

Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ–CAMTA1 Fusion Driven Sarcoma


{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Epithelioid Hemangioendothelioma
3. Molecular Alterations in EHE
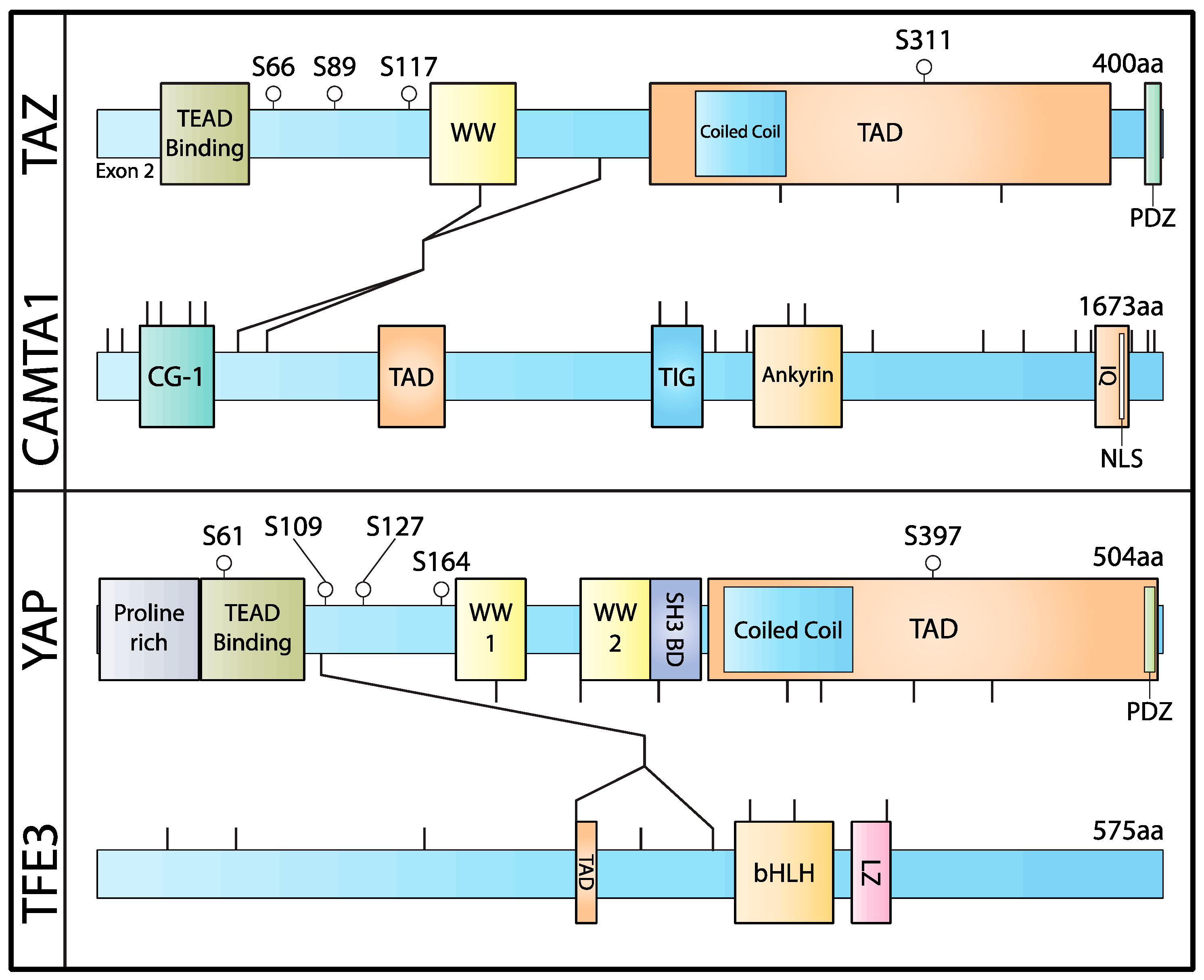
4. The Biology of WWTR1(TAZ)–CAMTA1 Fusion
5. Understanding the YAP1–TFE3 Fusion
6. C-Terminal Proteins Reshape the Chromatin Structure and Enhance Tumorigenesis
7. Novel Model Systems for Studying EHE Biology
8. Development of Targeted Therapies
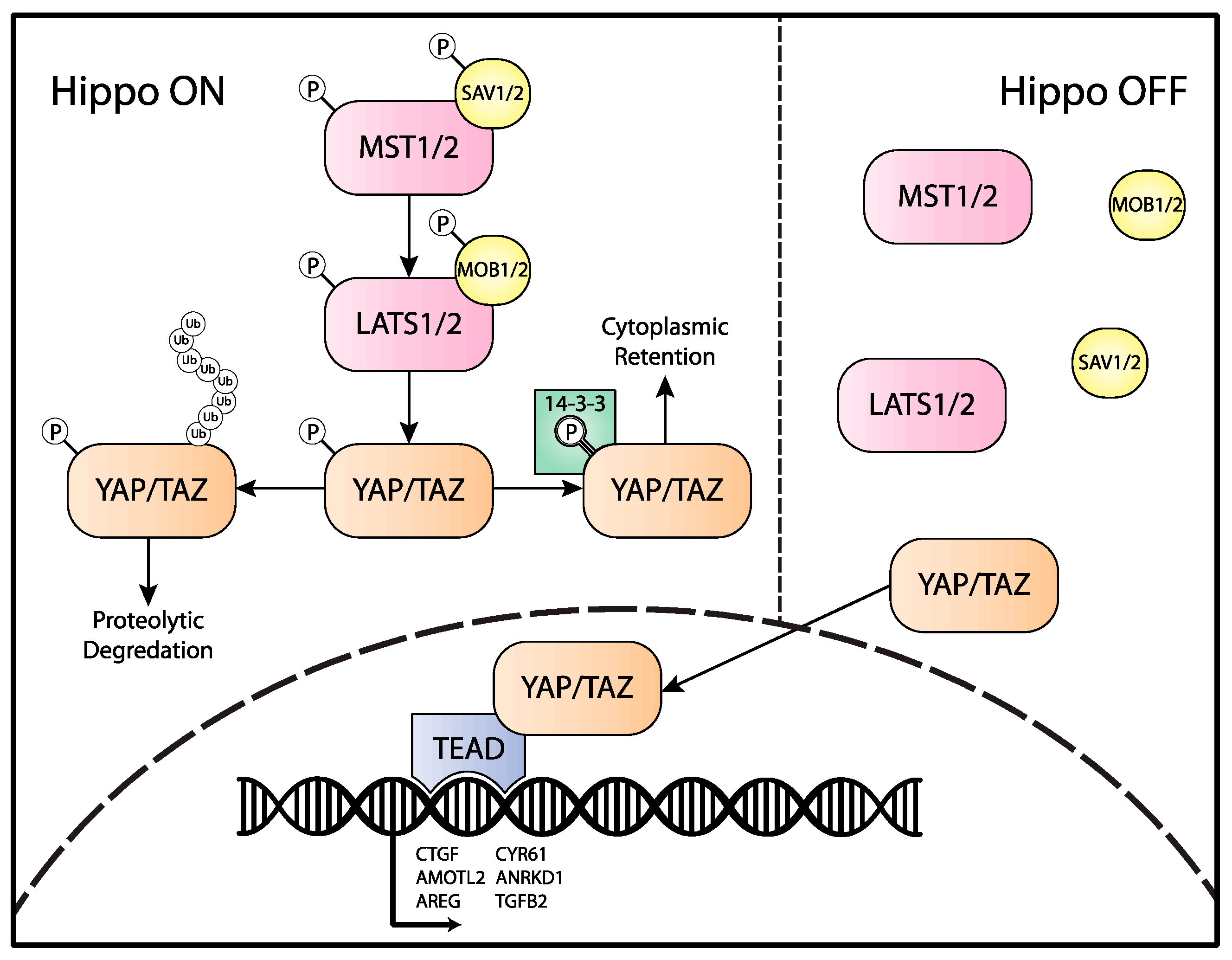
8.1. Leverage the “Hippo-Dependent” and “Hippo-Independent” Mechanisms Upstream of the Fusion
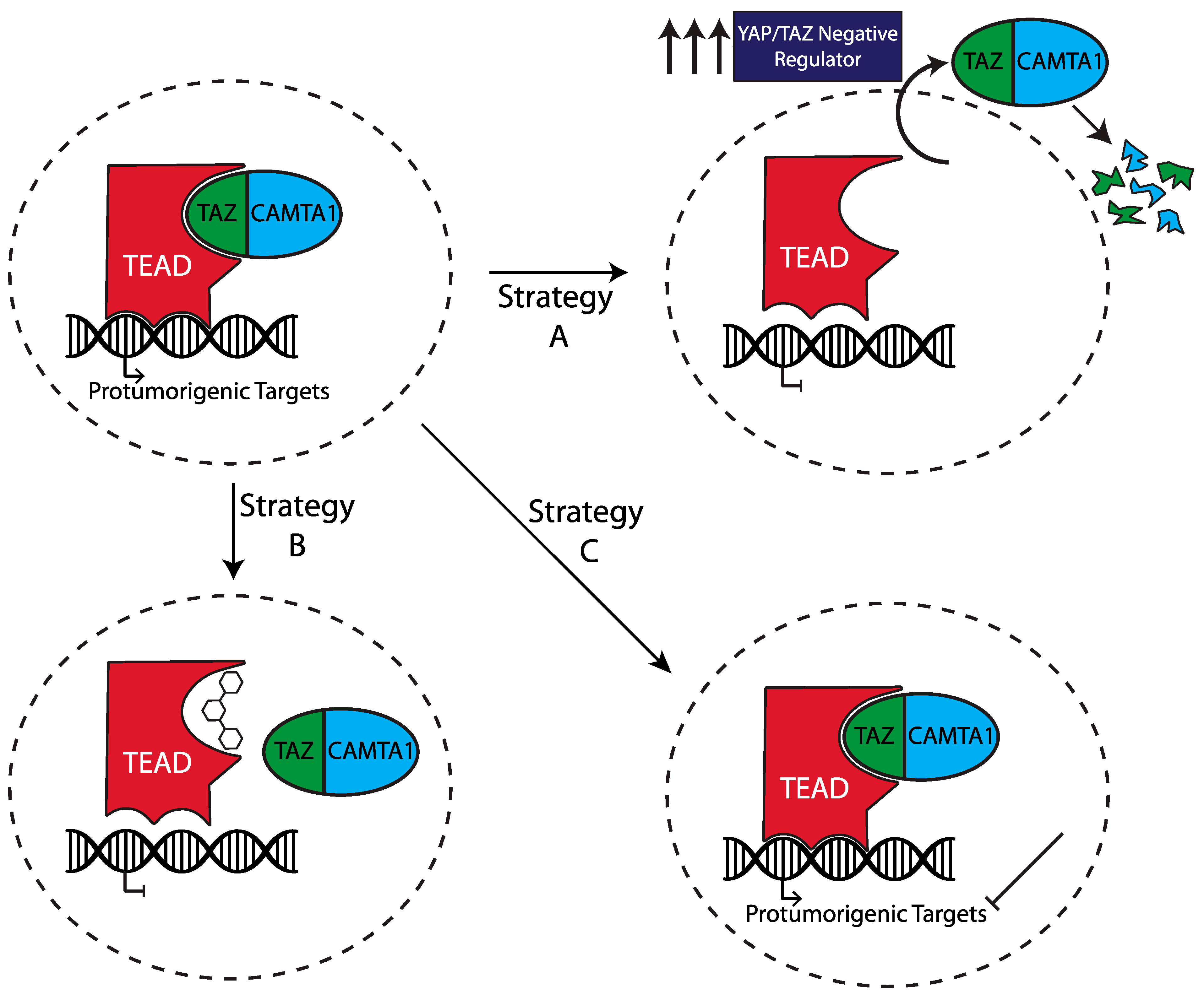
8.2. Use Drugs That Directly Act on the TC/TEAD Transcriptional Complex
8.3. Identifying and Targeting the Oncogenic Signaling Downstream of the Fusion
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, F.-X.; Zhao, B.; Guan, K.-L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Mak, K.K.; Topol, L.; Yun, K.; Hu, J.; Garrett, L.; Chen, Y.; Park, O.; Chang, J.; Simpson, R.M. Mammalian Mst1 and Mst2 Kinases Play Essential Roles in Organ Size Control and Tumor Suppression. Proc. Natl. Acad. Sci. USA 2010, 107, 1431–1436. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst Ortholog, Hippo, Restricts Growth and Cell Proliferation and Promotes Apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila Tumor Suppressor Gene Warts Encodes a Homolog of Human Myotonic Dystrophy Kinase and Is Required for the Control of Cell Shape and Proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, D.C.R.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. Salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell 2002, 110, 467–478. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. Hippo Encodes a Ste-20 Family Protein Kinase That Restricts Cell Proliferation and Promotes Apoptosis in Conjunction with Salvador and Warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying Tumor Suppressors in Genetic Mosaics: The Drosophila Lats Gene Encodes a Putative Protein Kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ Upstream Signals and Downstream Responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo Pathway Regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A Mechanical Checkpoint Controls Multicellular Growth through YAP/TAZ Regulation by Actin-Processing Factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, K.T.; Yamashita, K.; Sakurai, N.; Ohno, S. The Epithelial Circumferential Actin Belt Regulates YAP/TAZ through Nucleocytoplasmic Shuttling of Merlin. Cell Rep. 2017, 20, 1435–1447. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK Proteins and YAP-1 Signalling Downstream of Integrin Beta-1 in Myofibroblasts Promote Liver Fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef]
- Sabra, H.; Brunner, M.; Mandati, V.; Wehrle-Haller, B.; Lallemand, D.; Ribba, A.-S.; Chevalier, G.; Guardiola, P.; Block, M.R.; Bouvard, D. Β1 Integrin-Dependent Rac/Group I PAK Signaling Mediates YAP Activation of Yes-Associated Protein 1 (YAP1) via NF2/Merlin. J. Biol. Chem. 2017, 292, 19179–19197. [Google Scholar] [CrossRef] [Green Version]
