
Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment


Abstract
Simple Summary
Abstract
1. Introduction
2. Immune Cells
2.1. Immunotoxicity
2.2. Alteration of Immune Cell Infiltration and Polarization
2.2.1. Polarization of Th Cells
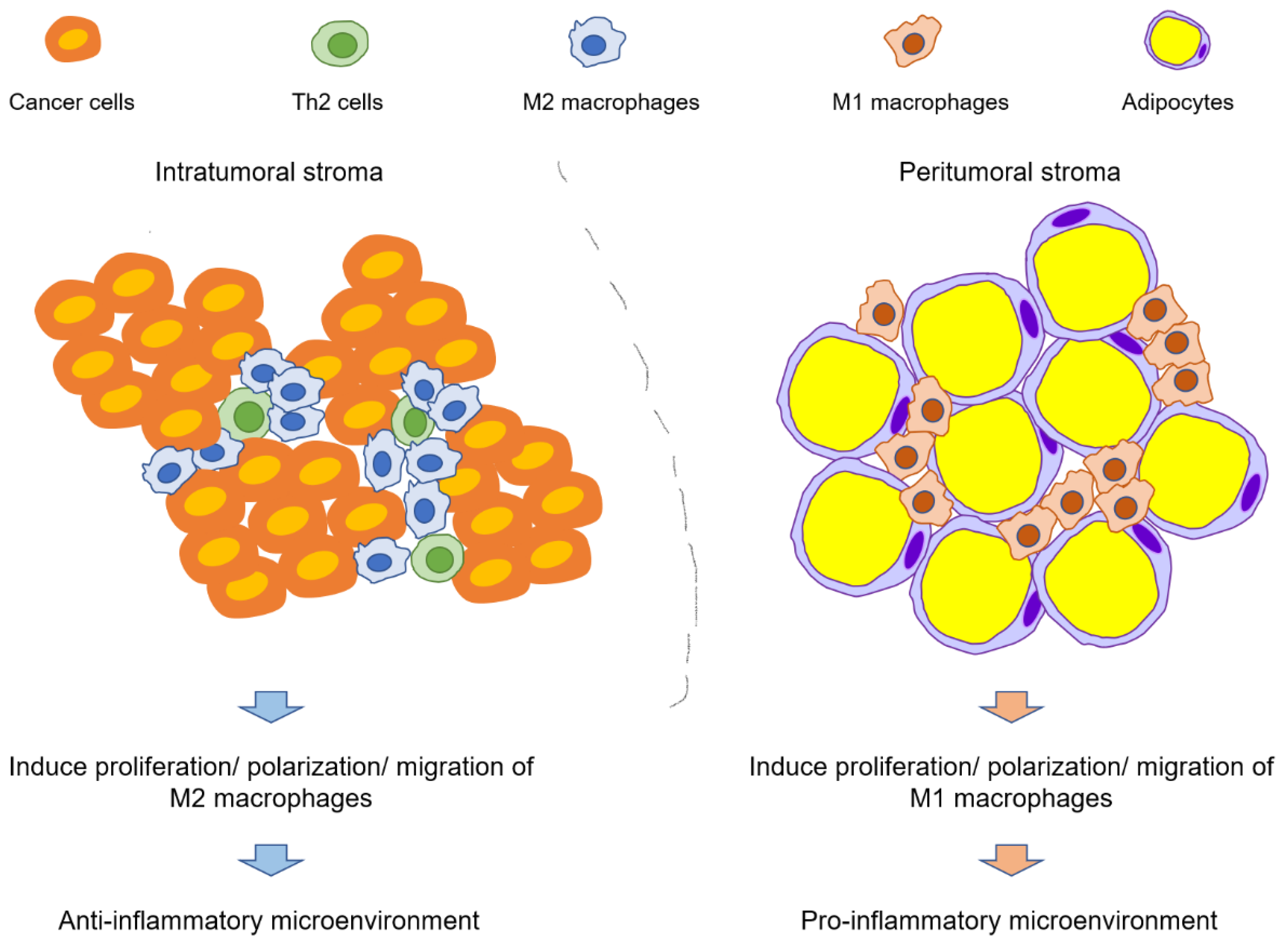
2.2.2. Infiltration and Polarization of Macrophages
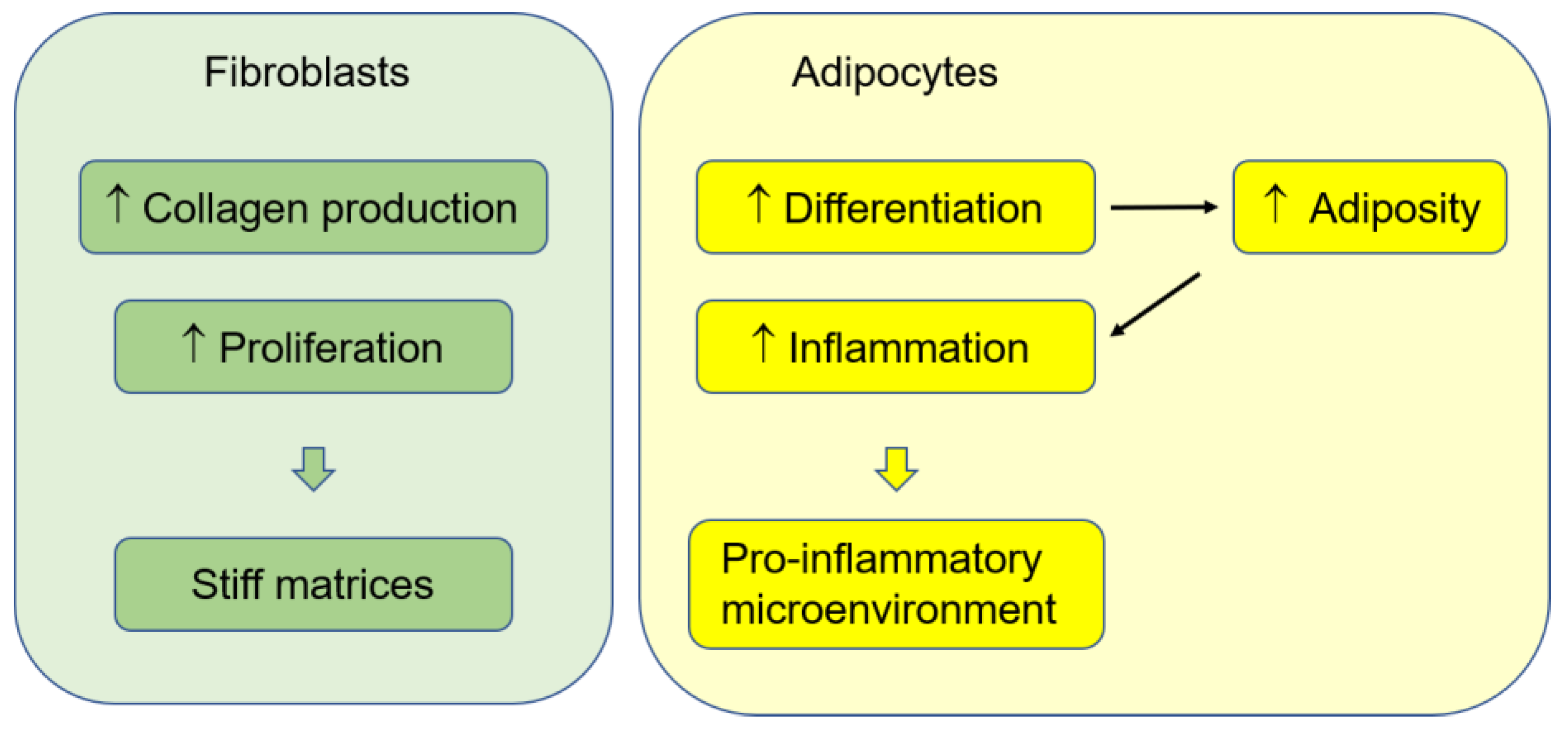
3. Fibroblasts and ECMs
4. Adipocytes
5. Conclusions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.Y.; Kim, Y.S.; Kim, Z.; Kim, H.Y.; Kim, H.J.; Park, S.; Bae, S.Y.; Yoon, K.H.; Lee, S.B.; Lee, S.K.; et al. Breast Cancer Statistics in Korea in 2017: Data from a Breast Cancer Registry. J. Breast Cancer 2020, 23, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.R.; Joubert, C.; Boniol, M.; Hery, C.; Ahn, S.H.; Won, Y.J.; Nishino, Y.; Sobue, T.; Chen, C.J.; You, S.L.; et al. Recent trends and patterns in breast cancer incidence among Eastern and Southeastern Asian women. Cancer Causes Control 2010, 21, 1777–1785. [Google Scholar] [CrossRef]
- Chia, K.S.; Reilly, M.; Tan, C.S.; Lee, J.; Pawitan, Y.; Adami, H.O.; Hall, P.; Mow, B. Profound changes in breast cancer incidence may reflect changes into a Westernized lifestyle: A comparative population-based study in Singapore and Sweden. Int. J. Cancer 2005, 113, 302–306. [Google Scholar] [CrossRef]
- Mousavi-Jarrrahi, S.H.; Kasaeian, A.; Mansori, K.; Ranjbaran, M.; Khodadost, M.; Mosavi-Jarrahi, A. Addressing the younger age at onset in breast cancer patients in Asia: An age-period-cohort analysis of fifty years of quality data from the international agency for research on cancer. ISRN Oncol. 2013, 2013, 429862. [Google Scholar] [CrossRef]
- Shen, Y.C.; Chang, C.J.; Hsu, C.; Cheng, C.C.; Chiu, C.F.; Cheng, A.L. Significant difference in the trends of female breast cancer incidence between Taiwanese and Caucasian Americans: Implications from age-period-cohort analysis. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 1986–1990. [Google Scholar] [CrossRef]
- Sung, H.N.; Rosenberg, P.S.; Chen, W.Q.; Hartman, M.; Lim, W.Y.; Chia, K.S.; Mang, O.W.K.; Chiang, C.J.; Kang, D.; Ngan, R.K.C.; et al. Female Breast Cancer Incidence Among Asian and Western Populations: More Similar Than Expected. J. Natl. Cancer Inst. 2015, 107, djv107. [Google Scholar] [CrossRef]
- Fan, L.; Goss, P.E.; Strasser-Weippl, K. Current Status and Future Projections of Breast Cancer in Asia. Breast Care 2015, 10, 372–378. [Google Scholar] [CrossRef]
- Lin, Z.K.; Wang, L.T.; Jia, Y.H.; Zhang, Y.F.; Dong, Q.X.; Huang, C.J. A Study on Environmental Bisphenol A Pollution in Plastics Industry Areas. Water Air Soil Pollut. 2017, 228, 98. [Google Scholar] [CrossRef]
- Abraham, A.; Chakraborty, P. A review on sources and health impacts of bisphenol A. Rev. Environ. Health 2020, 35, 201–210. [Google Scholar] [CrossRef]
- Vogel, S.A. The politics of plastics: The making and unmaking of bisphenol a “safety”. Am. J. Public Health 2009, 99 (Suppl. S3), S559–S566. [Google Scholar] [CrossRef] [PubMed]
- Almeida, S.; Raposo, A.; Almeida-Gonzalez, M.; Carrascosa, C. Bisphenol A: Food Exposure and Impact on Human Health. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1503–1517. [Google Scholar] [CrossRef] [PubMed]
- Banaderakhshan, R.; Kemp, P.; Breul, L.; Steinbichl, P.; Hartmann, C.; Furhacker, M. Bisphenol A and its alternatives in Austrian thermal paper receipts, and the migration from reusable plastic drinking bottles into water and artificial saliva using UHPLC-MS/MS. Chemosphere 2022, 286, 131842. [Google Scholar] [CrossRef] [PubMed]
- Healy, B.F.; English, K.R.; Jagals, P.; Sly, P.D. Bisphenol A exposure pathways in early childhood: Reviewing the need for improved risk assessment models. J. Expo. Sci. Environ. Epidemiol. 2015, 25, 544–556. [Google Scholar] [CrossRef]
- Rogers, L.D. What Does CLARITY-BPA Mean for Canadians? Int. J. Environ. Res. Public Health 2021, 18, 7001. [Google Scholar] [CrossRef]
- Lorber, M.; Schecter, A.; Paepke, O.; Shropshire, W.; Christensen, K.; Birnbaum, L. Exposure assessment of adult intake of bisphenol A (BPA) with emphasis on canned food dietary exposures. Environ. Int. 2015, 77, 55–62. [Google Scholar] [CrossRef]
- Fernandez, M.F.; Arrebola, J.P.; Taoufiki, J.; Navalon, A.; Ballesteros, O.; Pulgar, R.; Vilchez, J.L.; Olea, N. Bisphenol-A and chlorinated derivatives in adipose tissue of women. Reprod. Toxicol. 2007, 24, 259–264. [Google Scholar] [CrossRef]
- Ho, V.; Pelland-St-Pierre, L.; Gravel, S.; Bouchard, M.F.; Verner, M.A.; Labreche, F. Endocrine disruptors: Challenges and future directions in epidemiologic research. Environ. Res. 2022, 204, 111969. [Google Scholar] [CrossRef]
- Brophy, J.T.; Keith, M.M.; Watterson, A.; Park, R.; Gilbertson, M.; Maticka-Tyndale, E.; Beck, M.; Abu-Zahra, H.; Schneider, K.; Reinhartz, A.; et al. Breast cancer risk in relation to occupations with exposure to carcinogens and endocrine disruptors: A Canadian case-control study. Environ. Health 2012, 11, 87. [Google Scholar] [CrossRef]
