
Intestinal FXR Activation via Transgenic Chimera or Chemical Agonism Prevents Colitis-Associated and Genetically-Induced Colon Cancer

 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Mice
2.2. Colitis Carcinogenesis Model
2.3. Colitis Model
2.4. Intestinal Permeability Assay
2.5. Adenomatous Polyposis Coli ApcMin/+ Mice
2.6. BA Measurements
2.7. RNA Extraction and Quantitative Real Time qRT-PCR Analysis
2.8. Histology and Immunohistochemistry
2.9. Statistical Analysis
3. Results
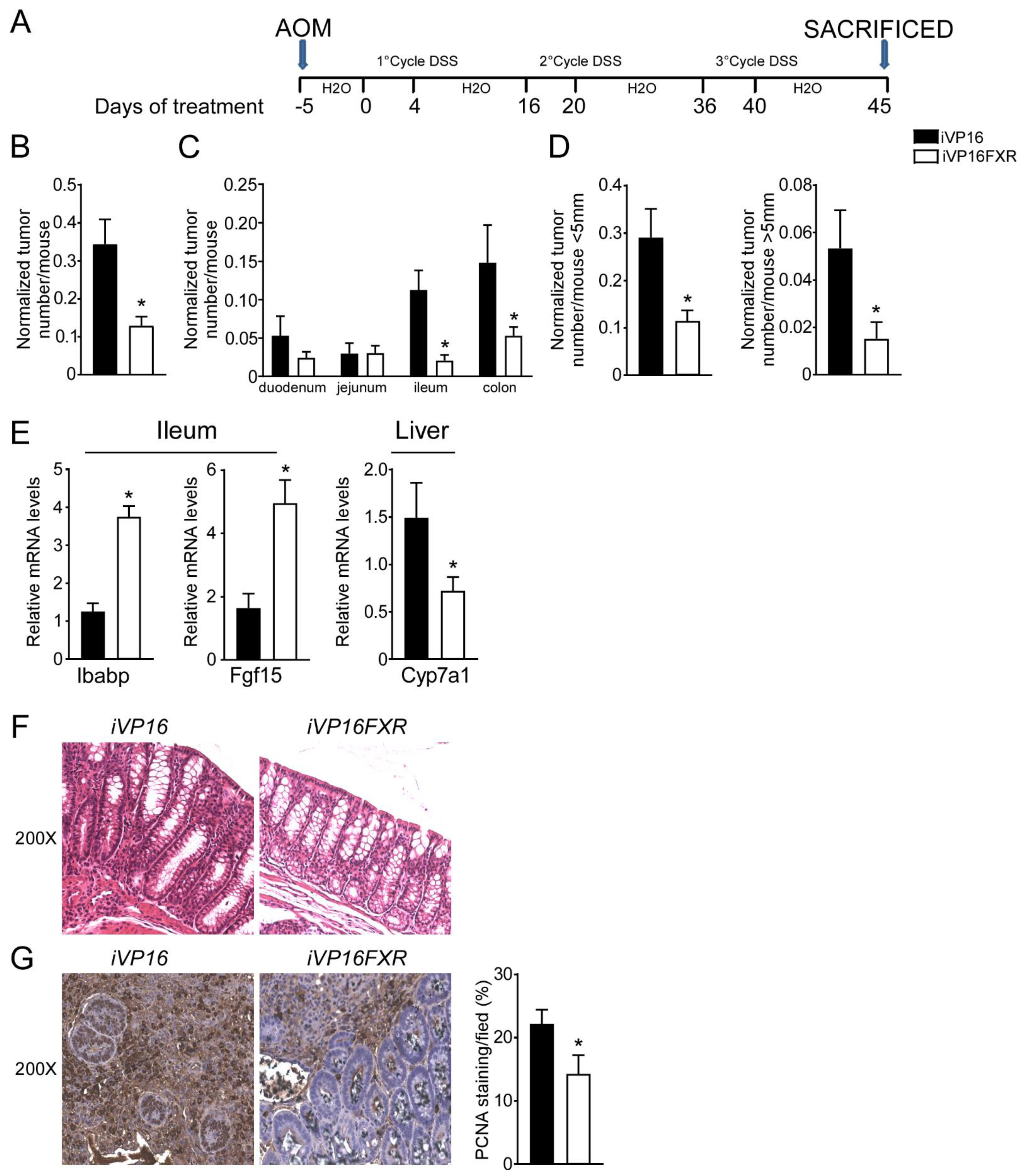
3.1. Intestinal Constitutive FXR Activation Protects from AOM/DSS-Induced Colorectal Cancer
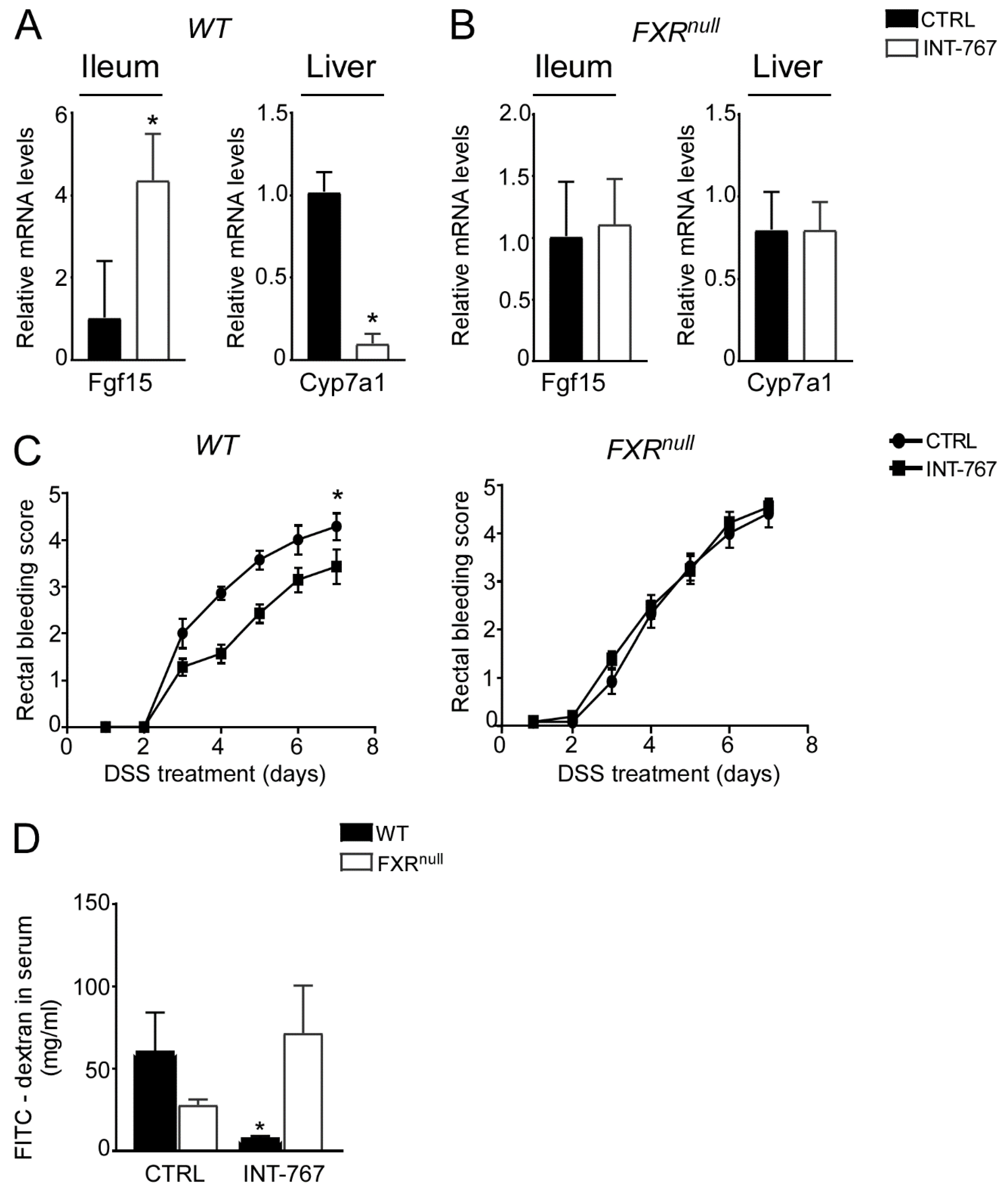
3.2. A Short-Term Administration of a Diet Enriched in INT-767 Activates FXR-Dependent Orchestration of BA Synthesis in the Gut-Liver Axis
3.3. INT-767 Protects from DSS-Induced Colitis via FXR Activation
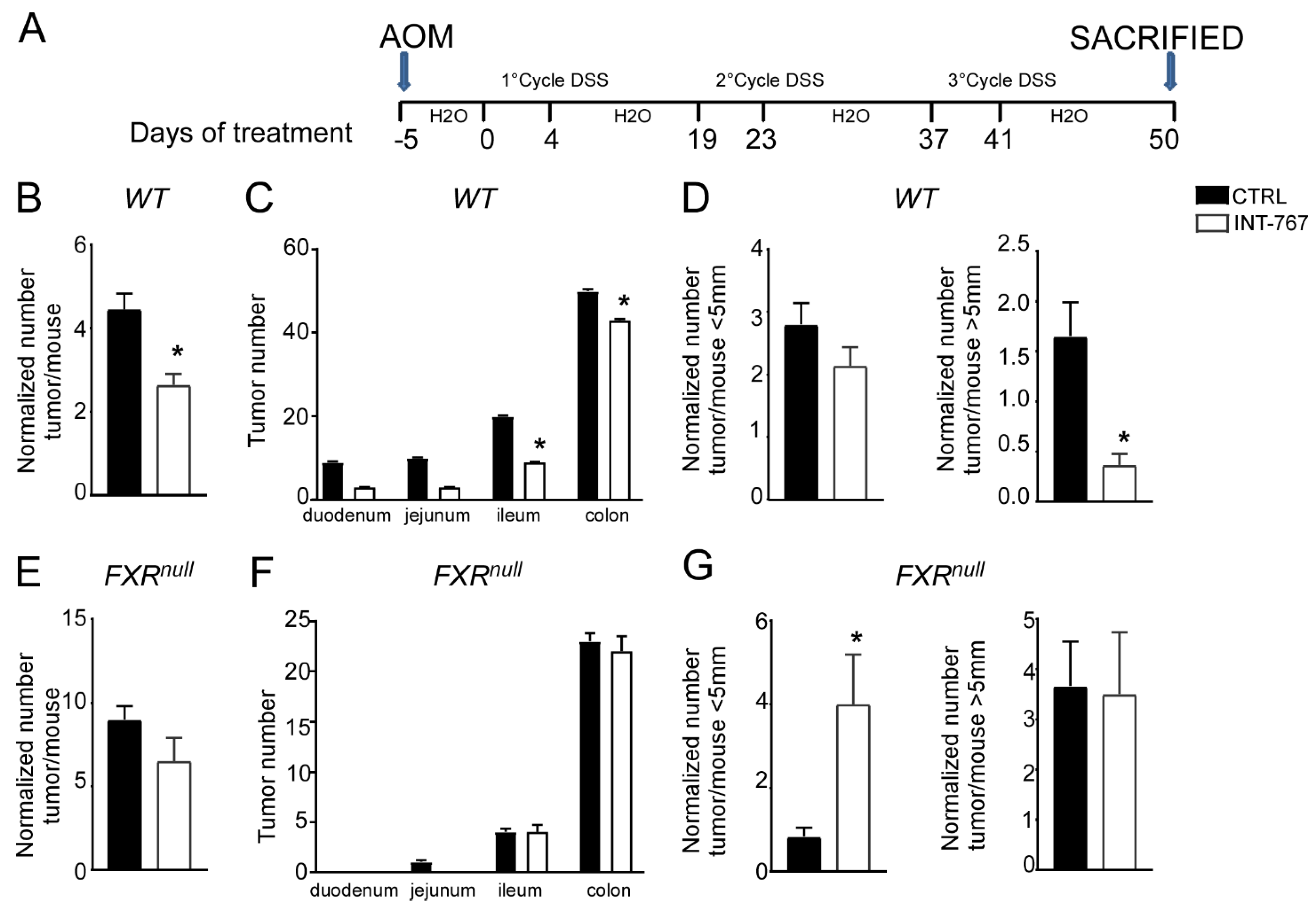
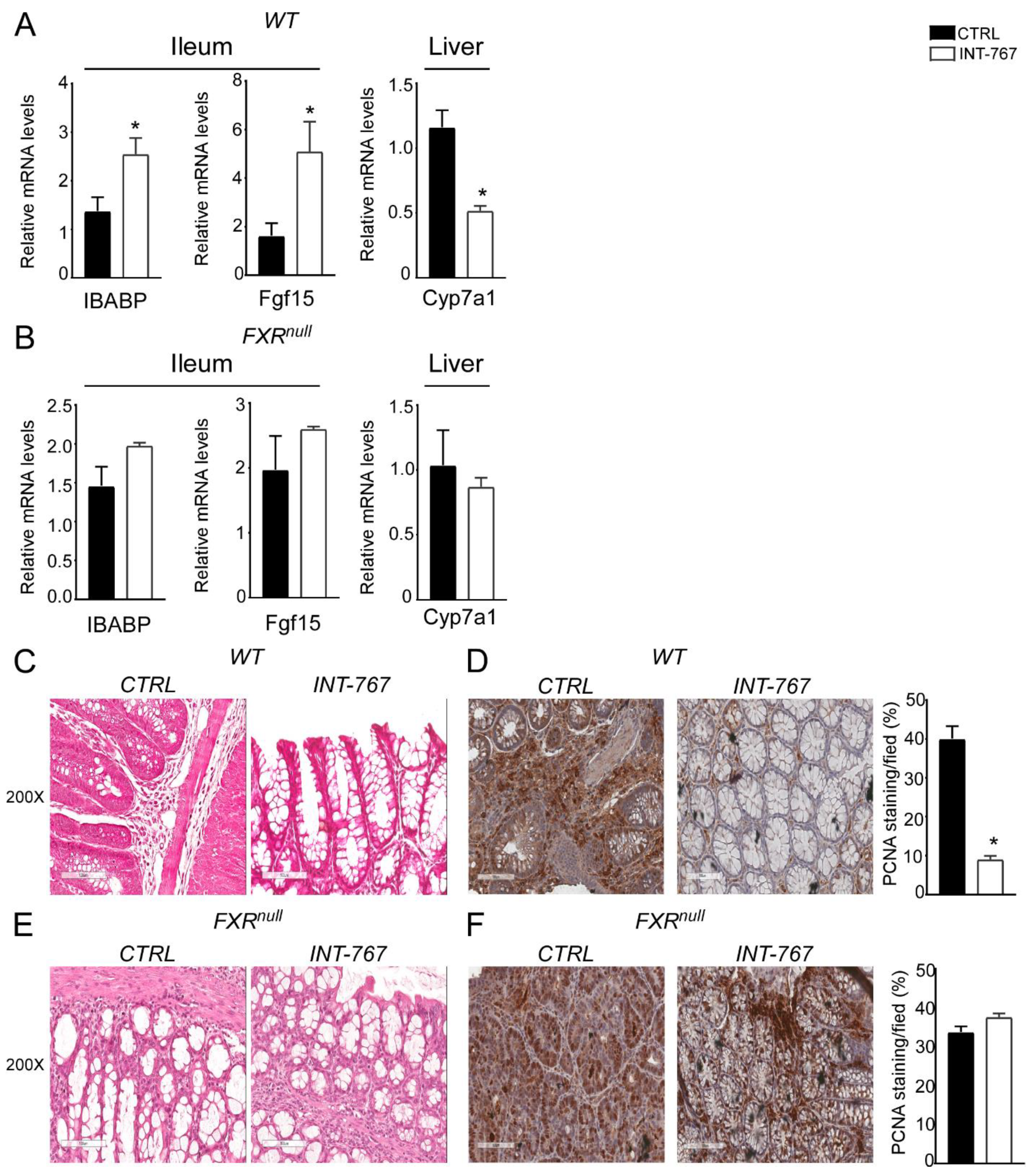
3.4. INT-767 Protects Wild-Type Mice from AOM/DSS-Induced Colorectal Cancer via FXR Activation
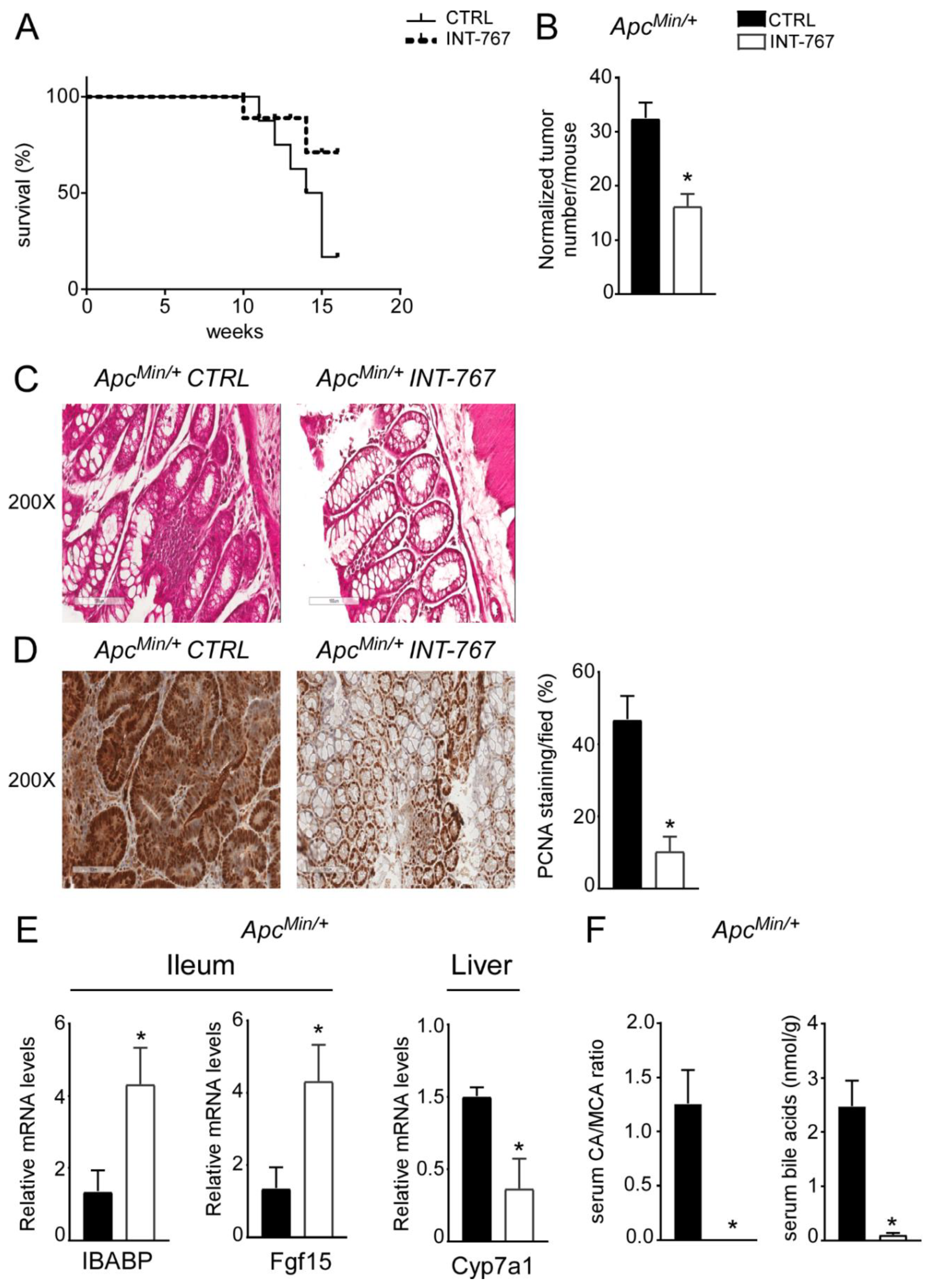
3.5. INT-767 Prevents Spontaneous Colorectal Carcinogenesis in Apcmin/+ Mice via Fxr-Dependent Orchestration of Ba Homeostasis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Rustgi, A.K. The genetics of hereditary colon cancer. Genes Dev. 2007, 21, 2525–2538. [Google Scholar] [CrossRef] [Green Version]
