
A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer


Abstract
Simple Summary
Abstract
1. Introduction
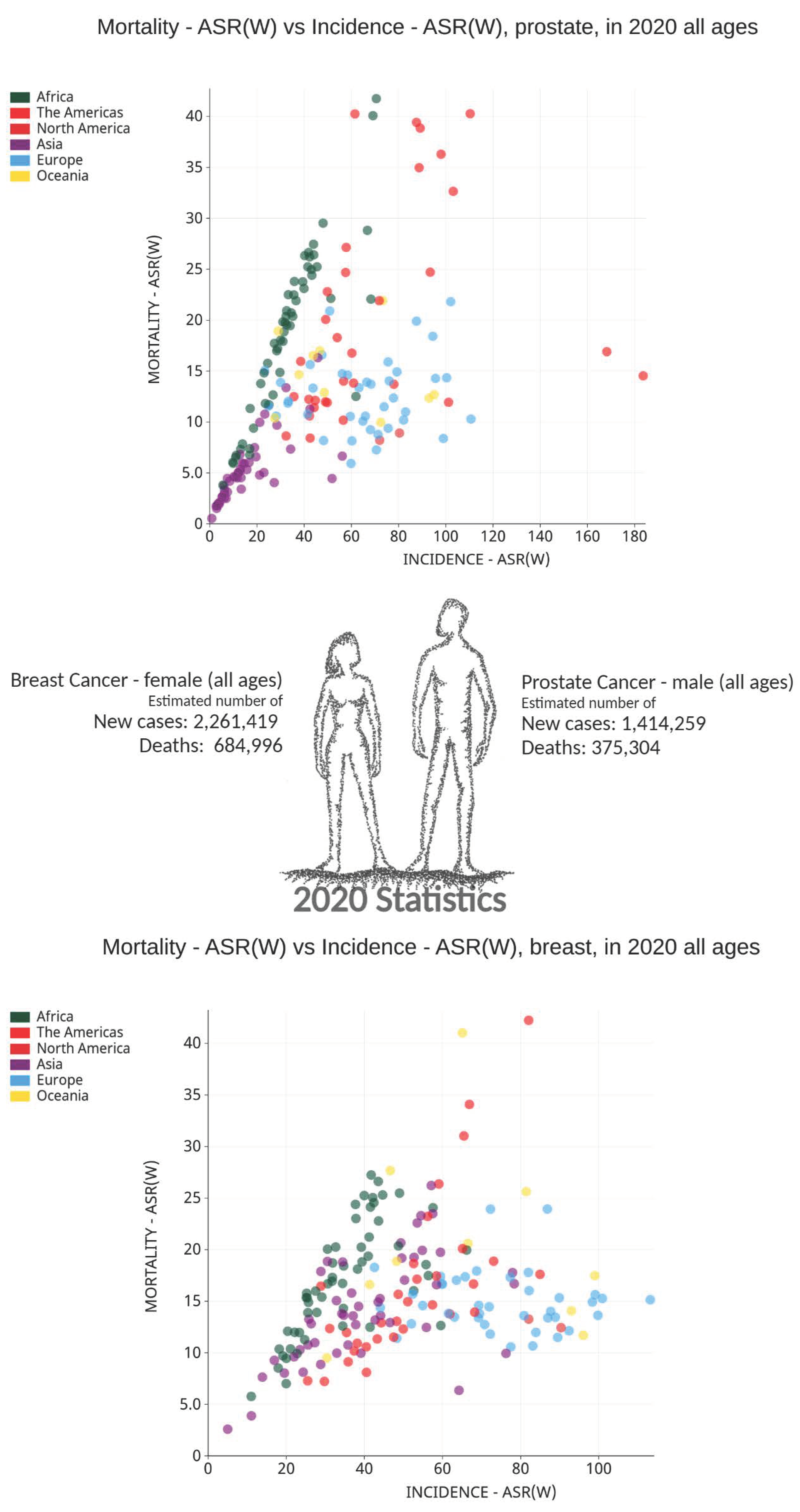
2. Cancer Burden


3. Physiology, Complexity, and Subtypes



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| The Main Type of Neoplasm | Subtype (s) |
|---|---|
| Glandular neoplasms | Acinar adenocarcinoma, Ductal adenocarcinoma, Intraductal carcinoma |
| Basal cell carcinoma | - |
| Urothelial carcinoma | - |
| Squamous neoplasms | Squamous cell carcinoma, Adenosquamous carcinoma |
| Neuroendocrine neoplasms | Small cell neuroendocrine carcinoma, large cell neuroendocrine carcinoma, Adenocarcinoma with neuroendocrine differentiation |
4. Pathology, Risk Factors, and Treatment
| Type | Therapy Type | Situation/Condition Used | Example(s) |
|---|---|---|---|
| Breast | Surgery | Depends on: size of the breast, size, and location of the tumor, spread to lymph nodes (e.g., sentinel lymph node biopsy or axillary lymph node dissection), prior treatments, breast reconstruction, a need to relieve symptoms of advanced disease | Breast-conserving surgery (lumpectomy, quadrantectomy, partial mastectomy, segmental mastectomy), mastectomy, lymph node surgeries, other (e.g., oophorectomy, deep inferior epigastric perforator (DIEP) flaps, noninvasive tissue oximetry, muscle-sparing (MS) free transverse rectus abdominis musculocutaneous (TRAM) flap, robot-assisted surgery) |
| Breast | Radiation therapy | Often after breast-conserving surgery, sometimes after mastectomy, treat metastases (e.g., bones, lungs, or brain), in addition to other treatments | External beam radiation therapy (EBRT), whole breast and tumor bed radiation, accelerated partial breast irradiation (APBI) like intraoperative radiation therapy (IORT), 3D-conformal radiotherapy (3D-CRT), intensity-modulated radiotherapy (IMRT), and brachytherapy (internal radiation—intracavitary brachytherapy, interstitial brachytherapy), lymph node radiation, chest wall radiation, hypo-fractionated radiation therapy |
| Breast | Chemotherapy | After surgery (i.e., adjuvant chemotherapy), prior to surgery (i.e., neoadjuvant chemotherapy), prevent recurrence, advanced (e.g., locally advanced cancers, residual disease) and/or metastatic disease, combination therapy, treatment response | Dose-dense chemotherapy, 5-fluorouracil, cyclophosphamide, platinum agents such as cisplatin and carboplatin, taxanes (e.g., paclitaxel, docetaxel, albumin-bound paclitaxel), anthracyclines (e.g., doxorubicin, epirubicin, pegylated liposomal doxorubicin), vinorelbine, capecitabine, gemcitabine, mitoxantrone, ixabepilone, eribulin |
| Breast | Hormone therapy/endocrine therapy | Treat HR + cancer, lower estrogen levels, often after surgery, as adjuvant therapy, as neoadjuvant therapy, treat metastatic disease in postmenopausal women, treat advanced cancer with no prior treatment with other hormone therapy or upon response failure of other hormone drugs, for ovarian ablation, for combination therapy (e.g., LHRH agonist, CDK 4/6 inhibitor, PI3K inhibitor) | Selective estrogen receptor modulators (SERM) such as tamoxifen and toremifene, anti-estrogen agents or selective estrogen receptor degrader (SERD) like fulvestrant, aromatase inhibitors that interfere with estrogen production (e.g., letrozole, anastrozole, exemestane), luteinizing-hormone releasing hormone (LHRH) agonists (e.g., goserelin, leuprolide), other drugs (progesterone-like drug „megestrol acetate “, high dose of estrogen, androgens) |
| Breast | Targeted therapy, Immunotherapy, & Other | Combination therapy, treat early-stage (aka neoadjuvant chemo) and/or advanced cancer | Monoclonal antibodies (e.g., trastuzumab and pertuzumab), antibody conjugates (e.g., Ado-trastuzumab emtansine, Fam-trastuzumab deruxtecan), PD-L1 immunotherapy (e.g., atezolizumab), HER2 kinase inhibitors (lapatinib, neratinib) mammalian target of rapamycin (mTOR) inhibitors (e.g., everolimus), cyclin-dependent kinase/CDK4-6 inhibitors (e.g., abemaciclib, palbociclib ribociclib), PI3K inhibitors (e.g., alpelisib), PARP inhibitors (e.g., olaparib, talazoparib), regenerative medicine (e.g., stem cell associated therapy/cytotherapy, gene therapy, tissue engineering) |
| Prostate | Active surveillance/observation | Disease risk, disease progression, every 3 to 6 months, based on Gleason score, low PSA level (<10 ng/mL), location and size of the lesion | Watch for any signs and symptoms, routine blood work, prostate-specific antigen (PSA) test, digital rectal exam (DRE), imaging tests (e.g., CT scan, MRI, bone scan) |
| Prostate | Surgery | Lymph node status, PSA level, prostate biopsy results, other factors, treat non-cancerous enlargement of the prostate (i.e., BPH) | Open radical prostatectomy (radical retropubic prostatectomy, radical perineal prostatectomy), laparoscopic prostatectomy (laparoscopic radical prostatectomy, Robotic-assisted laparoscopic radical prostatectomy), Transurethral resection of the prostate (TURP), pelvic lymph node dissection (PLND)/pelvic lymphadenectomy, nerve-sparing radical prostatectomy, orchiectomy |
| Prostate | Cryotherapy/cryosurgery | Recurrent cancer, radiation therapy | Cryoablation (freeze and kill cancer cells or most of the prostate) |
| Prostate | Radiation therapy | Stage of the disease, other factors, metastases to bone | External beam radiation therapy (EBRT), brachytherapy (internal radiation/seed implantation/interstitial radiation therapy), permanent (low dose rate) brachytherapy, temporary (high dose rate) brachytherapy, three-dimensional conformal radiation therapy (3D-CRT), intensity modulated radiation therapy (IMRT), image guided radiation therapy (IGRT), volumetric modulated arc therapy (VMAT), stereotactic body radiation therapy (SBRT), proton beam radiation therapy, systemic radiation therapy, radiopharmaceuticals (strontium-89, samarium-153, radium-223, iodine-125, palladium-103, iridium-192, cesium-137) |