- Keshavarz-Maleki, R.; Kaviani, A.; Omranipour, R.; Gholami, M.; Khoshayand, M.R.; Ostad, S.N.; Sabzevari, O. Bisphenol-A in biological samples of breast cancer mastectomy and mammoplasty patients and correlation with levels measured in urine and tissue. Sci. Rep. 2021, 11, 18411. [Google Scholar] [CrossRef]
- Weinhouse, C.; Anderson, O.S.; Bergin, I.L.; Vandenbergh, D.J.; Gyekis, J.P.; Dingman, M.A.; Yang, J.; Dolinoy, D.C. Dose-dependent incidence of hepatic tumors in adult mice following perinatal exposure to bisphenol A. Environ. Health Perspect. 2014, 122, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Liu, N.; Weng, S.F.; Wang, H.S. Bisphenol a Increases the Migration and Invasion of Triple-Negative Breast Cancer Cells via Oestrogen-related Receptor Gamma. Basic Clin. Pharmacol. Toxicol. 2016, 119, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Delfosse, V.; Grimaldi, M.; le Maire, A.; Bourguet, W.; Balaguer, P. Nuclear receptor profiling of bisphenol-A and its halogenated analogues. Vitam. Horm. 2014, 94, 229–251. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.S.; Wang, L.; Yang, N.F.; Wen, J.J.; Zhao, M.S.; Su, G.Y.; Zhang, J.F.; Weng, D. Peroxisome proliferator-activated receptor gamma (PPAR gamma) activation and metabolism disturbance induced by bisphenol A and its replacement analog bisphenol S using in vitro macrophages and in vivo mouse models. Environ. Int. 2020, 134, 105328. [Google Scholar] [CrossRef]
- Takayanagi, S.; Tokunaga, T.; Liu, X.; Okada, H.; Matsushima, A.; Shimohigashi, Y. Endocrine disruptor bisphenol A strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol. Lett. 2006, 167, 95–105. [Google Scholar] [CrossRef]
- Pupo, M.; Pisano, A.; Lappano, R.; Santolla, M.F.; De Francesco, E.M.; Abonante, S.; Rosano, C.; Maggiolini, M. Bisphenol A Induces Gene Expression Changes and Proliferative Effects through GPER in Breast Cancer Cells and Cancer-Associated Fibroblasts. Environ. Health Persp. 2012, 120, 1177–1182. [Google Scholar] [CrossRef]
- Sanchez, R.C.; Gomez, R.; Salazar, E.P. Bisphenol A Induces Migration through a GPER-, FAK-, Src-, and ERK2-Dependent Pathway in MDA-MB-231 Breast Cancer Cells. Chem. Res. Toxicol. 2016, 29, 285–295. [Google Scholar] [CrossRef]
- Pupo, M.; Maggiolini, M.; Musti, A.M. GPER Mediates Non-Genomic Effects of Estrogen. Methods. Mol. Biol. 2016, 1366, 471–488. [Google Scholar] [CrossRef]
- Jenkins, S.; Wang, J.; Eltoum, I.; Desmond, R.; Lamartiniere, C.A. Chronic oral exposure to bisphenol A results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ. Health Perspect. 2011, 119, 1604–1609. [Google Scholar] [CrossRef]
- Dumitrascu, M.C.; Mares, C.; Petca, R.C.; Sandru, F.; Popescu, R.I.; Mehedintu, C.; Petca, A. Carcinogenic effects of bisphenol A in breast and ovarian cancers. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Kwon, Y.; Godwin, A.K. Regulation of HGF and c-MET Interaction in Normal Ovary and Ovarian Cancer. Reprod. Sci. 2017, 24, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Pathak, H.B.; Godwin, A.K.; Kwon, Y. Epithelial-stromal communication via CXCL1-CXCR2 interaction stimulates growth of ovarian cancer cells through p38 activation. Cell. Oncol. 2021, 44, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Salemme, V.; Centonze, G.; Cavallo, F.; Defilippi, P.; Conti, L. The Crosstalk Between Tumor Cells and the Immune Microenvironment in Breast Cancer: Implications for Immunotherapy. Front. Oncol. 2021, 11, 610303. [Google Scholar] [CrossRef]
- De Marco, P.; Lappano, R.; De Francesco, E.M.; Cirillo, F.; Pupo, M.; Avino, S.; Vivacqua, A.; Abonante, S.; Picard, D.; Maggiolini, M. GPER signalling in both cancer-associated fibroblasts and breast cancer cells mediates a feedforward IL1 beta/IL1R1 response. Sci. Rep. 2016, 6, 1–14. [Google Scholar] [CrossRef]
- Pepermans, R.A.; Sharma, G.; Prossnitz, E.R. G Protein-Coupled Estrogen Receptor in Cancer and Stromal Cells: Functions and Novel Therapeutic Perspectives. Cells 2021, 10, 672. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, X.; Saredy, J.; Yuan, Z.; Yang, X.; Wang, H. Innate-adaptive immunity interplay and redox regulation in immune response. Redox. Biol. 2020, 37, 101759. [Google Scholar] [CrossRef]
- Rabb, H. The T cell as a bridge between innate and adaptive immune systems: Implications for the kidney. Kidney Int. 2002, 61, 1935–1946. [Google Scholar] [CrossRef]
- Lund, F.E.; Randall, T.D. Effector and regulatory B cells: Modulators of CD4+ T cell immunity. Nat. Rev. Immunol. 2010, 10, 236–247. [Google Scholar] [CrossRef]
- Ye, Y.; Xu, C.; Chen, F.; Liu, Q.; Cheng, N. Targeting Innate Immunity in Breast Cancer Therapy: A Narrative Review. Front. Immunol. 2021, 12, 771201. [Google Scholar] [CrossRef]