- Powell, S.M.; Zilz, N.; Beazer-Barclay, Y.; Bryan, T.M.; Hamilton, S.R.; Thibodeau, S.N.; Vogelstein, B.; Kinzler, K.W. APC mutations occur early during colorectal tumorigenesis. Nature 1992, 359, 235–237. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Su, L.K.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hedge, P.; McKechnie, D.; et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253, 661–665. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Nilbert, M.C.; Vogelstein, B.; Bryan, T.M.; Levy, D.B.; Smith, K.J.; Preisinger, A.C.; Hamilton, S.R.; Hedge, P.; Markham, A.; et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science 1991, 251, 1366–1370. [Google Scholar] [CrossRef] [Green Version]
- Khaderi, S.A.; Sussman, N.L. Screening for malignancy in primary sclerosing cholangitis (PSC). Curr. Gastroenterol. Rep. 2015, 17, 17. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal inflammation and cancer. Gastroenterology 2011, 140, 1807–1816. [Google Scholar] [CrossRef]
- Hill, M.J.; Melville, D.M.; Lennard-Jones, J.E.; Neale, K.; Ritchie, J.K. Faecal bile acids, dysplasia, and carcinoma in ulcerative colitis. Lancet 1987, 2, 185–186. [Google Scholar] [CrossRef]
- Bianchini, F.; Caderni, G.; Dolara, P.; Fantetti, L.; Kriebel, D. Effect of dietary fat, starch and cellulose on fecal bile acids in mice. J. Nutr. 1989, 119, 1617–1624. [Google Scholar] [CrossRef]