- Sero, J.E.; Bakal, C. Multiparametric Analysis of Cell Shape Demonstrates That β-PIX Directly Couples YAP Activation to Extracellular Matrix Adhesion. Cell Syst. 2017, 4, 84–96.e6. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, R.; Kim, N.-G.; Gumbiner, B.M. Regulation of Hippo Pathway by Mitogenic Growth Factors via Phosphoinositide 3-Kinase and Phosphoinositide-Dependent Kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, B.V.V.G.; Irvine, K.D. Regulation of Hippo Signaling by EGFR-MAPK Signaling through Ajuba Family Proteins. Dev. Cell 2013, 24, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ji, J.-Y.; Yu, M.; Overholtzer, M.; Smolen, G.A.; Wang, R.; Brugge, J.S.; Dyson, N.J.; Haber, D.A. YAP-Dependent Induction of Amphiregulin Identifies a Non-Cell-Autonomous Component of the Hippo Pathway. Nat. Cell Biol. 2009, 11, 1444–1450. [Google Scholar] [CrossRef]
- Bae, S.J.; Luo, X. Activation Mechanisms of the Hippo Kinase Signaling Cascade. Biosci. Rep. 2018, 38, BSR20171469. [Google Scholar] [CrossRef] [Green Version]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 Phosphorylates Hippo/MST Kinases to Regulate the Hippo-Salvador-Warts Tumor Suppressor Pathway. Dev. Cell 2011, 21, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Callus, B.A.; Verhagen, A.M.; Vaux, D.L. Association of Mammalian Sterile Twenty Kinases, Mst1 and Mst2, with HSalvador via C-Terminal Coiled-Coil Domains, Leads to Its Stabilization and Phosphorylation. FEBS J. 2006, 273, 4264–4276. [Google Scholar] [CrossRef]
- Praskova, M.; Xia, F.; Avruch, J. MOBKL1A/MOBKL1B Phosphorylation by MST1 and MST2 Inhibits Cell Proliferation. Curr. Biol. 2008, 18, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Yu, J.; Zheng, Y.; Chen, Q.; Zhang, N.; Pan, D. Spatial Organization of Hippo Signaling at the Plasma Membrane Mediated by the Tumor Suppressor Merlin/NF2. Cell 2013, 154, 1342–1355. [Google Scholar] [CrossRef] [Green Version]
- Hergovich, A.; Schmitz, D.; Hemmings, B.A. The Human Tumour Suppressor LATS1 Is Activated by Human MOB1 at the Membrane. Biochem. Biophys. Res. Commun. 2006, 345, 50–58. [Google Scholar] [CrossRef]
- Chan, E.H.Y.; Nousiainen, M.; Chalamalasetty, R.B.; Schäfer, A.; Nigg, E.A.; Silljé, H.H.W. The Ste20-like Kinase Mst2 Activates the Human Large Tumor Suppressor Kinase Lats1. Oncogene 2005, 24, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor Suppressor LATS1 Is a Negative Regulator of Oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.-Y.; Zhang, H.; Zhao, B.; Zha, Z.-Y.; Bai, F.; Pei, X.-H.; Zhao, S.; Xiong, Y.; Guan, K.-L. TAZ Promotes Cell Proliferation and Epithelial-Mesenchymal Transition and Is Inhibited by the Hippo Pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.-Y.; Zha, Z.-Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The Hippo Tumor Pathway Promotes TAZ Degradation by Phosphorylating a Phosphodegron and Recruiting the SCF{beta}-TrCP E3 Ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.W.; Lim, C.J.; Guo, K.; Ng, C.P.; Lee, I.; Hunziker, W.; Zeng, Q.; Hong, W. A Role for TAZ in Migration, Invasion, and Tumorigenesis of Breast Cancer Cells. Cancer Res. 2008, 68, 2592–2598. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Hong, W. Emerging Roles of TEAD Transcription Factors and Its Coactivators in Cancers. Cancer Biol. Ther. 2013, 14, 390–398. [Google Scholar] [CrossRef] [Green Version]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and Functional Similarity between the Vgll1-TEAD and the YAP-TEAD Complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [Green Version]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.G.; Hynes, R.O. The Hippo Pathway Target, YAP, Promotes Metastasis through Its TEAD-Interaction Domain. Proc. Natl. Acad. Sci. USA 2012, 109, E2441–E2450. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Tang, T.; Probst, G.; Konradi, A.; Jin, C.; Li, F.; Silvio Gutkind, J.; Fu, X.-D.; Guan, K.-L. Transcriptional Repression of Estrogen Receptor Alpha by YAP Reveals the Hippo Pathway as Therapeutic Target for ER+ Breast Cancer. Nat. Commun. 2022, 13, 1061. [Google Scholar] [CrossRef]
- Grieve, S.; Wajnberg, G.; Lees, M.; Chacko, S.; Weir, J.; Crapoulet, N.; Reiman, T. TAZ Functions as a Tumor Suppressor in Multiple Myeloma by Downregulating MYC. Blood Adv. 2019, 3, 3613–3625. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.D.; Huang, K.; Pacal, M.; McCurdy, S.R.; Lu, S.; Aubry, A.; Yu, T.; Wadosky, K.M.; Zhang, L.; Wang, T.; et al. Binary Pan-Cancer Classes with Distinct Vulnerabilities Defined by pro- or Anti-Cancer YAP/TEAD Activity. Cancer Cell 2021, 39, 1115–1134.e12. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Yang, B.; Nakaoka, H.J.; Yang, J.; Zhao, Y.; Le Nguyen, K.; Bishara, A.T.; Mandalia, T.K.; Wang, W. Hippo Signaling Dysfunction Induces Cancer Cell Addiction to YAP. Oncogene 2018, 37, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. A Combat with the YAP/TAZ-TEAD Oncoproteins for Cancer Therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef]
- Weiss, S.W.; Enzinger, F.M. Epithelioid Hemangioendothelioma: A Vascular Tumor Often Mistaken for a Carcinoma. Cancer 1982, 50, 970–981. [Google Scholar] [CrossRef]
- Sardaro, A.; Bardoscia, L.; Petruzzelli, M.F.; Portaluri, M. Epithelioid Hemangioendothelioma: An Overview and Update on a Rare Vascular Tumor. Oncol Rev. 2014, 8, 259. [Google Scholar] [CrossRef] [Green Version]
- Bagan, P.; Hassan, M.; Le Pimpec Barthes, F.; Peyrard, S.; Souilamas, R.; Danel, C.; Riquet, M. Prognostic Factors and Surgical Indications of Pulmonary Epithelioid Hemangioendothelioma: A Review of the Literature. Ann. Thorac. Surg. 2006, 82, 2010–2013. [Google Scholar] [CrossRef]
- Lau, K.; Massad, M.; Pollak, C.; Rubin, C.; Yeh, J.; Wang, J.; Edelman, G.; Yeh, J.; Prasad, S.; Weinberg, G. Clinical Patterns and Outcome in Epithelioid Hemangioendothelioma with or without Pulmonary Involvement: Insights from an Internet Registry in the Study of a Rare Cancer. Chest 2011, 140, 1312–1318. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, E.; Jadeja, B.; Xu, B.; Zhang, L.; Agaram, N.P.; Travis, W.; Singer, S.; Tap, W.D.; Antonescu, C.R. Prognostic Stratification of Clinical and Molecular Epithelioid Hemangioendothelioma Subsets. Mod. Pathol. 2020, 33, 591–602. [Google Scholar] [CrossRef]
- Amin, R.M.S.; Hiroshima, K.; Kokubo, T.; Nishikawa, M.; Narita, M.; Kuroki, M.; Nakatani, Y. Risk Factors and Independent Predictors of Survival in Patients with Pulmonary Epithelioid Haemangioendothelioma. Review of the Literature and a Case Report. Respirology 2006, 11, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Makhlouf, H.R.; Ishak, K.G.; Goodman, Z.D. Epithelioid Hemangioendothelioma of the Liver: A Clinicopathologic Study of 137 Cases. Cancer 1999, 85, 562–582. [Google Scholar] [CrossRef]
