikettu, J.; Martinez Nieto, G.; Silvennoinen, O. Inhibition of RNA Binding in SND1 Increases the Levels of miR-1-3p and Sensitizes Cancer Cells to Navitoclax. Cancers 2022, 14, 3100. https://doi.org/10.3390/cancers14133100
Lehmusvaara S, Haikarainen T, Saarikettu J, Martinez Nieto G, Silvennoinen O. Inhibition of RNA Binding in SND1 Increases the Levels of miR-1-3p and Sensitizes Cancer Cells to Navitoclax. Cancers. 2022; 14(13):3100. https://doi.org/10.3390/cancers14133100
Chicago/Turabian StyleLehmusvaara, Saara, Teemu Haikarainen, Juha Saarikettu, Guillermo Martinez Nieto, and Olli Silvennoinen. 2022. "Inhibition of RNA Binding in SND1 Increases the Levels of miR-1-3p and Sensitizes Cancer Cells to Navitoclax" Cancers 14, no. 13: 3100. https://doi.org/10.3390/cancers14133100
APA StyleLehmusvaara, S., Haikarainen, T., Saarikettu, J., Martinez Nieto, G., & Silvennoinen, O. (2022). Inhibition of RNA Binding in SND1 Increases the Levels of miR-1-3p and Sensitizes Cancer Cells to Navitoclax. Cancers, 14(13), 3100. https://doi.org/10.3390/cancers14133100