- Reddy, B.S.; Wynder, E.L. Metabolic epidemiology of colon cancer. Fecal bile acids and neutral sterols in colon cancer patients and patients with adenomatous polyps. Cancer 1977, 39, 2533–2539. [Google Scholar] [CrossRef]
- Caderni, G.; Bianchini, F.; Dolara, P.; Kriebel, D. Proliferative activity in the colon of the mouse and its modulation by dietary starch, fat, and cellulose. Cancer Res. 1989, 49, 1655–1659. [Google Scholar]
- Reddy, B.S. Diet and excretion of bile acids. Cancer Res. 1981, 41, 3766–3768. [Google Scholar]
- Reddy, B.S. Dietary fat and its relationship to large bowel cancer. Cancer Res. 1981, 41, 3700–3705. [Google Scholar]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Cariello, M.; Sabba, C.; Moschetta, A. Tissue-specific actions of FXR in metabolism and cancer. Biochim. Biophys. Acta 2015, 1851, 30–39. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Cariello, M.; Scialpi, N.; Peres, C.; Vetrano, S.; Fiorino, G.; Danese, S.; Ko, B.; Luo, J.; et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBiomedicine 2020, 54, 102719. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Degirolamo, C.; Modica, S.; Palasciano, G.; Moschetta, A. Bile acids and colon cancer: Solving the puzzle with nuclear receptors. Trends Mol. Med. 2011, 17, 564–572. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Moschetta, A. Bile acids and colon cancer: Is FXR the solution of the conundrum? Mol. Asp. Med. 2017, 56, 66–74. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, S.; Kim, Y.; Lee, Y.S.; Lee, S.; Lee, S.H.; Kweon, M.N. A High-Fat Diet Activates the BAs-FXR Axis and Triggers Cancer-Associated Fibroblast Properties in the Colon. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1141–1159. [Google Scholar] [CrossRef]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef] [Green Version]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Bajor, A.; Gillberg, P.G.; Abrahamsson, H. Bile acids: Short and long term effects in the intestine. Scand. J. Gastroenterol. 2010, 45, 645–664. [Google Scholar] [CrossRef]
- McGarr, S.E.; Ridlon, J.M.; Hylemon, P.B. Diet, anaerobic bacterial metabolism, and colon cancer: A review of the literature. J. Clin. Gastroenterol. 2005, 39, 98–109. [Google Scholar]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, T.; Shirakami, Y.; Mizutani, T.; Maruta, A.; Ideta, T.; Kubota, M.; Sakai, H.; Ibuka, T.; Genovese, S.; Fiorito, S.; et al. Novel FXR agonist nelumal A suppresses colitis and inflammation-related colorectal carcinogenesis. Sci. Rep. 2021, 11, 492. [Google Scholar] [CrossRef]
- De Gottardi, A.; Touri, F.; Maurer, C.A.; Perez, A.; Maurhofer, O.; Ventre, G.; Bentzen, C.L.; Niesor, E.J.; Dufour, J.F. The bile acid nuclear receptor FXR and the bile acid binding protein IBABP are differently expressed in colon cancer. Dig. Dis. Sci. 2004, 49, 982–989. [Google Scholar] [CrossRef]