- Antonescu, C. Malignant Vascular Tumors—An Update. Mod. Pathol. 2014, 27 (Suppl. S1), S30–S38. [Google Scholar] [CrossRef] [Green Version]
- Mentzel, T.; Beham, A.; Calonje, E.; Katenkamp, D.; Fletcher, C.D. Epithelioid Hemangioendothelioma of Skin and Soft Tissues: Clinicopathologic and Immunohistochemical Study of 30 Cases. Am. J. Surg. Pathol. 1997, 21, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Mendlick, M.R.; Nelson, M.; Pickering, D.; Johansson, S.L.; Seemayer, T.A.; Neff, J.R.; Vergara, G.; Rosenthal, H.; Bridge, J.A. Translocation t(1;3)(P36.3;Q25) Is a Nonrandom Aberration in Epithelioid Hemangioendothelioma. Am. J. Surg. Pathol. 2001, 25, 684–687. [Google Scholar] [CrossRef]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a Disease-Defining Gene Fusion in Epithelioid Hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar] [CrossRef]
- Errani, C.; Zhang, L.; Sung, Y.S.; Hajdu, M.; Singer, S.; Maki, R.G.; Healey, J.H.; Antonescu, C.R. A Novel WWTR1-CAMTA1 Gene Fusion Is a Consistent Abnormality in Epithelioid Hemangioendothelioma of Different Anatomic Sites. Genes Chromosomes Cancer 2011, 50, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, R.; Matsuyama, A.; Shiba, E.; Harada, H.; Yabuki, K.; Hisaoka, M. CAMTA1 Is a Useful Immunohistochemical Marker for Diagnosing Epithelioid Haemangioendothelioma. Histopathology 2015, 67, 827–835. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Le Loarer, F.; Mosquera, J.; Sboner, A.; Zhang, L.; Chen, C.; Chen, H.; Pathan, N.; Krausz, T.; Dickson, B.C. Novel YAP1-TFE3 Fusion Defines a Distinct Subset of Epithelioid Hemangioendothelioma. Genes Chromosomes Cancer 2013, 52, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Dermawan, J.K.; Azzato, E.M.; Billings, S.D.; Fritchie, K.J.; Aubert, S.; Bahrami, A.; Barisella, M.; Baumhoer, D.; Blum, V.; Bode, B.; et al. YAP1-TFE3-Fused Hemangioendothelioma: A Multi-Institutional Clinicopathologic Study of 24 Genetically-Confirmed Cases. Mod. Pathol. 2021, 34, 2211–2221. [Google Scholar] [CrossRef]
- Puls, F.; Niblett, A.; Clarke, J.; Kindblom, L.-G.; McCulloch, T. YAP1-TFE3 Epithelioid Hemangioendothelioma: A Case without Vasoformation and a New Transcript Variant. Virchows Arch. 2015, 466, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Dermawan, J.K.; Azzato, E.M.; McKenney, J.K.; Liegl-Atzwanger, B.; Rubin, B.P. YAP1-TFE3 Gene Fusion Variant in Clear Cell Stromal Tumour of Lung: Report of Two Cases in Support of a Distinct Entity. Histopathology 2021, 79, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Patton, A.; Bridge, J.A.; Liebner, D.; Chung, C.; Iwenofu, O.H. A YAP1::TFE3 Cutaneous Low-Grade Fibromyxoid Neoplasm: A Novel Entity! Genes Chromosomes Cancer 2022, 61, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A.; Stoehr, R.; Michal, M.; Christopoulos, P.; Winter, H.; Zhang, L.; Stenzinger, A.; Michal, M.; Mechtersheimer, G.; Antonescu, C.R. Recurrent YAP1-TFE3 Gene Fusions in Clear Cell Stromal Tumor of the Lung. Am. J. Surg. Pathol. 2021, 45, 1541–1549. [Google Scholar] [CrossRef]