- Gaudino, S.J.; Kumar, P. Cross-Talk Between Antigen Presenting Cells and T Cells Impacts Intestinal Homeostasis, Bacterial Infections, and Tumorigenesis. Front. Immunol. 2019, 10, 360. [Google Scholar] [CrossRef]
- Thompson, P.A.; Khatami, M.; Baglole, C.J.; Sun, J.; Harris, S.A.; Moon, E.Y.; Al-Mulla, F.; Al-Temaimi, R.; Brown, D.G.; Colacci, A.; et al. Environmental immune disruptors, inflammation and cancer risk. Carcinogenesis 2015, 36 (Suppl. S1), S232–S253. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.; Ahmed, S.A. The immune System Is a natural Target for Estrogen Action: Opposing Effects of Estrogen in Two Prototypical Autoimmune Diseases. Front. Immunol. 2016, 6, 635. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, J.; Qian, C.; Lou, J.; Lin, J.; Xu, A.; Xia, K.; Jin, L.; Liu, B.; Tao, H.; et al. Estrogen/ER in anti-tumor immunity regulation to tumor cell and tumor microenvironment. Cancer Cell Int. 2021, 21, 295. [Google Scholar] [CrossRef] [PubMed]
- Notas, G.; Kampa, M.; Castanas, E. G Protein-Coupled Estrogen Receptor in Immune Cells and Its Role in Immune-Related Diseases. Front. Endocrinol. 2020, 11, 579420. [Google Scholar] [CrossRef]
- Xu, J.; Huang, G.; Guo, T.L. Developmental Bisphenol A Exposure Modulates Immune-Related Diseases. Toxics 2016, 4, 23. [Google Scholar] [CrossRef]
- Mayer, A.; Zhang, Y.; Perelson, A.S.; Wingreen, N.S. Regulation of T cell expansion by antigen presentation dynamics. Proc. Natl. Acad. Sci. USA 2019, 116, 5914–5919. [Google Scholar] [CrossRef]
- Kaszubowska, L. Telomere shortening and ageing of the immune system. J. Physiol. Pharmacol. 2008, 59 (Suppl. S9), 169–186. [Google Scholar]
- Wagner, C.L.; Hanumanthu, V.S.; Talbot, C.C., Jr.; Abraham, R.S.; Hamm, D.; Gable, D.L.; Kanakry, C.G.; Applegate, C.D.; Siliciano, J.; Jackson, J.B.; et al. Short telomere syndromes cause a primary T cell immunodeficiency. J. Clin. Invest. 2018, 128, 5222–5234. [Google Scholar] [CrossRef]
- Dosset, M.; Castro, A.; Carter, H.; Zanetti, M. Telomerase and CD4 T Cell Immunity in Cancer. Cancers 2020, 12, 1687. [Google Scholar] [CrossRef]
- Qian, Y.; Ding, T.; Wei, L.; Cao, S.; Yang, L. Shorter telomere length of T-cells in peripheral blood of patients with lung cancer. Onco Targets Ther. 2016, 9, 2675–2682. [Google Scholar] [CrossRef][Green Version]
- Allsopp, R.C.; Morin, G.B.; DePinho, R.; Harley, C.B.; Weissman, I.L. Telomerase is required to slow telomere shortening and extend replicative lifespan of HSCs during serial transplantation. Blood 2003, 102, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Awada, Z.; Sleiman, F.; Mailhac, A.; Mouneimne, Y.; Tamim, H.; Zgheib, N.K. BPA exposure is associated with non-monotonic alteration in ESR1 promoter methylation in peripheral blood of men and shorter relative telomere length in peripheral blood of women. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Gostner, J.M.; Raggl, E.; Becker, K.; Uberall, F.; Schennach, H.; Pease, J.E.; Fuchs, D. Bisphenol A suppresses Th1-type immune response in human peripheral blood mononuclear cells in vitro. Immunol. Lett. 2015, 168, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Herz, C.; Tran, H.T.T.; Schlotz, N.; Michels, K.; Lamy, E. Low-dose levels of bisphenol A inhibit telomerase via ER/GPR30-ERK signalling, impair DNA integrity and reduce cell proliferation in primary PBMC. Sci. Rep. 2017, 7, 16631. [Google Scholar] [CrossRef]
- Tran, H.T.T.; Herz, C.; Lamy, E. Long-term exposure to “low-dose” bisphenol A decreases mitochondrial DNA copy number, and accelerates telomere shortening in human CD8 + T cells. Sci. Rep. 2020, 10, 15786. [Google Scholar] [CrossRef]
- Li, J.; Bach, A.; Crawford, R.B.; Phadnis-Moghe, A.S.; Chen, W.; D’Ingillo, S.; Kovalova, N.; Suarez-Martinez, J.E.; Zhou, J.; Kaplan, B.L.F.; et al. CLARITY-BPA: Effects of chronic bisphenol A exposure on the immune system: Part 2 - Characterization of lymphoproliferative and immune effector responses by splenic leukocytes. Toxicology 2018, 396–397, 54–67. [Google Scholar] [CrossRef]
- Lawlor, N.; Nehar-Belaid, D.; Grassmann, J.D.S.; Stoeckius, M.; Smibert, P.; Stitzel, M.L.; Pascual, V.; Banchereau, J.; Williams, A.; Ucar, D. Single Cell Analysis of Blood Mononuclear Cells Stimulated Through Either LPS or Anti-CD3 and Anti-CD28. Front. Immunol. 2021, 12, 636720. [Google Scholar] [CrossRef]
- Sugita-Konishi, Y.; Shimura, S.; Nishikawa, T.; Sunaga, F.; Naito, H.; Suzuki, Y. Effect of Bisphenol A on non-specific immunodefenses against non-pathogenic Escherichia coli. Toxicol. Lett. 2003, 136, 217–227. [Google Scholar] [CrossRef]
- Sawai, C.; Anderson, K.; Walser-Kuntz, D. Effect of bisphenol A on murine immune function: Modulation of interferon-gamma, IgG2a, and disease symptoms in NZB X NZW F1 mice. Environ. Health Perspect. 2003, 111, 1883–1887. [Google Scholar] [CrossRef]