- Maran, R.R.; Thomas, A.; Roth, M.; Sheng, Z.; Esterly, N.; Pinson, D.; Gao, X.; Zhang, Y.; Ganapathy, V.; Gonzalez, F.J.; et al. Farnesoid X receptor deficiency in mice leads to increased intestinal epithelial cell proliferation and tumor development. J. Pharm. Exp. 2009, 328, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Modica, S.; Murzilli, S.; Salvatore, L.; Schmidt, D.R.; Moschetta, A. Nuclear bile acid receptor FXR protects against intestinal tumorigenesis. Cancer Res. 2008, 68, 9589–9594. [Google Scholar] [CrossRef] [Green Version]
- Selmin, O.I.; Fang, C.; Lyon, A.M.; Doetschman, T.C.; Thompson, P.A.; Martinez, J.D.; Smith, J.W.; Lance, P.M.; Romagnolo, D.F. Inactivation of Adenomatous Polyposis Coli Reduces Bile Acid/Farnesoid X Receptor Expression through Fxr gene CpG Methylation in Mouse Colon Tumors and Human Colon Cancer Cells. J. Nutr. 2016, 146, 236–242. [Google Scholar] [CrossRef]
- Leclerc, D.; Deng, L.; Trasler, J.; Rozen, R. ApcMin/+ mouse model of colon cancer: Gene expression profiling in tumors. J. Cell Biochem. 2004, 93, 1242–1254. [Google Scholar] [CrossRef]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112.e1018. [Google Scholar] [CrossRef] [Green Version]