| Prostate | Chemotherapy | Metastases, failure of hormone therapy, castration-resistance, combination therapy, palliative chemotherapy | Docetaxel, cabazitaxel, paclitaxel, vinorelbine, doxorubicin, epirubicin, etoposide, mitoxantrone, estramustine, cisplatin, carboplatin |
| Prostate | Hormone therapy/androgen suppression therapy | Before radiation, initial treatment, metastases, advanced disease, cannot be cured by surgery or radiation, cancer remains or comes back after surgery/radiation, high Gleason score, castration-resistance, high PSA level, combination treatment, combined androgen blockade (CAB), triple androgen blockade (TAB), for orchiectomy | Androgen deprivation therapy (ADT), orchiectomy (i.e., surgical castration), medical castration (i.e., LHRH agonists/LHRH analogs/GnRH agonists) (e.g., leuprolide, triptorelin, goserelin, histrelin), LHRH antagonists (e.g., Degarelix), CYP17i inhibitor (e.g., abiraterone), anti-androgens (i.e., androgen receptor antagonists) (e.g., flutamide, bicalutamide, nilutamide, enzalutamide, apalutamide, darolutamide), other androgen suppressing drugs (e.g., estrogens, ketoconazole), 5-alpha reductase inhibitors (e.g., finasteride, dutasteride) |
| Prostate | Immunotherapy | Advanced prostate cancer, no response to hormone therapy, lynch syndrome | Vaccines (e.g., sipuleucel-T), immune checkpoint inhibitors such as PD-1j inhibitor (e.g., pembrolizumab) |
| Prostate | High intensity focused ultrasound (HIFU) | Recurrent cancer, after radiotherapy | Experimental treatment |
| Prostate | Other | Stage of the disease, other factors, metastases to bone, relieve pain | Bisphosphonates (e.g., zoledronic acid), antibodies (e.g., denosumab), pain medication, steroid drugs (aka corticosteroids) (e.g., prednisone, dexamethasone), regenerative medicine |
5. Future Landscape

6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hassanpour, S.H.; Dehghani, M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Venkatesulu, B.P.; Chandrasekar, V.T.; Girdhar, P.; Advani, P.; Sharma, A.; Elumalai, T.; Hsieh, C.E.; Elghazawy, H.I.; Verma, V.; Krishnan, S. A Systematic Review and Meta-Analysis of Cancer Patients Affected by a Novel Coronavirus. JNCI Cancer Spectr. 2021, 5, pkaa102. [Google Scholar] [CrossRef]
- Che, B.; Zhang, W.; Xu, S.; Yin, J.; He, J.; Huang, T.; Li, W.; Yu, Y.; Tang, K. Prostate Microbiota and Prostate Cancer: A New Trend in Treatment. Front. Oncol. 2021, 11, 805459. [Google Scholar] [CrossRef]
- Brand, J.S.; Colzani, E.; Johansson, A.L.V.; Giesecke, J.; Clements, M.; Bergh, J.; Hall, P.; Czene, K. Infection-related hospitalizations in breast cancer patients: Risk and impact on prognosis. J. Infect. 2016, 72, 650–658. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Cancer-Research-UK. Worldwide Cancer Incidence Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/worldwide-cancer/incidence (accessed on 1 March 2020).
- WHO. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 20 November 2021).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789. [Google Scholar] [CrossRef]
- Ferlay, J.E.M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.fr/today (accessed on 20 November 2021).
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Sauter, E.R. Reliable Biomarkers to Identify New and Recurrent Cancer. Eur. J. Breast Health 2017, 13, 162–167. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Bhattacharyya, G.S.; Doval, D.C.; Desai, C.J.; Chaturvedi, H.; Sharma, S.; Somashekhar, S.P. Overview of Breast Cancer and Implications of Overtreatment of Early-Stage Breast Cancer: An Indian Perspective. JCO Glob. Oncol. 2020, 789–798. [Google Scholar] [CrossRef]
- Bellanger, M.; Zeinomar, N.; Tehranifar, P.; Terry, M.B. Are Global Breast Cancer Incidence and Mortality Patterns Related to Country-Specific Economic Development and Prevention Strategies? J. Glob. Oncol. 2018, 1–16. [Google Scholar] [CrossRef]
- Sun, L.; Legood, R.; Dos-Santos-Silva, I.; Gaiha, S.M.; Sadique, Z. Global treatment costs of breast cancer by stage: A systematic review. PLoS ONE 2018, 13, e0207993. [Google Scholar] [CrossRef]
- Cancer.ca. Breast Cancer. Available online: https://www.cancer.ca/en/cancer-information/cancer-type/breast/breast-cancer/?region=sk (accessed on 30 March 2020).
- Akram, M.; Iqbal, M.; Daniyal, M.; Khan, A.U. Awareness and current knowledge of breast cancer. Biol. Res. 2017, 50, 33. [Google Scholar] [CrossRef]
- Yalaza, M.; İnan, A.; Bozer, M. Male Breast Cancer. J. Breast Health 2016, 12, 1–8. [Google Scholar] [CrossRef]
- Giordano, S.H. Breast Cancer in Men. N. Engl. J. Med. 2018, 378, 2311–2320. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef]
- Coleman, M.P.; Quaresma, M.; Berrino, F.; Lutz, J.M.; De Angelis, R.; Capocaccia, R.; Baili, P.; Rachet, B.; Gatta, G.; Hakulinen, T.; et al. Cancer survival in five continents: A worldwide population-based study (CONCORD). Lancet Oncol. 2008, 9, 730–756. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Lorenzo, G.; Hughes, T.J.R.; Dominguez-Frojan, P.; Reali, A.; Gomez, H. Computer simulations suggest that prostate enlargement due to benign prostatic hyperplasia mechanically impedes prostate cancer growth. Proc. Natl. Acad. Sci. USA 2019, 116, 1152–1161. [Google Scholar] [CrossRef]
- ProstateCancer.ca. Statistics. Available online: https://www.prostatecancer.ca/prostate-cancer/about-prostate-cancer/statistics (accessed on 5 April 2020).