- Koike, E.; Yanagisawa, R.; Win-Shwe, T.T.; Takano, H. Exposure to low-dose bisphenol A during the juvenile period of development disrupts the immune system and aggravates allergic airway inflammation in mice. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418774897. [Google Scholar] [CrossRef]
- Yanagisawa, R.; Koike, E.; Win-Shwe, T.T.; Takano, H. Oral exposure to low dose bisphenol A aggravates allergic airway inflammation in mice. Toxicol. Rep. 2019, 6, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Takamoto, M.; Sugane, K. Bisphenol A promotes IL-4 production by Th2 cells. Int. Arch. Allergy Immunol. 2003, 132, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, T.; Uemura, Y.; Jiao, S.; Wang, D.; Lin, Z.; Narita, Y.; Suzuki, M.; Hirosawa, N.; Ichihara, Y.; et al. Bisphenol A in combination with TNF-alpha selectively induces Th2 cell-promoting dendritic cells in vitro with an estrogen-like activity. Cell. Mol. Immunol. 2010, 7, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Arreola, M.I.; Nava-Castro, K.E.; Rio-Araiza, V.H.D.; Perez-Sanchez, N.Y.; Morales-Montor, J. A single neonatal administration of Bisphenol A induces higher tumour weight associated to changes in tumour microenvironment in the adulthood. Sci. Rep. 2017, 7, 10573. [Google Scholar] [CrossRef]
- Lee, H.W.; Ha, S.K.; Kim, Y. Bisphenol A disrupts inflammatory responses via Nod-like receptor protein 3 pathway in macrophages. Appl. Biol. Chem. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Kim, J.; Jeong, H. Suppression of inducible nitric oxide synthase and tumor necrosis factor-A expression by bisphenol a via nuclear factor-KB inactivation in macrophages. Toxicol. Sci. 2003, 72, 153–154. [Google Scholar]
- Byun, J.A.; Heo, Y.; Kim, Y.O.; Pyo, M.Y. Bisphenol A-induced downregulation of murine macrophage activities in vitro and ex vivo. Environ. Toxicol. Pharmacol. 2005, 19, 19–24. [Google Scholar] [CrossRef]
- Lu, X.; Li, M.; Wu, C.; Zhou, C.; Zhang, J.; Zhu, Q.; Shen, T. Bisphenol A promotes macrophage proinflammatory subtype polarization via upregulation of IRF5 expression in vitro. Toxicol. Vitr. 2019, 60, 97–106. [Google Scholar] [CrossRef]
- Liu, Y.; Mei, C.; Liu, H.; Wang, H.; Zeng, G.; Lin, J.; Xu, M. Modulation of cytokine expression in human macrophages by endocrine-disrupting chemical Bisphenol-A. Biochem. Biophys. Res. Commun. 2014, 451, 592–598. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.S.; Piao, Y.J.; Moon, W.K. Bisphenol A Promotes the Invasive and Metastatic Potential of Ductal Carcinoma In Situ and Protumorigenic Polarization of Macrophages. Toxicol. Sci. 2019, 170, 283–295. [Google Scholar] [CrossRef]
- Teixeira, D.; Marques, C.; Pestana, D.; Faria, A.; Norberto, S.; Calhau, C.; Monteiro, R. Effects of xenoestrogens in human M1 and M2 macrophage migration, cytokine release, and estrogen-related signaling pathways. Environ. Toxicol. 2016, 31, 1496–1509. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak-Wrona, W.; Rusak, M.; Nowak, K.; Dabrowska, M.; Radziwon, P.; Jablonska, E. Effect of bisphenol A on human neutrophils immunophenotype. Sci. Rep. 2020, 10, 3083. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jonsson, F. Expression, Role, and Regulation of Neutrophil Fcgamma Receptors. Front. Immunol. 2019, 10, 1958. [Google Scholar] [CrossRef] [PubMed]
- Bense, R.D.; Sotiriou, C.; Piccart-Gebhart, M.J.; Haanen, J.B.A.G.; van Vugt, M.A.T.M.; de Vries, E.G.E.; Schroder, C.P.; Fehrmann, R.S.N. Relevance of Tumor-Infiltrating Immune Cell Composition and Functionality for Disease Outcome in Breast Cancer. J. Natl. Cancer Inst. 2017, 109, djv107. [Google Scholar] [CrossRef] [PubMed]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Brit. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Zhang, S.C.; Hu, Z.Q.; Long, J.H.; Zhu, G.M.; Wang, Y.; Jia, Y.; Zhou, J.; Ouyang, Y.; Zeng, Z. Clinical Implications of Tumor-Infiltrating Immune Cells in Breast Cancer. J. Cancer 2019, 10, 6175–6184. [Google Scholar] [CrossRef]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef]
- Almatroodi, S.A.; McDonald, C.F.; Darby, I.A.; Pouniotis, D.S. Characterization of M1/M2 Tumour-Associated Macrophages (TAMs) and Th1/Th2 Cytokine Profiles in Patients with NSCLC. Cancer Microenviron. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- Liu, J.Y.; Geng, X.F.; Hou, J.X.; Wu, G.S. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 1–7. [Google Scholar] [CrossRef]
- Spellberg, B.; Edwards, J.E., Jr. Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 2001, 32, 76–102. [Google Scholar] [CrossRef]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: A mechanism of tumor evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.L.; Pedroza-Gonzalez, A.; Wu, T.C.; Aspord, C.; Shafer, S.; Marches, F.; Gallegos, M.; Burton, E.; Hori, T.; Tanaka, Y.; et al. Promotion of breast cancer development via Th2 polarization by TSLP. J. Immunol. 2011, 186, 165. [Google Scholar]