- Cariello, M.; Peres, C.; Zerlotin, R.; Porru, E.; Sabba, C.; Roda, A.; Moschetta, A. Long-term Administration of Nuclear Bile Acid Receptor FXR Agonist Prevents Spontaneous Hepatocarcinogenesis in Abcb4(-/-) Mice. Sci. Rep. 2017, 7, 11203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Di Tullio, G.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012, 142, 355–365.e4. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Ohkusa, T.; Kajiura, K.; Kanno, J.; Sakamoto, S. Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 1996, 39, 87–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgar, S.; Karlsson, L.; Rehnstrom, E.; Karlsson, A.; Utkovic, H.; Jansson, L.; Michaelsson, E. Validation of murine dextran sulfate sodium-induced colitis using four therapeutic agents for human inflammatory bowel disease. Int. Immunopharmacol. 2008, 8, 836–844. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, E.J.; Grady, W.M.; Lieberman, D.; Seufferlein, T.; Sung, J.J.; Boelens, P.G.; van de Velde, C.J.; Watanabe, T. Colorectal cancer. Nat. Rev. Dis. Primers 2015, 1, 15065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Font-Burgada, J.; Sun, B.; Karin, M. Obesity and Cancer: The Oil that Feeds the Flame. Cell Metab. 2016, 23, 48–62. [Google Scholar] [CrossRef] [Green Version]
- Gadaleta, R.M.; Scialpi, N.; Peres, C.; Cariello, M.; Ko, B.; Luo, J.; Porru, E.; Roda, A.; Sabba, C.; Moschetta, A. Suppression of Hepatic Bile Acid Synthesis by a non-tumorigenic FGF19 analogue Protects Mice from Fibrosis and Hepatocarcinogenesis. Sci. Rep. 2018, 8, 17210. [Google Scholar] [CrossRef]