- Suurmeijer, A.J.; Dickson, B.C.; Swanson, D.; Sung, Y.S.; Zhang, L.; Antonescu, C.R. Variant WWTR1 Gene Fusions in Epithelioid Hemangioendothelioma—A Genetic Subset Associated with Cardiac Involvement. Genes Chromosomes Cancer 2020, 59, 389–395. [Google Scholar] [CrossRef]
- Seligson, N.D.; Awasthi, A.; Millis, S.Z.; Turpin, B.K.; Meyer, C.F.; Grand’Maison, A.; Liebner, D.A.; Hays, J.L.; Chen, J.L. Common Secondary Genomic Variants Associated with Advanced Epithelioid Hemangioendothelioma. JAMA Netw. Open 2019, 2, e1912416. [Google Scholar] [CrossRef]
- Bas-Orth, C.; Tan, Y.-W.; Oliveira, A.M.; Bengtson, C.P.; Bading, H. The Calmodulin-Binding Transcription Activator CAMTA1 Is Required for Long-Term Memory Formation in Mice. Learn. Mem. 2016, 23, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Huentelman, M.J.; Papassotiropoulos, A.; Craig, D.W.; Hoerndli, F.J.; Pearson, J.V.; Huynh, K.-D.; Corneveaux, J.; Hänggi, J.; Mondadori, C.R.; Buchmann, A. Calmodulin-Binding Transcription Activator 1 (CAMTA1) Alleles Predispose Human Episodic Memory Performance. Hum. Mol. Genet. 2007, 16, 1469–1477. [Google Scholar] [CrossRef] [Green Version]
- Schraivogel, D.; Weinmann, L.; Beier, D.; Tabatabai, G.; Eichner, A.; Zhu, J.Y.; Anton, M.; Sixt, M.; Weller, M.; Beier, C.P. CAMTA1 Is a Novel Tumour Suppressor Regulated by MiR-9/9* in Glioblastoma Stem Cells. EMBO J. 2011, 30, 4309–4322. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.-J.; Li, Y.; Wang, S.-D.; Wang, X.-S.; Fang, F.; Wang, W.-Y.; Lv, P.; Zhao, D.-H.; Wei, F.; Qi, L. Long Noncoding RNA LncCAMTA1 Promotes Proliferation and Cancer Stem Cell-like Properties of Liver Cancer by Inhibiting CAMTA1. Int. J. Mol. Sci. 2016, 17, 1617. [Google Scholar] [CrossRef]
- Henrich, K.-O.; Bauer, T.; Schulte, J.; Ehemann, V.; Deubzer, H.; Gogolin, S.; Muth, D.; Fischer, M.; Benner, A.; König, R. CAMTA1, a 1p36 Tumor Suppressor Candidate, Inhibits Growth and Activates Differentiation Programs in Neuroblastoma Cells. Cancer Res. 2011, 71, 3142–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henrich, K.-O.; Fischer, M.; Mertens, D.; Benner, A.; Wiedemeyer, R.; Brors, B.; Oberthuer, A.; Berthold, F.; Wei, J.S.; Khan, J. Reduced Expression of CAMTA1 Correlates with Adverse Outcome in Neuroblastoma Patients. Clin. Cancer Res. 2006, 12, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbashina, V.; Salazar, P.; Holland, E.C.; Rosenblum, M.K.; Ladanyi, M. Allelic Losses at 1p36 and 19q13 in Gliomas: Correlation with Histologic Classification, Definition of a 150-Kb Minimal Deleted Region on 1p36, and Evaluation of CAMTA1 as a Candidate Tumor Suppressor Gene. Clin. Cancer Res. 2005, 11, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Yang, C.; He, Y.; Gong, B.; Yin, C.; Feng, J.; Chen, L.; Tang, J.; Chen, Y. CAMTA1, a Novel Antitumor Gene, Regulates Proliferation and the Cell Cycle in Glioma by Inhibiting AKT Phosphorylation. Cell. Signal. 2021, 79, 109882. [Google Scholar] [CrossRef] [PubMed]