- Zhang, Q.; Qin, J.; Zhong, L.; Gong, L.; Zhang, B.; Zhang, Y.; Gao, W.Q. CCL5-Mediated Th2 Immune Polarization Promotes Metastasis in Luminal Breast Cancer. Cancer Res. 2015, 75, 4312–4321. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pan, Y.; Gou, P.; Zhou, C.; Ma, L.; Liu, Q.; Du, Y.; Yang, J.; Wang, Q. Effect of xanthohumol on Th1/Th2 balance in a breast cancer mouse model. Oncol. Rep. 2018, 39, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Valeta-Magara, A.; Gadi, A.; Volta, V.; Walters, B.; Arju, R.; Giashuddin, S.; Zhong, H.; Schneider, R.J. Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells through a Complex Chemokine Network. Cancer Res. 2019, 79, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Tiainen, S.; Tumelius, R.; Rilla, K.; Hamalainen, K.; Tammi, M.; Tammi, R.; Kosma, V.M.; Oikari, S.; Auvinen, P. High numbers of macrophages, especially M2-like (CD163-positive), correlate with hyaluronan accumulation and poor outcome in breast cancer. Histopathology 2015, 66, 873–883. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limon, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef]
- Nonaka, K.; Saio, M.; Umemura, N.; Kikuchi, A.; Takahashi, T.; Osada, S.; Yoshida, K. Th1 polarization in the tumor microenvironment upregulates the myeloid-derived suppressor-like function of macrophages. Cell. Immunol. 2021, 369, 104437. [Google Scholar] [CrossRef]
- Dong, C.; Flavell, R.A. Cell fate decision: T-helper 1 and 2 subsets in immune responses. Arthritis. Res. 2000, 2, 179–188. [Google Scholar] [CrossRef]
- Johansson, M.; Denardo, D.G.; Coussens, L.M. Polarized immune responses differentially regulate cancer development. Immunol. Rev. 2008, 222, 145–154. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Arreola, M.I.; Moreno-Mendoza, N.A.; Nava-Castro, K.E.; Segovia-Mendoza, M.; Perez-Torres, A.; Garay-Canales, C.A.; Morales-Montor, J. The Endocrine Disruptor Compound Bisphenol-A (BPA) Regulates the Intra-Tumoral Immune Microenvironment and Increases Lung Metastasis in an Experimental Model of Breast Cancer. Int. J. Mol. Sci. 2022, 23, 2523. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Lanzavecchia, A. The instructive role of dendritic cells on T-cell responses. Arthritis Res. 2002, 4 (Suppl. S3), S127–S132. [Google Scholar] [CrossRef]
- Qi, H.; Denning, T.L.; Soong, L. Differential induction of interleukin-10 and interleukin-12 in dendritic cells by microbial toll-like receptor activators and skewing of T-cell cytokine profiles. Infect. Immun. 2003, 71, 3337–3342. [Google Scholar] [CrossRef] [PubMed]
- Schoedon, G.; Troppmair, J.; Adolf, G.; Huber, C.; Niederwieser, A. Interferon-gamma enhances biosynthesis of pterins in peripheral blood mononuclear cells by induction of GTP-cyclohydrolase I activity. J. Interferon. Res. 1986, 6, 697–703. [Google Scholar] [CrossRef]
- Zhai, L.; Spranger, S.; Binder, D.C.; Gritsina, G.; Lauing, K.L.; Giles, F.J.; Wainwright, D.A. Molecular Pathways: Targeting IDO1 and Other Tryptophan Dioxygenases for Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 5427–5433. [Google Scholar] [CrossRef]
- Scribner, K.C.; Behbod, F.; Porter, W.W. Regulation of DCIS to invasive breast cancer progression by Singleminded-2s (SIM2s). Oncogene 2013, 32, 2631–2639. [Google Scholar] [CrossRef]
- Vogel, D.Y.S.; Heijnen, P.D.A.M.; Breur, M.; de Vries, H.E.; Tool, A.T.J.; Amor, S.; Dijkstra, C.D. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J. Neuroinflamm. 2014, 11, 23. [Google Scholar] [CrossRef]
- Karunarathne, W.; Molagoda, I.M.N.; Choi, Y.H.; Park, S.R.; Lee, S.; Kim, G.Y. Bisphenol A: A potential Toll-like receptor 4/myeloid differentiation factor 2 complex agonist. Environ. Pollut. 2021, 278, 116829. [Google Scholar] [CrossRef]
- Chavez-Galan, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR+ macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Poczobutt, J.M.; De, S.; Yadav, V.K.; Nguyen, T.T.; Li, H.; Sippel, T.R.; Weiser-Evans, M.C.; Nemenoff, R.A. Expression Profiling of Macrophages Reveals Multiple Populations with Distinct Biological Roles in an Immunocompetent Orthotopic Model of Lung Cancer. J. Immunol. 2016, 196, 2847–2859. [Google Scholar] [CrossRef] [PubMed]
- Gregory, K.J.; Morin, S.M.; Kubosiak, A.; Ser-Dolansky, J.; Schalet, B.J.; Jerry, D.J.; Schneider, S.S. The use of patient-derived breast tissue explants to study macrophage polarization and the effects of environmental chemical exposure. Immunol. Cell Biol. 2020, 98, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Boyd, N.F.; Dite, G.S.; Stone, J.; Gunasekara, A.; English, D.R.; McCredie, M.R.; Giles, G.G.; Tritchler, D.; Chiarelli, A.; Yaffe, M.J.; et al. Heritability of mammographic density, a risk factor for breast cancer. N. Engl. J. Med. 2002, 347, 886–894. [Google Scholar] [CrossRef]