- Kim, I.; Morimura, K.; Shah, Y.; Yang, Q.; Ward, J.M.; Gonzalez, F.J. Spontaneous hepatocarcinogenesis in farnesoid X receptor-null mice. Carcinogenesis 2007, 28, 940–946. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.; Huang, X.; Yi, T.; Yen, Y.; Moore, D.D.; Huang, W. Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res. 2007, 67, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Xu, Z.; Guo, J.; Zheng, J.; Sun, X.; Yu, J. Farnesoid X receptor activation induces antitumour activity in colorectal cancer by suppressing JAK2/STAT3 signalling via transactivation of SOCS3 gene. J. Cell Mol. Med. 2020, 24, 14549–14560. [Google Scholar] [CrossRef]
- Guo, J.; Zheng, J.; Mu, M.; Chen, Z.; Xu, Z.; Zhao, C.; Yang, K.; Qin, X.; Sun, X.; Yu, J. GW4064 enhances the chemosensitivity of colorectal cancer to oxaliplatin by inducing pyroptosis. Biochem. Biophys. Res. Commun. 2021, 548, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Mauad, T.H.; van Nieuwkerk, C.M.; Dingemans, K.P.; Smit, J.J.; Schinkel, A.H.; Notenboom, R.G.; van den Bergh Weerman, M.A.; Verkruisen, R.P.; Groen, A.K.; Oude Elferink, R.P.; et al. Mice with homozygous disruption of the mdr2 P-glycoprotein gene. A novel animal model for studies of nonsuppurative inflammatory cholangitis and hepatocarcinogenesis. Am. J. Pathol. 