- Tanas, M.R.; Ma, S.; Jadaan, F.O.; Ng, C.K.Y.; Weigelt, B.; Reis-Filho, J.S.; Rubin, B.P. Mechanism of Action of a WWTR1(TAZ)-CAMTA1 Fusion Oncoprotein. Oncogene 2016, 35, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Szulzewsky, F.; Arora, S.; Hoellerbauer, P.; King, C.; Nathan, E.; Chan, M.; Cimino, P.J.; Ozawa, T.; Kawauchi, D.; Pajtler, K.W.; et al. Comparison of Tumor-Associated YAP1 Fusions Identifies a Recurrent Set of Functions Critical for Oncogenesis. Genes Dev. 2020, 34, 1051–1064. [Google Scholar] [CrossRef]
- Merritt, N.; Garcia, K.; Rajendran, D.; Lin, Z.-Y.; Zhang, X.; Mitchell, K.A.; Borcherding, N.; Fullenkamp, C.; Chimenti, M.S.; Gingras, A.-C.; et al. TAZ-CAMTA1 and YAP-TFE3 Alter the TAZ/YAP Transcriptome by Recruiting the ATAC Histone Acetyltransferase Complex. eLife 2021, 10, e62857. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.-Y.; Guan, K.-L. A Coordinated Phosphorylation by Lats and CK1 Regulates YAP Stability through SCF(Beta-TRCP). Genes Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef] [Green Version]
- Steingrimsson, E.; Tessarollo, L.; Pathak, B.; Hou, L.; Arnheiter, H.; Copeland, N.G.; Jenkins, N.A. Mitf and Tfe3, Two Members of the Mitf-Tfe Family of BHLH-Zip Transcription Factors, Have Important but Functionally Redundant Roles in Osteoclast Development. Proc. Natl. Acad. Sci. USA 2002, 99, 4477–4482. [Google Scholar] [CrossRef] [Green Version]
- Seavey, C.N.; Rubin, B.P. Letter by Seavey and Rubin Regarding Article, “Sustained Activation of Endothelial YAP1 Causes Epithelioid Hemangioendothelioma”. Arter. Thromb. Vasc. Biol. 2021, 41, e491–e492. [Google Scholar] [CrossRef]
- Suganuma, T.; Gutiérrez, J.L.; Li, B.; Florens, L.; Swanson, S.K.; Washburn, M.P.; Abmayr, S.M.; Workman, J.L. ATAC Is a Double Histone Acetyltransferase Complex That Stimulates Nucleosome Sliding. Nat. Struct. Mol. Biol. 2008, 15, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Ciurciu, A.; Komonyi, O.; Pankotai, T.; Boros, I.M. The Drosophila Histone Acetyltransferase Gcn5 and Transcriptional Adaptor Ada2a Are Involved in Nucleosomal Histone H4 Acetylation. Mol. Cell Biol. 2006, 26, 9413–9423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guelman, S.; Suganuma, T.; Florens, L.; Swanson, S.K.; Kiesecker, C.L.; Kusch, T.; Anderson, S.; Yates, J.R., 3rd; Washburn, M.P.; Abmayr, S.M.; et al. Host Cell Factor and an Uncharacterized SANT Domain Protein Are Stable Components of ATAC, a Novel DAda2A/DGcn5-Containing Histone Acetyltransferase Complex in Drosophila. Mol. Cell Biol. 2006, 26, 871–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 Acetylation Co-Occur at Many Gene Regulatory Elements, While H3K14ac Marks a Subset of Inactive Inducible Promoters in Mouse Embryonic Stem Cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]
- Shogren-Knaak, M.; Ishii, H.; Sun, J.-M.; Pazin, M.J.; Davie, J.R.; Peterson, C.L. Histone H4-K16 Acetylation Controls Chromatin Structure and Protein Interactions. Science 2006, 311, 844–847. [Google Scholar] [CrossRef] [Green Version]
- Guelman, S.; Kozuka, K.; Mao, Y.; Pham, V.; Solloway, M.J.; Wang, J.; Wu, J.; Lill, J.R.; Zha, J. The Double-Histone-Acetyltransferase Complex ATAC Is Essential for Mammalian Development. Mol. Cell Biol. 2009, 29, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Mi, W.; Guan, H.; Lyu, J.; Zhao, D.; Xi, Y.; Jiang, S.; Andrews, F.H.; Wang, X.; Gagea, M.; Wen, H. YEATS2 Links Histone Acetylation to Tumorigenesis of Non-Small Cell Lung Cancer. Nat. Commun. 2017, 8, 1088. [Google Scholar] [CrossRef]