- Li, T.; Sun, L.; Miller, N.; Nicklee, T.; Woo, J.; Hulse-Smith, L.; Tsao, M.S.; Khokha, R.; Martin, L.; Boyd, N. The association of measured breast tissue characteristics with mammographic density and other risk factors for breast cancer. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 343–349. [Google Scholar] [CrossRef]
- Ursin, G.; Hovanessian-Larsen, L.; Parisky, Y.R.; Pike, M.C.; Wu, A.H. Greatly increased occurrence of breast cancers in areas of mammographically dense tissue. Breast Cancer Res. 2005, 7, R605–R608. [Google Scholar] [CrossRef]
- Northey, J.J.; Barrett, A.S.; Acerbi, I.; Hayward, M.K.; Talamantes, S.; Dean, I.S.; Mouw, J.K.; Ponik, S.M.; Lakins, J.N.; Huang, P.J.; et al. Stiff stroma increases breast cancer risk by inducing the oncogene ZNF217. J. Clin. Investig. 2020, 130, 5721–5737. [Google Scholar] [CrossRef]
- Kendall, R.T.; Feghali-Bostwick, C.A. Fibroblasts in fibrosis: Novel roles and mediators. Front. Pharmacol. 2014, 5, 123. [Google Scholar] [CrossRef]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Houthuijzen, J.M.; Jonkers, J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metast. Rev. 2018, 37, 577–597. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y. Possible Beneficial Effects of N-Acetylcysteine for Treatment of Triple-Negative Breast Cancer. Antioxidants 2021, 10, 169. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rigiracciolo, D.C.; Belfiore, A.; Maggiolini, M.; De Francesco, E.M. Cancer associated fibroblasts: Role in breast cancer and potential as therapeutic targets. Expert. Opin. Ther. Tar. 2020, 24, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Sprague, B.L.; Trentham-Dietz, A.; Hedman, C.J.; Wang, J.; Hemming, J.D.; Hampton, J.M.; Buist, D.S.; Aiello Bowles, E.J.; Sisney, G.S.; Burnside, E.S. Circulating serum xenoestrogens and mammographic breast density. Breast Cancer Res. 2013, 15, R45. [Google Scholar] [CrossRef]
- Wormsbaecher, C.; Hindman, A.R.; Avendano, A.; Cortes-Medina, M.; Jones, C.E.; Bushman, A.; Onua, L.; Kovalchin, C.E.; Murphy, A.R.; Helber, H.L.; et al. In utero estrogenic endocrine disruption alters the stroma to increase extracellular matrix density and mammary gland stiffness. Breast Cancer Res. 2020, 22, 41. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, L.; Wu, X.; Hou, L.; Li, Z.; Ju, J.; Li, Q.; Qin, W.; Li, J.; Zhang, Q.; et al. Bisphenol A, an environmental estrogen-like toxic chemical, induces cardiac fibrosis by activating the ERK1/2 pathway. Toxicol. Lett. 2016, 250–251, 1–9. [Google Scholar] [CrossRef]
- Wen, X.; Zhu, M.; Li, Z.H.; Li, T.; Xu, X.W. Dual effects of bisphenol A on wound healing, involvement of estrogen receptor beta. Ecotoxicol. Environ. Saf. 2022, 231, 113207. [Google Scholar] [CrossRef]
- Arpino, V.; Brock, M.; Gill, S.E. The role of TIMPs in regulation of extracellular matrix proteolysis. Matrix Biol. 2015, 44–46, 247–254. [Google Scholar] [CrossRef]
- Gao, J.; Song, T.; Che, D.; Li, C.; Jiang, J.; Pang, J.; Yang, Y.; Goma; Li, P. The effect of bisphenol a exposure onto endothelial and decidualized stromal cells on regulation of the invasion ability of trophoblastic spheroids in in vitro co-culture model. Biochem. Biophys. Res. Commun. 2019, 516, 506–514. [Google Scholar] [CrossRef]
- Markiewicz, M.; Znoyko, S.; Stawski, L.; Ghatnekar, A.; Gilkeson, G.; Trojanowska, M. A role for estrogen receptor-alpha and estrogen receptor-beta in collagen biosynthesis in mouse skin. J. Investig. Dermatol. 2013, 133, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Kalamajski, S.; Oldberg, A. The role of small leucine-rich proteoglycans in collagen fibrillogenesis. Matrix Biol. 2010, 29, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, T.F.R.; Colleta, S.J.; Zuccari, D.A.P.D.; Vilamaior, P.S.L.; Leonel, E.C.R.; Taboga, S.R. Hormone receptor expression in aging mammary tissue and carcinoma from a rodent model after xenoestrogen disruption. Life Sci. 2021, 285, 120010. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Yang, Y.; Li, Q.; Feng, Y.; Liu, T.; Guo, W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.Y.; Wang, X.N.; Wu, N.N.; He, S.Q.; Yi, W.J.; Xiang, S.Y.; Zhang, P.W.; Xie, X.; Ying, C.J. Bisphenol A induces proliferative effects on both breast cancer cells and vascular endothelial cells through a shared GPER-dependent pathway in hypoxia. Environ. Pollut. 2017, 231, 1609–1620. [Google Scholar] [CrossRef]
- Gao, Y.H.; Chen, X.S.; He, Q.; Gimple, R.C.; Liao, Y.J.; Wang, L.; Wu, R.; Xie, Q.; Rich, J.N.; Shen, K.W.; et al. Adipocytes promote breast tumorigenesis through TAZ-dependent secretion of Resistin. Proc. Natl. Acad. Sci. USA 2020, 117, 33295–33304. [Google Scholar] [CrossRef]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body Mass Index and Breast Cancer Risk According to Postmenopausal Estrogen-Progestin Use and Hormone Receptor Status. Epidemiol. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef]