- Cancer.ca. Prostate Cancer Statistics. Available online: https://www.cancer.ca/en/cancer-information/cancer-type/prostate/statistics/?region=ab (accessed on 5 April 2020).
- Cancer.org. Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer.html (accessed on 5 April 2020).
- Cancer.ca. Cancer 101. Available online: https://www.cancer.ca/en/cancer-information/cancer-101/what-is-cancer/?region=sk (accessed on 30 March 2020).
- Zhao, S.G.; Chen, W.S.; Das, R.; Chang, S.L.; Tomlins, S.A.; Chou, J.; Quigley, D.A.; Dang, H.X.; Barnard, T.J.; Mahal, B.A.; et al. Clinical and Genomic Implications of Luminal and Basal Subtypes Across Carcinomas. Clin. Cancer Res. 2019, 25, 2450–2457. [Google Scholar] [CrossRef]
- Hinck, L.; Näthke, I. Changes in cell and tissue organization in cancer of the breast and colon. Curr. Opin. Cell Biol. 2014, 26, 87–95. [Google Scholar] [CrossRef]
- Dimri, G.; Band, H.; Band, V. Mammary epithelial cell transformation: Insights from cell culture and mouse models. Breast Cancer Res. 2005, 7, 171. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Cellular and Molecular Mechanisms Underlying Prostate Cancer Development: Therapeutic Implications. Medicines 2019, 6, 82. [Google Scholar] [CrossRef]
- Tang, D.G. Cancers of the breast and prostate: A stem cell perspective. Endocr.-Relat. Cancer 2015, 22, E9–E11. [Google Scholar] [CrossRef][Green Version]
- Gatti, V.; Bongiorno-Borbone, L.; Fierro, C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Peschiaroli, A. p63 at the Crossroads between Stemness and Metastasis in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 2683. [Google Scholar] [CrossRef]
- Steurer, S.; Riemann, C.; Büscheck, F.; Luebke, A.M.; Kluth, M.; Hube-Magg, C.; Hinsch, A.; Höflmayer, D.; Weidemann, S.; Fraune, C.; et al. p63 expression in human tumors and normal tissues: A tissue microarray study on 10,200 tumors. Biomark. Res. 2021, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Chenlo, M.; Aliyev, E.; Rodrigues, J.S.; Vieiro-Balo, P.; Blanco Freire, M.N.; Cameselle-Teijeiro, J.M.; Alvarez, C.V. Sequential Colocalization of ERa, PR, and AR Hormone Receptors Using Confocal Microscopy Enables New Insights into Normal Breast and Prostate Tissue and Cancers. Cancers 2020, 12, 3591. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, Y.; Rohr, J.; Shang, L.; Ma, J. p63 expression is associated with high histological grade, aberrant p53 expression and TP53 mutation in HER2-positive breast carcinoma. J. Clin. Pathol. 2021, 74, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Risbridger, G.P.; Davis, I.D.; Birrell, S.N.; Tilley, W.D. Breast and prostate cancer: More similar than different. Nat. Rev. Cancer 2010, 10, 205–212. [Google Scholar] [CrossRef] [PubMed]
- McGuire, K.P. Breast Anatomy and Physiology. In Breast Disease: Diagnosis and Pathology; Aydiner, A., İğci, A., Soran, A., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–14. [Google Scholar]
- Martaindale, S.R. Breast MR Imaging: Atlas of Anatomy, Physiology, Pathophysiology, and Breast Imaging Reporting and Data Systems Lexicon. Magn. Reson. Imaging Clin. N. Am. 2018, 26, 179–190. [Google Scholar] [CrossRef]
- Sharma, G.N.; Dave, R.; Sanadya, J.; Sharma, P.; Sharma, K.K. Various types and management of breast cancer: An overview. J. Adv. Pharm. Technol. Res. 2010, 1, 109–126. [Google Scholar]
- Bazira, P.J.; Ellis, H.; Mahadevan, V. Anatomy and physiology of the breast. Surgery 2022, 40, 79–83. [Google Scholar] [CrossRef]
- Suami, H.; Pan, W.-R.; Mann, G.B.; Taylor, G.I. The Lymphatic Anatomy of the Breast and its Implications for Sentinel Lymph Node Biopsy: A Human Cadaver Study. Ann. Surg. Oncol. 2008, 15, 863–871. [Google Scholar] [CrossRef]
- Townsend, C.M.; Beauchamp, R.D.; Evers, B.M.; Mattox, K.L. Sabiston Textbook of Surgery E-Book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Javed, A.; Lteif, A. Development of the human breast. Semin. Plast. Surg. 2013, 27, 5–12. [Google Scholar] [CrossRef]
- Macias, H.; Hinck, L. Mammary gland development. Wiley Interdiscip. Reviews. Dev. Biol. 2012, 1, 533–557. [Google Scholar] [CrossRef]
- Ellis, H.; Mahadevan, V. Anatomy and physiology of the breast. Surgery 2013, 31, 11–14. [Google Scholar] [CrossRef]
- Owens, M.B.; Hill, A.D.; Hopkins, A.M. Ductal barriers in mammary epithelium. Tissue Barriers 2013, 1, e25933. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dzięgelewska, Ż.; Gajewska, M. Stromal-Epithelial Interactions during Mammary Gland Development. In Stromal Cells-Structure, Function, and Therapeutic Implications; Valarmathi, M.T., Ed.; IntechOpen Limited: London, UK, 2018; p. 260. [Google Scholar]
- Pellacani, D.; Tan, S.; Lefort, S.; Eaves, C.J. Transcriptional regulation of normal human mammary cell heterogeneity and its perturbation in breast cancer. EMBO J. 2019, 38, e100330. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.H.; Peaker, M.; Wilde, C.J. Local control of mammary development and function. Rev. Reprod. 1998, 3, 104–112. [Google Scholar] [CrossRef]
- Tharmapalan, P.; Mahendralingam, M.; Berman, H.K.; Khokha, R. Mammary stem cells and progenitors: Targeting the roots of breast cancer for prevention. EMBO J. 2019, 38, e100852. [Google Scholar] [CrossRef]
- Fleming, J.M.; Long, E.L.; Ginsburg, E.; Gerscovich, D.; Meltzer, P.S.; Vonderhaar, B.K. Interlobular and intralobular mammary stroma: Genotype may not reflect phenotype. BMC Cell Biol. 2008, 9, 46. [Google Scholar] [CrossRef]
- Cancer.org. Breast Cancer. Available online: https://www.cancer.org/cancer/breast-cancer.html (accessed on 5 April 2020).
- Cafasso, J.; Bien, M. Fibrocystic Breast Disease. Available online: http://www.healthline.com/health/fibrocystic-breast-disease#overview1 (accessed on 30 January 2019).
- NIH-NCI. Breast Cancer—Patient Version. Available online: https://www.cancer.gov/types/breast/patient/breast-treatment-pdq (accessed on 20 March 2019).
- PDQ Adult Treatment Editorial Board. Breast Cancer Treatment (Adult) (PDQ(R)): Patient Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Wilson, N.; Ironside, A.; Diana, A.; Oikonomidou, O. Lobular Breast Cancer: A Review. Front. Oncol. 2021, 10, 591399. [Google Scholar] [CrossRef]
- MayoClinic.org. Inflammatory Breast Cancer. Available online: https://www.mayoclinic.org/diseases-conditions/inflammatory-breast-cancer/symptoms-causes/syc-20355413 (accessed on 30 March 2020).