- Seavey, C.N.; Pobbati, A.V.; Hallett, A.; Ma, S.; Reynolds, J.P.; Kanai, R.; Lamar, J.M.; Rubin, B.P. WWTR1(TAZ)-CAMTA1 Gene Fusion Is Sufficient to Dysregulate YAP/TAZ Signaling and Drive Epithelioid Hemangioendothelioma Tumorigenesis. Genes Dev. 2021, 35, 512–527. [Google Scholar] [CrossRef]
- Oswald, J.; Boxberger, S.; Jørgensen, B.; Feldmann, S.; Ehninger, G.; Bornhäuser, M.; Werner, C. Mesenchymal Stem Cells Can Be Differentiated into Endothelial Cells in Vitro. Stem. Cells 2004, 22, 377–384. [Google Scholar] [CrossRef]
- Yu, Q.C.; Song, W.; Wang, D.; Zeng, Y.A. Identification of Blood Vascular Endothelial Stem Cells by the Expression of Protein C Receptor. Cell Res. 2016, 26, 1079–1098. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, T.; Naito, H.; Suehiro, J.-I.; Lin, Y.; Kawaji, H.; Iba, T.; Kouno, T.; Ishikawa-Kato, S.; Furuno, M.; Takara, K.; et al. CD157 Marks Tissue-Resident Endothelial Stem Cells with Homeostatic and Regenerative Properties. Cell Stem Cell 2018, 22, 384–397.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Driskill, J.H.; Zheng, Y.; Wu, B.-K.; Wang, L.; Cai, J.; Rakheja, D.; Dellinger, M.; Pan, D. WWTR1(TAZ)-CAMTA1 Reprograms Endothelial Cells to Drive Epithelioid Hemangioendothelioma. Genes Dev. 2021, 35, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.; Buchdunger, E.; Druker, B.J. The Development of Imatinib as a Therapeutic Agent for Chronic Myeloid Leukemia. Blood 2005, 105, 2640–2653. [Google Scholar] [CrossRef] [Green Version]
- Hochhaus, A.; Larson, R.A.; Guilhot, F.; Radich, J.P.; Branford, S.; Hughes, T.P.; Baccarani, M.; Deininger, M.W.; Cervantes, F.; Fujihara, S. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N. Engl. J. Med. 2017, 376, 917–927. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J. Kinase Mutations and Imatinib Response in Patients with Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamar, J.M.; Motilal Nehru, V.; Weinberg, G. Epithelioid Hemangioendothelioma as a Model of YAP/TAZ-Driven Cancer: Insights from a Rare Fusion Sarcoma. Cancers 2018, 10, 229. [Google Scholar] [CrossRef] [Green Version]
- Chevreau, C.; Le Cesne, A.; Ray-Coquard, I.; Italiano, A.; Cioffi, A.; Isambert, N.; Robin, Y.M.; Fournier, C.; Clisant, S.; Chaigneau, L.; et al. Sorafenib in Patients with Progressive Epithelioid Hemangioendothelioma: A Phase 2 Study by the French Sarcoma Group (GSF/GETO). Cancer 2013, 119, 2639–2644. [Google Scholar] [CrossRef]
- Park, M.S.; Ravi, V.; Araujo, D.M. Inhibiting the VEGF-VEGFR Pathway in Angiosarcoma, Epithelioid Hemangioendothelioma, and Hemangiopericytoma/Solitary Fibrous Tumor. Curr. Opin. Oncol. 2010, 22, 351–355. [Google Scholar] [CrossRef]
- Agulnik, M.; Yarber, J.L.; Okuno, S.H.; von Mehren, M.; Jovanovic, B.D.; Brockstein, B.E.; Evens, A.M.; Benjamin, R.S. An Open-Label, Multicenter, Phase II Study of Bevacizumab for the Treatment of Angiosarcoma and Epithelioid Hemangioendotheliomas. Ann. Oncol. 2013, 24, 257–263. [Google Scholar] [CrossRef]