- Bulun, S.E.; Chen, D.; Moy, I.; Brooks, D.C.; Zhao, H. Aromatase, breast cancer and obesity: A complex interaction. Trends Endocrinol. Metab. 2012, 23, 83–89. [Google Scholar] [CrossRef]
- Wang, X.; Simpson, E.R.; Brown, K.A. Aromatase overexpression in dysfunctional adipose tissue links obesity to postmenopausal breast cancer. J. Steroid Biochem. Mol. Biol. 2015, 153, 35–44. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Goncalves, M.D.; Cantley, L.C. Obesity and Cancer Mechanisms: Cancer Metabolism. J. Clin. Oncol. 2016, 34, 4277–4283. [Google Scholar] [CrossRef]
- Saxena, N.K.; Vertino, P.M.; Anania, F.A.; Sharma, D. leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 promoter via activation of Stat3. J. Biol. Chem. 2007, 282, 13316–13325. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.D.; Mortimer, J.E.; Natarajan, R.; Dietze, E.C.; Seewaldt, V.L. Metabolic Health, Insulin, and Breast Cancer: Why Oncologists Should Care About Insulin. Front. Endocrinol. 2020, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.K.; Nguyen, T.T.; Thanh, V.V.; Quang, T.L.; Minh, L.B.; Pham, V.H.; Ngoc, V.T.N.; et al. The Effects of Adipocytes on the Regulation of Breast Cancer in the Tumor Microenvironment: An Update. Cells 2019, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- Vaysse, C.; Lomo, J.; Garred, O.; Fjeldheim, F.; Lofteroed, T.; Schlichting, E.; McTiernan, A.; Frydenberg, H.; Husoy, A.; Lundgren, S.; et al. Inflammation of mammary adipose tissue occurs in overweight and obese patients exhibiting early-stage breast cancer. NPJ Breast Cancer 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Lehuede, C.; Laurent, V.; Dirat, B.; Dauvillier, S.; Bochet, L.; Le Gonidec, S.; Escourrou, G.; Valet, P.; Muller, C. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012, 324, 142–151. [Google Scholar] [CrossRef]
- Subbaramaiah, K.; Howe, L.R.; Bhardwaj, P.; Du, B.H.; Gravaghi, C.; Yantiss, R.K.; Zhou, X.K.; Blaho, V.A.; Hla, T.; Yang, P.Y.; et al. Obesity Is Associated with Inflammation and Elevated Aromatase Expression in the Mouse Mammary Gland. Cancer Prev. Res. 2011, 4, 329–346. [Google Scholar] [CrossRef]
- Kolb, R.; Sutterwala, F.S.; Zhang, W. Obesity and cancer: Inflammation bridges the two. Curr. Opin. Pharmacol. 2016, 29, 77–89. [Google Scholar] [CrossRef]
- Maliniak, M.L.; Cheriyan, A.M.; Sherman, M.E.; Liu, Y.; Gogineni, K.; Liu, J.Q.; He, J.B.; Krishnamurti, U.; Miller-Kleinhenz, J.; Ashiqueali, R.; et al. Detection of crown-like structures in breast adipose tissue and clinical outcomes among African-American and White women with breast cancer. Breast Cancer Res. 2020, 22, 1–13. [Google Scholar] [CrossRef]
- Lee, K.; Kruper, L.; Dieli-Conwright, C.M.; Mortimer, J.E. The Impact of Obesity on Breast Cancer Diagnosis and Treatment. Curr. Oncol. Rep. 2019, 21, 41. [Google Scholar] [CrossRef]
- Chang, M.C.; Eslami, Z.; Ennis, M.; Goodwin, P.J. Crown-like structures in breast adipose tissue of breast cancer patients: Associations with CD68 expression, obesity, metabolic factors and prognosis. NPJ Breast Cancer 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Lin, L.L.; Kuhn, C.; Ditsch, N.; Kolben, T.; Czogalla, B.; Beyer, S.; Trillsch, F.; Schmoeckel, E.; Mayr, D.; Mahner, S.; et al. Breast adipose tissue macrophages (BATMs) have a stronger correlation with breast cancer survival than breast tumor stroma macrophages (BTSMs). Breast Cancer Res. 2021, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Naomi, R.; Yazid, M.D.; Bahari, H.; Keong, Y.Y.; Rajandram, R.; Embong, H.; Teoh, S.H.; Halim, S.; Othman, F. Bisphenol A (BPA) Leading to Obesity and Cardiovascular Complications: A Compilation of Current In Vivo Study. Int. J. Mol. Sci. 2022, 23, 2969. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Li, M.; Liu, A.; Wu, C.; Li, D.; Deng, Q.; Zhang, B.; Du, J.; Gao, X.; Hong, Y. Bisphenol A and the Risk of Obesity a Systematic Review with Meta-Analysis of the Epidemiological Evidence. Dose Response 2020, 18, 1559325820916949. [Google Scholar] [CrossRef] [PubMed]
- Ohlstein, J.F.; Strong, A.L.; McLachlan, J.A.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Bisphenol A enhances adipogenic differentiation of human adipose stromal/stem cells. J. Mol. Endocrinol. 2014, 53, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, A.; Shidfar, F.; Hedayati, M.; Neshatbini Tehrani, A.; Farshad, A.A.; Mohammadi, S. Bisphenol A enhances adipogenic signaling pathways in human mesenchymal stem cells. Genes Environ. 2020, 42, 13. [Google Scholar] [CrossRef]
- Junge, K.M.; Leppert, B.; Jahreis, S.; Wissenbach, D.K.; Feltens, R.; Grutzmann, K.; Thurmann, L.; Bauer, T.; Ishaque, N.; Schick, M.; et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin. Epigenetics. 2018, 10, 58. [Google Scholar] [CrossRef]