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef]
- Leslie, S.W.; Soon-Sutton, T.L.; Sajjad, H.; Siref, L.E. Prostate Cancer; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022; Volume 2022. [Google Scholar]
- Toivanen, R.; Shen, M.M. Prostate organogenesis: Tissue induction, hormonal regulation and cell type specification. Development 2017, 144, 1382–1398. [Google Scholar] [CrossRef]
- Cancer.ca. Prostate Cancer. Available online: https://www.cancer.ca/en/cancer-information/cancer-type/prostate/prostate-cancer/?region=on (accessed on 25 March 2020).
- Inamura, K. Prostatic cancers: Understanding their molecular pathology and the 2016 WHO classification. Oncotarget 2018, 9, 14723–14737. [Google Scholar] [CrossRef]
- Brawer, M.K. Prostatic intraepithelial neoplasia: An overview. Rev. Urol. 2005, 7 (Suppl. S3), S11–S18. [Google Scholar] [PubMed]
- Kim, H.L.; Yang, X.J. Prevalence of high-grade prostatic intraepithelial neoplasia and its relationship to serum prostate specific antigen. Int. Braz. J. Urol. 2002, 28, 413–416. [Google Scholar] [PubMed]
- Godoy, G.; Taneja, S.S. Contemporary clinical management of isolated high-grade prostatic intraepithelial neoplasia. Prostate Cancer Prostatic Dis. 2008, 11, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, J.; Lampert, E.; Schlanger, S.; DePriest, A.D.; Hu, Q.; Gomez, E.C.; Murakam, M.; Glenn, S.T.; Conroy, J.; et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur. Urol. 2017, 71, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Sowalsky, A.G. Molecular correlates of intermediate- and high-risk localized prostate cancer. Urol. Oncol. 2018, 36, 368–374. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Prajapati, A.; Gupta, S.; Mistry, B.; Gupta, S. Prostate Stem Cells in the Development of Benign Prostate Hyperplasia and Prostate Cancer: Emerging Role and Concepts. BioMed Res. Int. 2013, 2013, 107954. [Google Scholar] [CrossRef]
- Barron, D.A.; Rowley, D.R. The reactive stroma microenvironment and prostate cancer progression. Endocr.-Relat. Cancer 2012, 19, R187. [Google Scholar] [CrossRef]
- Salem, O.; Hansen, C.G. The Hippo Pathway in Prostate Cancer. Cells 2019, 8, 370. [Google Scholar] [CrossRef]
- Zhang, B.; Kwon, O.-J.; Henry, G.; Malewska, A.; Wei, X.; Zhang, L.; Brinkley, W.; Zhang, Y.; Castro, P.D.; Titus, M. Non-cell-autonomous regulation of prostate epithelial homeostasis by androgen receptor. Mol. Cell 2016, 63, 976–989. [Google Scholar] [CrossRef]
- Packer, J.R.; Maitland, N.J. The molecular and cellular origin of human prostate cancer. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2016, 1863, 1238–1260. [Google Scholar] [CrossRef] [PubMed]
- Sathianathen, N.J.; Konety, B.R.; Crook, J.; Saad, F.; Lawrentschuk, N. Landmarks in prostate cancer. Nat. Rev. Urol. 2018, 15, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, M.G.; Phillips, S. 6.24—Incontinence (Benign Prostatic Hyperplasia/Prostate Dysfunction). In Comprehensive Medicinal Chemistry II; Taylor, J.B., Triggle, D.J., Eds.; Elsevier: Oxford, UK, 2007; pp. 551–573. [Google Scholar]
- Langhammer, R. Metabolomic Imaging for Human Prostate Cancer Detection Using MR Spectroscopy at 7T. Doctoral Dissertation, Universität Würzburg, Würzburg, Germany, 2018. [Google Scholar]
- Cancer.gov. TCGA’s Study of Prostate Carcinoma. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/studied-cancers/prostate (accessed on 20 April 2020).
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur. Urol. 2016, 70, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Eliyatkın, N.; Yalçın, E.; Zengel, B.; Aktaş, S.; Vardar, E. Molecular Classification of Breast Carcinoma: From Traditional, Old-Fashioned Way to A New Age, and A New Way. J. Breast Health 2015, 11, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef] [PubMed]
- Picornell, A.C.; Echavarria, I.; Alvarez, E.; López-Tarruella, S.; Jerez, Y.; Hoadley, K.; Parker, J.S.; del Monte-Millán, M.; Ramos-Medina, R.; Gayarre, J.; et al. Breast cancer PAM50 signature: Correlation and concordance between RNA-Seq and digital multiplexed gene expression technologies in a triple negative breast cancer series. BMC Genom. 2019, 20, 452. [Google Scholar] [CrossRef]
- Mian, O.Y.; Tendulkar, R.D.; Abazeed, M.E. The evolving role of molecular profiling in prostate cancer: Basal and luminal subtyping transcends tissue of origin. Transl. Cancer Res. 2017, 6, S1441–S1445. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Goncalves, A.; Birnbaum, D. The therapeutic response of ER+/HER2− breast cancers differs according to the molecular Basal or Luminal subtype. NPJ Breast Cancer 2020, 6, 8. [Google Scholar] [CrossRef]
- BreastCancer.org. Molecular Subtypes of Breast Cancer. Available online: https://www.breastcancer.org/symptoms/types/molecular-subtypes (accessed on 3 April 2020).
- Cejalvo, J.M.; Pascual, T.; Fernández-Martínez, A.; Brasó-Maristany, F.; Gomis, R.R.; Perou, C.M.; Muñoz, M.; Prat, A. Clinical implications of the non-luminal intrinsic subtypes in hormone receptor-positive breast cancer. Cancer Treat. Rev. 2018, 67, 63–70. [Google Scholar] [CrossRef]
- Hashmi, A.A.; Hashmi, K.A.; Irfan, M.; Khan, S.M.; Edhi, M.M.; Ali, J.P.; Hashmi, S.K.; Asif, H.; Faridi, N.; Khan, A. Ki67 index in intrinsic breast cancer subtypes and its association with prognostic parameters. BMC Res. Notes 2019, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Mapelli, S.N.; Albino, D.; Mello-Grand, M.; Shinde, D.; Scimeca, M.; Bonfiglio, R.; Bonanno, E.; Chiorino, G.; Garcia-Escudero, R.; Catapano, C.V.; et al. A Novel Prostate Cell Type-Specific Gene Signature to Interrogate Prostate Tumor Differentiation Status and Monitor Therapeutic Response (Running Title: Phenotypic Classification of Prostate Tumors). Cancers 2020, 12, 176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of Luminal and Basal Subtyping of Prostate Cancer With Prognosis and Response to Androgen Deprivation Therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Rider, L.; Cramer, S.D. SPOP the mutation. eLife 2015, 4, e11760. [Google Scholar] [CrossRef]
- Adamo, P.; Ladomery, M.R. The oncogene ERG: A key factor in prostate cancer. Oncogene 2016, 35, 403–414. [Google Scholar] [CrossRef]
- Mani, R.-S. The emerging role of speckle-type POZ protein (SPOP) in cancer development. Drug Discov. Today 2014, 19, 1498–1502. [Google Scholar] [CrossRef]
- Arora, K.; Barbieri, C.E. Molecular Subtypes of Prostate Cancer. Curr. Oncol. Rep. 2018, 20, 58. [Google Scholar] [CrossRef]
- Inno, A.; Bogina, G.; Turazza, M.; Bortesi, L.; Duranti, S.; Massocco, A.; Zamboni, G.; Carbognin, G.; Alongi, F.; Salgarello, M.; et al. Neuroendocrine Carcinoma of the Breast: Current Evidence and Future Perspectives. Oncologist 2016, 21, 28–32. [Google Scholar] [CrossRef]
- Priemer, D.S.; Montironi, R.; Wang, L.; Williamson, S.R.; Lopez-Beltran, A.; Cheng, L. Neuroendocrine Tumors of the Prostate: Emerging Insights from Molecular Data and Updates to the 2016 World Health Organization Classification. Endocr. Pathol. 2016, 27, 123–135. [Google Scholar] [CrossRef]
- Trevisi, E.; La Salvia, A.; Daniele, L.; Brizzi, M.P.; De Rosa, G.; Scagliotti, G.V.; Di Maio, M. Neuroendocrine breast carcinoma: A rare but challenging entity. Med. Oncol. 2020, 37, 70. [Google Scholar] [CrossRef]
- Cancer.org. Second Cancers after Pancreatic Neuroendocrine Tumors. Available online: https://www.cancer.org/cancer/pancreatic-neuroendocrine-tumor/after-treatment/second-cancers.html (accessed on 25 April 2020).