- Azad, T.; Janse van Rensburg, H.J.; Lightbody, E.D.; Neveu, B.; Champagne, A.; Ghaffari, A.; Kay, V.R.; Hao, Y.; Shen, H.; Yeung, B.; et al. A LATS Biosensor Screen Identifies VEGFR as a Regulator of the Hippo Pathway in Angiogenesis. Nat. Commun. 2018, 9, 1061. [Google Scholar] [CrossRef]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K Positively Regulates YAP and TAZ in Mammary Tumorigenesis through Multiple Signaling Pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Stacchiotti, S.; Simeone, N.; Lo Vullo, S.; Baldi, G.G.; Brunello, A.; Vincenzi, B.; Palassini, E.; Dagrada, G.; Collini, P.; Morosi, C.; et al. Activity of Sirolimus in Patients with Progressive Epithelioid Hemangioendothelioma: A Case-Series Analysis within the Italian Rare Cancer Network. Cancer 2021, 127, 569–576. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the Mevalonate Pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Subramaniam, A.; Zheng, J.; Yalamanchili, S.; Conley, A.P.; Ratan, R.; Somaiah, N.; Livingston, J.A.; Zarzour, M.A.; Araujo, D.M.; Benjamin, R.S.; et al. Modulation of YAP/ TAZ by Statins to Improve Survival in Epithelioid Hemangioendothelioma (EHE). J. Clin. Oncol. 2020, 38, e23527. [Google Scholar] [CrossRef]
- Che, K.; Pobbati, A.V.; Seavey, C.N.; Fedorov, Y.; Komar, A.A.; Burtscher, A.; Ma, S.; Rubin, B.P. Aurintricarboxylic Acid Is a Canonical Disruptor of the TAZ-TEAD Transcriptional Complex. PLoS ONE 2022, 17, e0266143. [Google Scholar] [CrossRef]
- A Phase I Study of IAG933 in Patients with Advanced Mesothelioma and Other Solid Tumors. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04857372 (accessed on 3 May 2022).
- Oral TEAD Inhibitor Targeting the Hippo Pathway in Subjects with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT05228015 (accessed on 3 May 2022).
- Study to Evaluate VT3989 in Patients with Metastatic Solid Tumors Enriched for Tumors with NF2 Gene Mutations. Available online: https://clinicaltrials.gov/ct2/show/NCT04665206 (accessed on 3 May 2022).
- Ma, S.; Kanai, R.; Pobbati, A.V.; Li, S.; Che, K.; Seavey, C.N.; Hallett, A.; Burtscher, A.; Lamar, J.M.; Rubin, B.P. The TAZ-CAMTA1 Fusion Protein Promotes Tumorigenesis via Connective Tissue Growth Factor and Ras-MAPK Signaling in Epithelioid Hemangioendothelioma. Clin. Cancer Res. 2022, clincanres.0421.2022. [Google Scholar] [CrossRef]
- Trametinib in Treating Patients with Epithelioid Hemangioendothelioma That Is Metastatic, Locally Advanced, or Cannot Be Removed by Surgery. Available online: https://clinicaltrials.gov/ct2/show/NCT03148275 (accessed on 3 May 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seavey, C.N.; Pobbati, A.V.; Rubin, B.P. Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ–CAMTA1 Fusion Driven Sarcoma. Cancers 2022, 14, 2980. https://doi.org/10.3390/cancers14122980
Seavey CN, Pobbati AV, Rubin BP. Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ–CAMTA1 Fusion Driven Sarcoma. Cancers. 2022; 14(12):2980. https://doi.org/10.3390/cancers14122980
Chicago/Turabian StyleSeavey, Caleb N., Ajaybabu V. Pobbati, and Brian P. Rubin. 2022. "Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ–CAMTA1 Fusion Driven Sarcoma" Cancers 14, no. 12: 2980. https://doi.org/10.3390/cancers14122980
APA StyleSeavey, C. N., Pobbati, A. V., & Rubin, B. P. (2022). Unraveling the Biology of Epithelioid Hemangioendothelioma, a TAZ–CAMTA1 Fusion Driven Sarcoma. Cancers, 14(12), 2980. https://doi.org/10.3390/cancers14122980