- Volberg, V.; Harley, K.; Calafat, A.M.; Dave, V.; McFadden, J.; Eskenazi, B.; Holland, N. Maternal bisphenol a exposure during pregnancy and its association with adipokines in Mexican-American children. Environ. Mol. Mutagen. 2013, 54, 621–628. [Google Scholar] [CrossRef]
- Desai, M.; Ferrini, M.G.; Jellyman, J.K.; Han, G.; Ross, M.G. In vivo and in vitro bisphenol A exposure effects on adiposity. J. Dev. Orig. Health Dis. 2018, 9, 678–687. [Google Scholar] [CrossRef]
- Kaneda, M.; Okano, M.; Hata, K.; Sado, T.; Tsujimoto, N.; Li, E.; Sasaki, H. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 2004, 429, 900–903. [Google Scholar] [CrossRef]
- Longo, M.; Zatterale, F.; Naderi, J.; Nigro, C.; Oriente, F.; Formisano, P.; Miele, C.; Beguinot, F. Low-dose Bisphenol-A Promotes Epigenetic Changes at Ppargamma Promoter in Adipose Precursor Cells. Nutrients 2020, 12, 3498. [Google Scholar] [CrossRef]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics. Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y. Immuno-Resolving Ability of Resolvins, Protectins, and Maresins Derived from Omega-3 Fatty Acids in Metabolic Syndrome. Mol. Nutr. Food Res. 2020, 64, e1900824. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, M.; Wang, J.; Xu, M.; Sun, J.; Ding, L.; Lv, X.; Ma, Q.; Bi, Y.; Liu, R.; et al. Bisphenol A Promotes Adiposity and Inflammation in a Nonmonotonic Dose-response Way in 5-week-old Male and Female C57BL/6J Mice Fed a Low-calorie Diet. Endocrinology 2016, 157, 2333–2345. [Google Scholar] [CrossRef] [PubMed]
- Savastano, S.; Tarantino, G.; D’Esposito, V.; Passaretti, F.; Cabaro, S.; Liotti, A.; Liguoro, D.; Perruolo, G.; Ariemma, F.; Finelli, C.; et al. Bisphenol-A plasma levels are related to inflammatory markers, visceral obesity and insulin-resistance: A cross-sectional study on adult male population. J. Transl. Med. 2015, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.K.; Jeong, I.K.; Jung Oh, T.; Ahn, H.Y.; Kim, H.H.; Park, Y.J.; Jang, H.C.; Park, K.S. Long-term oral exposure to bisphenol A induces glucose intolerance and insulin resistance. J. Endocrinol. 2015, 226, 35–42. [Google Scholar] [CrossRef]
- Valentino, R.; D’Esposito, V.; Passaretti, F.; Liotti, A.; Cabaro, S.; Longo, M.; Perruolo, G.; Oriente, F.; Beguinot, F.; Formisano, P. Bisphenol-A impairs insulin action and up-regulates inflammatory pathways in human subcutaneous adipocytes and 3T3-L1 cells. PLoS ONE 2013, 8, e82099. [Google Scholar] [CrossRef]
- Cimmino, I.; Oriente, F.; D’Esposito, V.; Liguoro, D.; Liguoro, P.; Ambrosio, M.R.; Cabaro, S.; D’Andrea, F.; Beguinot, F.; Formisano, P.; et al. Low-dose Bisphenol-A regulates inflammatory cytokines through GPR30 in mammary adipose cells. J. Mol. Endocrinol. 2019, 63, 273–283. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Cell Type | Subtype | Function in TME | Effect of BPA |
|---|---|---|---|
| CD8+ cells (CTLs) | Eliminate cancer cells | Reduces telomere length [52,54,55] Suppresses proliferation [55] | |
| CD4+ cells (Th cells) | Th1 | Produce pro-inflammatory immune microenvironments (secrete IL-2, IFN-γ) Induce CTL response Induce M1 macrophage polarization Eliminate cancer cells | Inhibits polarization/function (inhibits IFN-γ production) in allergen-stimulated conditions [53,59] |
| Th2 | Produce anti-inflammatory immune microenvironment (secrete IL-4, IL-10) Induce humoral immune response Induce M2 macrophage polarization Increase cancer growth and migration | Enhances polarization/function in allergic disease models [60,61] and in parasite infection [62] Induces polarization by dendritic cells upon maturation with BPA/TNF-α [63] | |
| Treg | Induce anti-inflammatory immune microenvironment | Derives large proportion in breast tumors grown on BPA-exposed mice [64] | |
| Macrophages | M1 | Induce pro-inflammatory microenvironment Eliminate cancer cells | Inhibits polarization in RAW 264.7 macrophages [65,66] and ex vivo culture of mouse peritoneal macrophages [67] Promotes polarization in mouse peritoneal macrophages [68] and THP1 cells [69] |
| M2 | Induce anti-inflammatory microenvironment Increase tumor promotion | Derives large proportion in breast tumors grown on BPA-exposed mice [64] Derives large content in BPA-exposed before and during cancer formation in mice [70] Induces migration in human PBMCs [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, Y. Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment. Cancers 2022, 14, 3021. https://doi.org/10.3390/cancers14123021
Kwon Y. Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment. Cancers. 2022; 14(12):3021. https://doi.org/10.3390/cancers14123021
Chicago/Turabian StyleKwon, Youngjoo. 2022. "Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment" Cancers 14, no. 12: 3021. https://doi.org/10.3390/cancers14123021
APA StyleKwon, Y. (2022). Potential Pro-Tumorigenic Effect of Bisphenol A in Breast Cancer via Altering the Tumor Microenvironment. Cancers, 14(12), 3021. https://doi.org/10.3390/cancers14123021