- Brierley, J.; O’Sullivan, B.; Asamura, H.; Byrd, D.; Huang, S.H.; Lee, A.; Piñeros, M.; Mason, M.; Moraes, F.Y.; Rösler, W.; et al. Global Consultation on Cancer Staging: Promoting consistent understanding and use. Nat. Rev. Clin. Oncol. 2019, 16, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Yu, Q.; Zhang, T.; Tie, W.; Li, J.; Zhou, X. Updates on mechanistic insights and targeting of tumour metastasis. J. Cell. Mol. Med. 2020, 24, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Brierley, J.; Gospodarowicz, M.; O’Sullivan, B. The principles of cancer staging. Ecancermedicalscience 2016, 10, ed61. [Google Scholar] [CrossRef] [PubMed]
- Gress, D.M.; Edge, S.B.; Greene, F.L.; Washington, M.K.; Asare, E.A.; Brierley, J.D.; Byrd, D.R.; Compton, C.C.; Jessup, J.M.; Winchester, D.P. Principles of cancer staging. In AJCC Cancer Staging Manual, 8th ed.; Springer: New York, NY, USA, 2017; pp. 3–30. [Google Scholar]
- Fidler, I.J.; Kripke, M.L. The challenge of targeting metastasis. Cancer Metastasis Rev. 2015, 34, 635–641. [Google Scholar] [CrossRef]
- Chen, N.; Zhou, Q. The evolving Gleason grading system. Chin. J. Cancer Res. Chung-Kuo Yen Cheng Yen Chiu 2016, 28, 58–64. [Google Scholar] [CrossRef] [PubMed]
- MayoClinic.org. Breast Cancer. Available online: https://www.mayoclinic.org/diseases-conditions/breast-cancer/symptoms-causes/syc-20352470 (accessed on 30 March 2020).
- Bellanger, M.; Barry, K.; Rana, J.; Regnaux, J.-P. Cost-Effectiveness of Lifestyle-Related Interventions for the Primary Prevention of Breast Cancer: A Rapid Review. Front. Med. 2020, 6, 325. [Google Scholar] [CrossRef]
- BCCancer.bc.ca. Diagnosis & Staging. Available online: http://www.bccancer.bc.ca/health-info/types-of-cancer/breast-cancer#Diagnosis--&--staging (accessed on 30 March 2019).
- PDQ Adult Treatment Editorial Board. Financial Toxicity (Financial Distress) and Cancer Treatment (PDQ®): Patient Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Ekwueme, D.U.; Allaire, B.T.; Parish, W.J.; Thomas, C.C.; Poehler, D.; Guy Jr, G.P.; Aldridge, A.P.; Lahoti, S.R.; Fairley, T.L.; Trogdon, J.G. Estimation of breast cancer incident cases and medical care costs attributable to alcohol consumption among insured women aged<45 years in the US. Am. J. Prev. Med. 2017, 53, S47–S54. [Google Scholar]
- Ataollahi, M.R.; Sharifi, J.; Paknahad, M.R.; Paknahad, A. Breast cancer and associated factors: A review. J. Med. Life 2015, 8, 6–11. [Google Scholar]
- Ding, D.; Lawson, K.D.; Kolbe-Alexander, T.L.; Finkelstein, E.A.; Katzmarzyk, P.T.; Van Mechelen, W.; Pratt, M.; Lancet Physical Activity Series 2 Executive Committee. The economic burden of physical inactivity: A global analysis of major non-communicable diseases. Lancet 2016, 388, 1311–1324. [Google Scholar] [CrossRef]
- Torre, L.A.; Islami, F.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer in Women: Burden and Trends. Cancer Epidemiol. Biomark. Prev. 2017, 26, 444–457. [Google Scholar] [CrossRef]
- Luengo-Fernandez, R.; Leal, J.; Gray, A.; Sullivan, R. Economic burden of cancer across the European Union: A population-based cost analysis. Lancet Oncol. 2013, 14, 1165–1174. [Google Scholar] [CrossRef]
- Pearce, A.; Sharp, L.; Hanly, P.; Barchuk, A.; Bray, F.; de Camargo Cancela, M.; Gupta, P.; Meheus, F.; Qiao, Y.-L.; Sitas, F.; et al. Productivity losses due to premature mortality from cancer in Brazil, Russia, India, China, and South Africa (BRICS): A population-based comparison. Cancer Epidemiol. 2018, 53, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, G.K.; Zhao, X.; Band, H.; Band, V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol. Ther. 2010, 10, 955–960. [Google Scholar] [CrossRef]
- Gao, R.; Davis, A.; McDonald, T.O.; Sei, E.; Shi, X.; Wang, Y.; Tsai, P.-C.; Casasent, A.; Waters, J.; Zhang, H.; et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nature Genet. 2016, 48, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Kader, T.; Hill, P.; Rakha, E.A.; Campbell, I.G.; Gorringe, K.L. Atypical ductal hyperplasia: Update on diagnosis, management, and molecular landscape. Breast Cancer Res. 2018, 20, 39. [Google Scholar] [CrossRef]
- Saotome, M.; Poduval, D.B.; Nair, R.; Cooper, M.; Takaku, M. GATA3 Truncation Mutants Alter EMT Related Gene Expression via Partial Motif Recognition in Luminal Breast Cancer Cells. Front. Genet. 2022, 13, 820532. [Google Scholar] [CrossRef]
- Xiao, W.; Zhang, G.; Chen, B.; Chen, X.; Wen, L.; Lai, J.; Li, X.; Li, M.; Liu, H.; Liu, J.; et al. Characterization of Frequently Mutated Cancer Genes and Tumor Mutation Burden in Chinese Breast Cancer. Front. Oncol. 2021, 11, 1107. [Google Scholar] [CrossRef]
- Annunziato, S.; de Ruiter, J.R.; Henneman, L.; Brambillasca, C.S.; Lutz, C.; Vaillant, F.; Ferrante, F.; Drenth, A.P.; van der Burg, E.; Siteur, B.; et al. Comparative oncogenomics identifies combinations of driver genes and drug targets in BRCA1-mutated breast cancer. Nat. Commun. 2019, 10, 397. [Google Scholar] [CrossRef]
- Lima, Z.S.; Ghadamzadeh, M.; Arashloo, F.T.; Amjad, G.; Ebadi, M.R.; Younesi, L. Recent advances of therapeutic targets based on the molecular signature in breast cancer: Genetic mutations and implications for current treatment paradigms. J. Hematol. Oncol. 2019, 12, 38. [Google Scholar] [CrossRef]
- Kreuzaler, P.; Clarke, M.A.; Brown, E.J.; Wilson, C.H.; Kortlever, R.M.; Piterman, N.; Littlewood, T.; Evan, G.I.; Fisher, J. Heterogeneity of Myc expression in breast cancer exposes pharmacological vulnerabilities revealed through executable mechanistic modeling. Proc. Natl. Acad. Sci. USA 2019, 116, 22399–22408. [Google Scholar] [CrossRef]
- Risom, T.; Wang, X.; Liang, J.; Zhang, X.; Pelz, C.; Campbell, L.G.; Eng, J.; Chin, K.; Farrington, C.; Narla, G.; et al. Deregulating MYC in a model of HER2+ breast cancer mimics human intertumoral heterogeneity. J. Clin. Investig. 2020, 130, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, A.; Lindström, L.S.; Li, J.; Harrell, J.C.; Darai-Ramqvist, E.; Sifakis, E.G.; Foukakis, T.; Perou, C.M.; Czene, K.; Bergh, J.; et al. The long-term prognostic and predictive capacity of cyclin D1 gene amplification in 2305 breast tumours. Breast Cancer Res. 2019, 21, 34. [Google Scholar] [CrossRef] [PubMed]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Ding, Z. Fibroblast growth factor receptors in breast cancer. Tumor Biol. 2017, 39, 1010428317698370. [Google Scholar] [CrossRef]
- Li, K.; Liao, N.; Chen, B.; Zhang, G.; Wang, Y.; Guo, L.; Wei, G.; Jia, M.; Wen, L.; Ren, C.; et al. Genetic mutation profile of Chinese HER2-positive breast cancers and genetic predictors of responses to Neoadjuvant anti-HER2 therapy. Breast Cancer Res. Treat. 2020, 183, 321–332. [Google Scholar] [CrossRef]
- Godoy-Ortiz, A.; Sanchez-Muñoz, A.; Chica Parrado, M.R.; Álvarez, M.; Ribelles, N.; Rueda Dominguez, A.; Alba, E. Deciphering HER2 Breast Cancer Disease: Biological and Clinical Implications. Front. Oncol. 2019, 9, 1124. [Google Scholar] [CrossRef]
- Dey, P.; Rathod, M.; De, A. Targeting stem cells in the realm of drug-resistant breast cancer. Breast Cancer 2019, 11, 115–135. [Google Scholar] [CrossRef]
- Cancer.net. Breast Cancer—Statistics. Available online: https://www.cancer.net/cancer-types/breast-cancer/statistics/2015 (accessed on 30 March 2019).