1994, 145, 1237–1245. [Google Scholar] [PubMed]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cariello, M.; Zerlotin, R.; Pasculli, E.; Piccinin, E.; Peres, C.; Porru, E.; Roda, A.; Gadaleta, R.M.; Moschetta, A. Intestinal FXR Activation via Transgenic Chimera or Chemical Agonism Prevents Colitis-Associated and Genetically-Induced Colon Cancer. Cancers 2022, 14, 3081. https://doi.org/10.3390/cancers14133081
Cariello M, Zerlotin R, Pasculli E, Piccinin E, Peres C, Porru E, Roda A, Gadaleta RM, Moschetta A. Intestinal FXR Activation via Transgenic Chimera or Chemical Agonism Prevents Colitis-Associated and Genetically-Induced Colon Cancer. Cancers. 2022; 14(13):3081. https://doi.org/10.3390/cancers14133081
Chicago/Turabian StyleCariello, Marica, Roberta Zerlotin, Emanuela Pasculli, Elena Piccinin, Claudia Peres, Emanuele Porru, Aldo Roda, Raffaella Maria Gadaleta, and Antonio Moschetta. 2022. "Intestinal FXR Activation via Transgenic Chimera or Chemical Agonism Prevents Colitis-Associated and Genetically-Induced Colon Cancer" Cancers 14, no. 13: 3081. https://doi.org/10.3390/cancers14133081
APA StyleCariello, M., Zerlotin, R., Pasculli, E., Piccinin, E., Peres, C., Porru, E., Roda, A., Gadaleta, R. M., & Moschetta, A. (2022). Intestinal FXR Activation via Transgenic Chimera or Chemical Agonism Prevents Colitis-Associated and Genetically-Induced Colon Cancer. Cancers, 14(13), 3081. https://doi.org/10.3390/cancers14133081