- Wang, G.; Zhao, D.; Spring, D.J.; DePinho, R.A. Genetics and biology of prostate cancer. Genes Dev. 2018, 32, 1105–1140. [Google Scholar] [CrossRef]
- Shen, M.M.; Abate-Shen, C. Molecular genetics of prostate cancer: New prospects for old challenges. Genes Dev. 2010, 24, 1967–2000. [Google Scholar] [CrossRef]
- Powers, E.; Karachaliou, G.S.; Kao, C.; Harrison, M.R.; Hoimes, C.J.; George, D.J.; Armstrong, A.J.; Zhang, T. Novel therapies are changing treatment paradigms in metastatic prostate cancer. J. Hematol. Oncol. 2020, 13, 144. [Google Scholar] [CrossRef]
- Zander, A.R.; Kröger, N.; Schmoor, C.; Krüger, W.; Möbus, V.; Frickhofen, N.; Metzner, B.; Schultze, W.; Berdel, W.E.; Koenigsmann, M.; et al. High-Dose Chemotherapy With Autologous Hematopoietic Stem-Cell Support Compared With Standard-Dose Chemotherapy in Breast Cancer Patients With 10 or More Positive Lymph Nodes: First Results of a Randomized Trial. J. Clin. Oncol. 2004, 22, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- UNM. Breast Cancer- ASCT. Available online: http://cancer.unm.edu/cancer/cancer-info/cancer-treatment/stem-cell-transplantation/autologous-stem-cell-transplant/cancers-treated-with-asct/breast-cancer-asct/asct-stage-iv-breast-cancer/ (accessed on 20 March 2019).
- Shamash, J.; Jacob, J.; Agrawal, S.; Powles, T.; Mutsvangwa, K.; Wilson, P.; Stebbing, J. Whole Blood Stem Cell Reinfusion and Escalated Dose Melphalan in Castration-Resistant Prostate Cancer: A Phase 1 Study. Clin. Cancer Res. 2012, 18, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- JHU. Side Effects from Breast Cancer Treatment: Johns Hopkins Breast Center. Available online: https://www.hopkinsmedicine.org/kimmel_cancer_center/centers/breast_cancer_program/treatment_and_services/survivorship/side_effects.html (accessed on 30 March 2019).
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Taitt, H.E. Global Trends and Prostate Cancer: A Review of Incidence, Detection, and Mortality as Influenced by Race, Ethnicity, and Geographic Location. Am. J. Men’s Health 2018, 12, 1807–1823. [Google Scholar] [CrossRef] [PubMed]
- Rizos, C.; Papassava, M.; Golias, C.; Charalabopoulos, K. Alcohol consumption and prostate cancer: A mini review. Exp. Oncol. 2010, 32, 66–70. [Google Scholar] [PubMed]
- Denmeade, S.R.; Lin, X.S.; Isaacs, J.T. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate 1996, 28, 251–265. [Google Scholar] [CrossRef]
- Di, Y. Flaxseed Lignan Supplementation as Possible Adjuvant Therapy for Prostate and Breast Cancer. Doctorate Thesis, University of Saskatchewan, Saskatoon, SK, Canada, 2017. [Google Scholar]
- Kozlowski, J.M.; Ellis, W.J.; Grayhack, J.T. Advanced prostatic carcinoma. Early versus late endocrine therapy. Urol. Clin. N. Am. 1991, 18, 15–24. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Garrett-Mayer, E.S.; Yang, Y.C.; de Wit, R.; Tannock, I.F.; Eisenberger, M. A contemporary prognostic nomogram for men with hormone-refractory metastatic prostate cancer: A TAX327 study analysis. Clin. Cancer Res. 2007, 13, 6396–6403. [Google Scholar] [CrossRef]
- Amaral, T.M.; Macedo, D.; Fernandes, I.; Costa, L. Castration-resistant prostate cancer: Mechanisms, targets, and treatment. Prostate Cancer 2012, 2012, 327253. [Google Scholar] [CrossRef]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- Bubendorf, L.; Kononen, J.; Koivisto, P.; Schraml, P.; Moch, H.; Gasser, T.C.; Willi, N.; Mihatsch, M.J.; Sauter, G.; Kallioniemi, O.P. Survey of gene amplifications during prostate cancer progression by high-throughout fluorescence in situ hybridization on tissue microarrays. Cancer Res. 1999, 59, 803–806. [Google Scholar] [PubMed]
- Taplin, M.E.; Bubley, G.J.; Shuster, T.D.; Frantz, M.E.; Spooner, A.E.; Ogata, G.K.; Keer, H.N.; Balk, S.P. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N. Engl. J. Med. 1995, 332, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Veldscholte, J.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Berrevoets, C.; Claassen, E.; van Rooij, H.C.; Trapman, J.; Brinkmann, A.O.; Mulder, E. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem. Biophys. Res. Commun. 1990, 173, 534–540. [Google Scholar] [CrossRef]
- Harris, W.P.; Mostaghel, E.A.; Nelson, P.S.; Montgomery, B. Androgen deprivation therapy: Progress in understanding mechanisms of resistance and optimizing androgen depletion. Nat. Clin. Pract. Urol. 2009, 6, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.A.; Guns, E.S.; Lubik, A.A.; Adomat, H.H.; Hendy, S.C.; Wood, C.A.; Ettinger, S.L.; Gleave, M.E.; Nelson, C.C. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008, 68, 6407–6415. [Google Scholar] [CrossRef]
- Beltran, H.; Romanel, A.; Conteduca, V.; Casiraghi, N.; Sigouros, M.; Franceschini, G.M.; Orlando, F.; Fedrizzi, T.; Ku, S.Y.; Dann, E.; et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J. Clin. Investig. 2020, 130, 1653–1668. [Google Scholar] [CrossRef]
- Lin, Y.; Zhao, X.; Miao, Z.; Ling, Z.; Wei, X.; Pu, J.; Hou, J.; Shen, B. Data-driven translational prostate cancer research: From biomarker discovery to clinical decision. J. Transl. Med. 2020, 18, 119. [Google Scholar] [CrossRef]
- Haas, G.P.; Delongchamps, N.; Brawley, O.W.; Wang, C.Y.; de la Roza, G. The worldwide epidemiology of prostate cancer: Perspectives from autopsy studies. Can. J. Urol. 2008, 15, 3866–3871. [Google Scholar]
- Datta, D.; Aftabuddin, M.; Gupta, D.K.; Raha, S.; Sen, P. Human Prostate Cancer Hallmarks Map. Sci. Rep. 2016, 6, 30691. [Google Scholar] [CrossRef]
- Mazaris, E.; Tsiotras, A. Molecular pathways in prostate cancer. Nephro-Urol. Mon. 2013, 5, 792–800. [Google Scholar] [CrossRef]
- Impact of Prostate Cancer Multifocality on Its Biology and Treatment. J. Endourol. 2010, 24, 799–804. [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Katsogiannou, M.; Ziouziou, H.; Karaki, S.; Andrieu, C.; Henry de Villeneuve, M.; Rocchi, P. The hallmarks of castration-resistant prostate cancers. Cancer Treat. Rev. 2015, 41, 588–597. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Nguyen, T.T.; Tien, N.L.B.; Tran, D.-K.; Jeong, J.-H.; Anh, P.G.; Thanh, V.V.; Truong, D.T.; Dinh, T.C. Recent Progress of Stem Cell Therapy in Cancer Treatment: Molecular Mechanisms and Potential Applications. Cells 2020, 9, 563. [Google Scholar] [CrossRef]
- Segaliny, A.I.; Cheng, J.L.; Farhoodi, H.P.; Toledano, M.; Yu, C.C.; Tierra, B.; Hildebrand, L.; Liu, L.; Liao, M.J.; Cho, J.; et al. Combinatorial targeting of cancer bone metastasis using mRNA engineered stem cells. eBioMedicine 2019, 45, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.; Akhondi, M.; Mousavi, N.S.; Haghparast, N.; Ghodsi, A.; Baharvand, H.; Ebrahimi, M.; Hassani, S.-N. Pluripotent Stem Cells: Cancer Study, Therapy, and Vaccination. Stem Cell Rev. Rep. 2021, 17, 1975–1992. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Cancer.ca. Treatments for Breast Cancer. Available online: https://www.cancer.ca/en/cancer-information/cancer-type/breast/treatment/?region=bc (accessed on 5 April 2020).
- Cancer.ca. Treatments for Prostate Cancer. Available online: https://www.cancer.ca/en/cancer-information/cancer-type/prostate/treatment/?region=ab (accessed on 25 April 2020).
- Cancer.org. Treating Breast Cancer. Available online: https://www.cancer.org/cancer/breast-cancer/treatment.html (accessed on 5 April 2020).
- Cancer.org. Treating Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/treating.html (accessed on 5 April 2020).
- Litwin, M.S.; Tan, H.-J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Sumanasuriya, S.; De Bono, J. Treatment of Advanced Prostate Cancer-A Review of Current Therapies and Future Promise. Cold Spring Harb. Perspect. Med. 2018, 8, a030635. [Google Scholar] [CrossRef]
- Chen, K.; Beeraka, N.M.; Sinelnikov, M.Y.; Zhang, J.; Song, D.; Gu, Y.; Li, J.; Reshetov, I.V.; Startseva, O.I.; Liu, J.; et al. Patient Management Strategies in Perioperative, Intraoperative, and Postoperative Period in Breast Reconstruction With DIEP-Flap: Clinical Recommendations. Front. Surg. 2022, 9. [Google Scholar] [CrossRef]
- Lindelauf, A.; Vranken, N.P.A.; Rutjens, V.G.H.; Schols, R.M.; Heijmans, J.H.; Weerwind, P.W.; van der Hulst, R. Economic Analysis of Noninvasive Tissue Oximetry for Postoperative Monitoring of Deep Inferior Epigastric Perforator Flap Breast Reconstruction: A Review. Surg. Innov. 2020, 27, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.P.; Koupparis, A.J. Expanding the indications of robotic surgery in urology: A systematic review of the literature. Arab J. Urol. 2018, 16, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Donnely, E.; Griffin, M.F.; Butler, P.E. Robotic Surgery: A Novel Approach for Breast Surgery and Reconstruction. Plast. Reconstr. Surg. Glob. Open 2020, 8, e2578. [Google Scholar] [CrossRef] [PubMed]
- Bishop, S.N.; Selber, J.C. Minimally invasive robotic breast reconstruction surgery. Gland. Surg. 2021, 10, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, V.; Beheshtizadeh, N.; Gharibshahian, M.; Sabouri, L.; Varzandeh, M.; Rezaei, N. Recent advances in regenerative medicine strategies for cancer treatment. Biomed. Pharmacother. 2021, 141, 111875. [Google Scholar] [CrossRef]
- Sadanandan, N.; Shear, A.; Brooks, B.; Saft, M.; Cabantan, D.A.G.; Kingsbury, C.; Zhang, H.; Anthony, S.; Wang, Z.-J.; Salazar, F.E.; et al. Treating Metastatic Brain Cancers With Stem Cells. Front. Mol. Neurosci. 2021, 14, 749716. [Google Scholar] [CrossRef]
- Christodoulou, I.; Goulielmaki, M.; Devetzi, M.; Panagiotidis, M.; Koliakos, G.; Zoumpourlis, V. Mesenchymal stem cells in preclinical cancer cytotherapy: A systematic review. Stem Cell Res. Ther. 2018, 9, 336. [Google Scholar] [CrossRef]
- PDQ Adult Treatment Editorial Board. Prostate Cancer Treatment (PDQ®): Patient Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: Global cancer incidence and the role of prevention 2020–2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef]
- Brenner, D.R.; Weir, H.K.; Demers, A.A.; Ellison, L.F.; Louzado, C.; Shaw, A.; Turner, D.; Woods, R.R.; Smith, L.M. Projected estimates of cancer in Canada in 2020. Can. Med. Assoc. J. 2020, 192, E199–E205. [Google Scholar] [CrossRef]
- Thurston, D.E.; Carter, S. Immuno-oncology agents for cancer therapy. Pharm. J. 2020, 304. [Google Scholar] [CrossRef]
- Curtius, K.; Wright, N.A.; Graham, T.A. An evolutionary perspective on field cancerization. Nat. Rev. Cancer 2018, 18, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. Multifocal epithelial tumors and field cancerization: Stroma as a primary determinant. J. Clin. Investig. 2014, 124, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Cancer.org. The History of Cancer: Cancer Treatment-Surgery. Available online: https://www.cancer.org/cancer/cancer-basics/history-of-cancer/cancer-treatment-surgery.html (accessed on 30 June 2020).
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Cabanos, H.F.; Hata, A.N. Emerging Insights into Targeted Therapy-Tolerant Persister Cells in Cancer. Cancers 2021, 13, 2666. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef] [PubMed]
- De Silva, F.; Alcorn, J. The Elusive Road Towards Effective Cancer Prevention and Treatment, 1st ed.; Taylor & Francis, CRC Press: Boca Raton, FL, USA, 2022. [Google Scholar]
- Beaver, J.A.; Hazarika, M.; Mulkey, F.; Mushti, S.; Chen, H.; He, K.; Sridhara, R.; Goldberg, K.B.; Chuk, M.K.; Chi, D.-C.; et al. Patients with melanoma treated with an anti-PD-1 antibody beyond RECIST progression: A US Food and Drug Administration pooled analysis. Lancet Oncol. 2018, 19, 229–239. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef]
- Zhang, Z.; Qin, S.; Chen, Y.; Zhou, L.; Yang, M.; Tang, Y.; Zuo, J.; Zhang, J.; Mizokami, A.; Nice, E.C.; et al. Inhibition of NPC1L1 disrupts adaptive responses of drug-tolerant persister cells to chemotherapy. EMBO Mol. Med. 2022, 14, e14903. [Google Scholar] [CrossRef]
- Fu, L.; Chen, Z.-S. Redox signaling-governed drug-tolerant persister cancer cell: A key spark of treatment failure. Signal Transduct. Target. Ther. 2022, 7, 89. [Google Scholar] [CrossRef]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Schell, T.D.; Amin, S.; Robertson, G.P. Drug-tolerant persister cells in cancer therapy resistance. Cancer Res. 2022; Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mikubo, M.; Inoue, Y.; Liu, G.; Tsao, M.-S. Mechanism of Drug Tolerant Persister Cancer Cells: The Landscape and Clinical Implication for Therapy. J. Thorac. Oncol. 2021, 16, 1798–1809. [Google Scholar] [CrossRef]
- Qin, S.; Jiang, J.; Lu, Y.; Nice, E.C.; Huang, C.; Zhang, J.; He, W. Emerging role of tumor cell plasticity in modifying therapeutic response. Signal Transduct. Target. Ther. 2020, 5, 228. [Google Scholar] [CrossRef]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hütter, J.-C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- De Conti, G.; Dias, M.H.; Bernards, R. Fighting Drug Resistance through the Targeting of Drug-Tolerant Persister Cells. Cancers 2021, 13, 1118. [Google Scholar] [CrossRef]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; KaiWai Cheung, T.; Kim, H.J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237.e13. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef]
- Huang, W.; Sun, X.; Li, Y.; He, Z.; Li, L.; Deng, Z.; Huang, X.; Han, S.; Zhang, T.; Zhong, J.; et al. Discovery and Identification of Small Molecules as Methuosis Inducers with in Vivo Antitumor Activities. J. Med. Chem. 2018, 61, 5424–5434. [Google Scholar] [CrossRef]
- Song, S.; Zhang, Y.; Ding, T.; Ji, N.; Zhao, H. The Dual Role of Macropinocytosis in Cancers: Promoting Growth and Inducing Methuosis to Participate in Anticancer Therapies as Targets. Front. Oncol. 2021, 10, 570108. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Orlikova, B.; Dicato, M.; Diederich, M. 1000 Ways to die: Natural compounds modulate non-canonical cell death pathways in cancer cells. Phytochem. Rev. 2014, 13, 277–293. [Google Scholar] [CrossRef]
- Jan, R.; Chaudhry, G.-E.S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef] [PubMed]
- Missiroli, S.; Perrone, M.; Genovese, I.; Pinton, P.; Giorgi, C. Cancer metabolism and mitochondria: Finding novel mechanisms to fight tumours. eBioMedicine 2020, 59. [Google Scholar] [CrossRef]
- Rapoport, B.L.; Anderson, R. Realizing the Clinical Potential of Immunogenic Cell Death in Cancer Chemotherapy and Radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef]
- Fulda, S.; Kögel, D. Cell death by autophagy: Emerging molecular mechanisms and implications for cancer therapy. Oncogene 2015, 34, 5105–5113. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Ritter, M.; Bresgen, N.; Kerschbaum, H.H. From Pinocytosis to Methuosis—Fluid Consumption as a Risk Factor for Cell Death. Front. Cell Dev. Biol. 2021, 9, 651982. [Google Scholar] [CrossRef]
- De Silva, S.F.; Alcorn, J. Flaxseed Lignans as Important Dietary Polyphenols for Cancer Prevention and Treatment: Chemistry, Pharmacokinetics, and Molecular Targets. Pharmaceuticals 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Fayazzadeh, E.; Li, X.; Koirala, N.; Wadera, A.; Lang, M.; Zernic, M.; Panick, C.; Nesbitt, P.; McLennan, G. Quiescent hepatic stellate cells induce toxicity and sensitivity to doxorubicin in cancer cells through a caspase-independent cell death pathway: Central role of apoptosis-inducing factor. J. Cell. Physiol. 2020, 235, 6167–6182. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hua, P.; He, C.; Chen, M. Non-apoptotic cell death-based cancer therapy: Molecular mechanism, pharmacological modulators, and nanomedicine. Acta Pharm. Sin. B, 2022; in press. [Google Scholar] [CrossRef]
- Butler, M.; van der Meer, L.T.; van Leeuwen, F.N. Amino Acid Depletion Therapies: Starving Cancer Cells to Death. Trends Endocrinol. Metab. 2021, 32, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Díaz-García, D.; Cenariu, D.; Pérez, Y.; Cruz, P.; del Hierro, I.; Prashar, S.; Fischer-Fodor, E.; Gómez-Ruiz, S. Modulation of the mechanism of apoptosis in cancer cell lines by treatment with silica-based nanostructured materials functionalized with different metallodrugs. Dalton Trans. 2018, 47, 12284–12299. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.-Y.; Lin, L.-T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- N’Guessan, K.F.; Patel, P.H.; Qi, X. SapC-DOPS – a Phosphatidylserine-targeted Nanovesicle for selective Cancer therapy. Cell Commun. Signal. 2020, 18, 6. [Google Scholar] [CrossRef]
- Jia, S.; Ge, S.; Fan, X.; Leong, K.W.; Ruan, J. Promoting reactive oxygen species generation: A key strategy in nanosensitizer-mediated radiotherapy. Nanomedicine 2021, 16, 759–778. [Google Scholar] [CrossRef]
- András Szarka, O.K.; Lőrincz, T.; Bánhegyi, G. Vitamin C and Cell Death. Antioxid. Redox Signal. 2021, 34, 831–844. [Google Scholar] [CrossRef]
- Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880–26913. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Mbah, N.E.; Overmeyer, J.H.; Sarver, J.G.; George, S.; Trabbic, C.J.; Erhardt, P.W.; Maltese, W.A. The JNK signaling pathway plays a key role in methuosis (non-apoptotic cell death) induced by MOMIPP in glioblastoma. BMC Cancer 2019, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Ince, T.A. Normal cell phenotypes of breast epithelial cells provide the foundation of a breast cancer taxonomy. Expert Rev. Anticancer Ther. 2014, 14, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Laconi, E.; Marongiu, F.; DeGregori, J. Cancer as a disease of old age: Changing mutational and microenvironmental landscapes. Br. J. Cancer 2020, 122, 943–952. [Google Scholar] [CrossRef]
- Cao, Y. Tumorigenesis as a process of gradual loss of original cell identity and gain of properties of neural precursor/progenitor cells. Cell Biosci. 2017, 7, 61. [Google Scholar] [CrossRef]
- Mitrus, I.; Bryndza, E.; Sochanik, A.; Szala, S. Evolving models of tumor origin and progression. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2012, 33, 911–917. [Google Scholar] [CrossRef][Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Silva, F.; Alcorn, J. A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer. Cancers 2022, 14, 2954. https://doi.org/10.3390/cancers14122954
De Silva F, Alcorn J. A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer. Cancers. 2022; 14(12):2954. https://doi.org/10.3390/cancers14122954
Chicago/Turabian StyleDe Silva, Franklyn, and Jane Alcorn. 2022. "A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer" Cancers 14, no. 12: 2954. https://doi.org/10.3390/cancers14122954
APA StyleDe Silva, F., & Alcorn, J. (2022). A Tale of Two Cancers: A Current Concise Overview of Breast and Prostate Cancer. Cancers, 14(12), 2954. https://doi.org/10.3390/cancers14122954